

Comparative Binding Ability of Human Monoclonal Antibodies against Omicron Variants of SARS-CoV-2: An In Silico Investigation


Abstract

1. Introduction
2. Materials and Methods
2.1. Data Mining
2.2. Homology Modelling and Validation
2.3. Molecular Docking and Determination of Biophysical Interactions
2.4. Determination of Binding Affinity
2.5. Analysis of Conformational Stability, Molecular Motion, and Dynamics
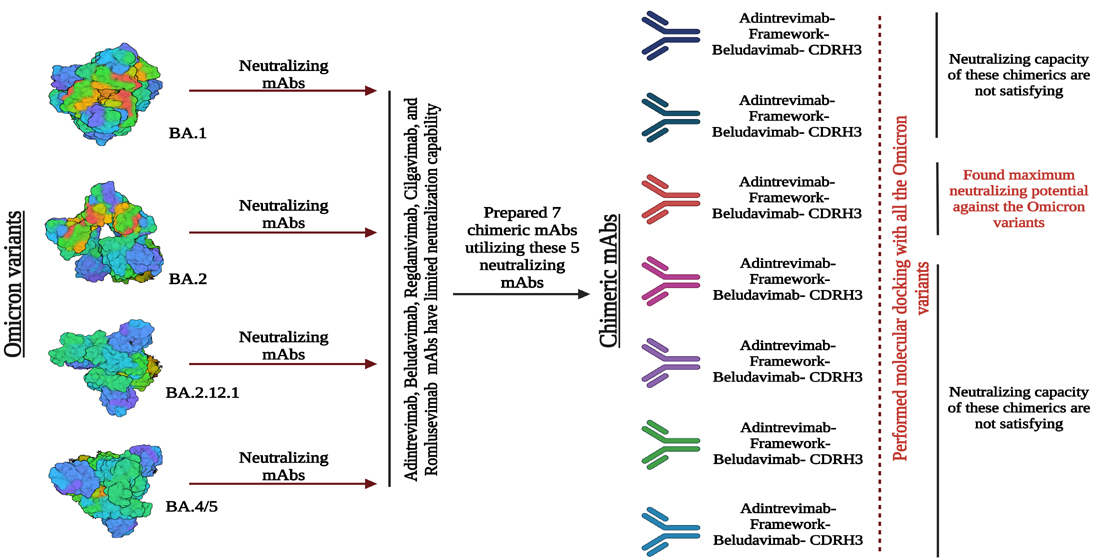
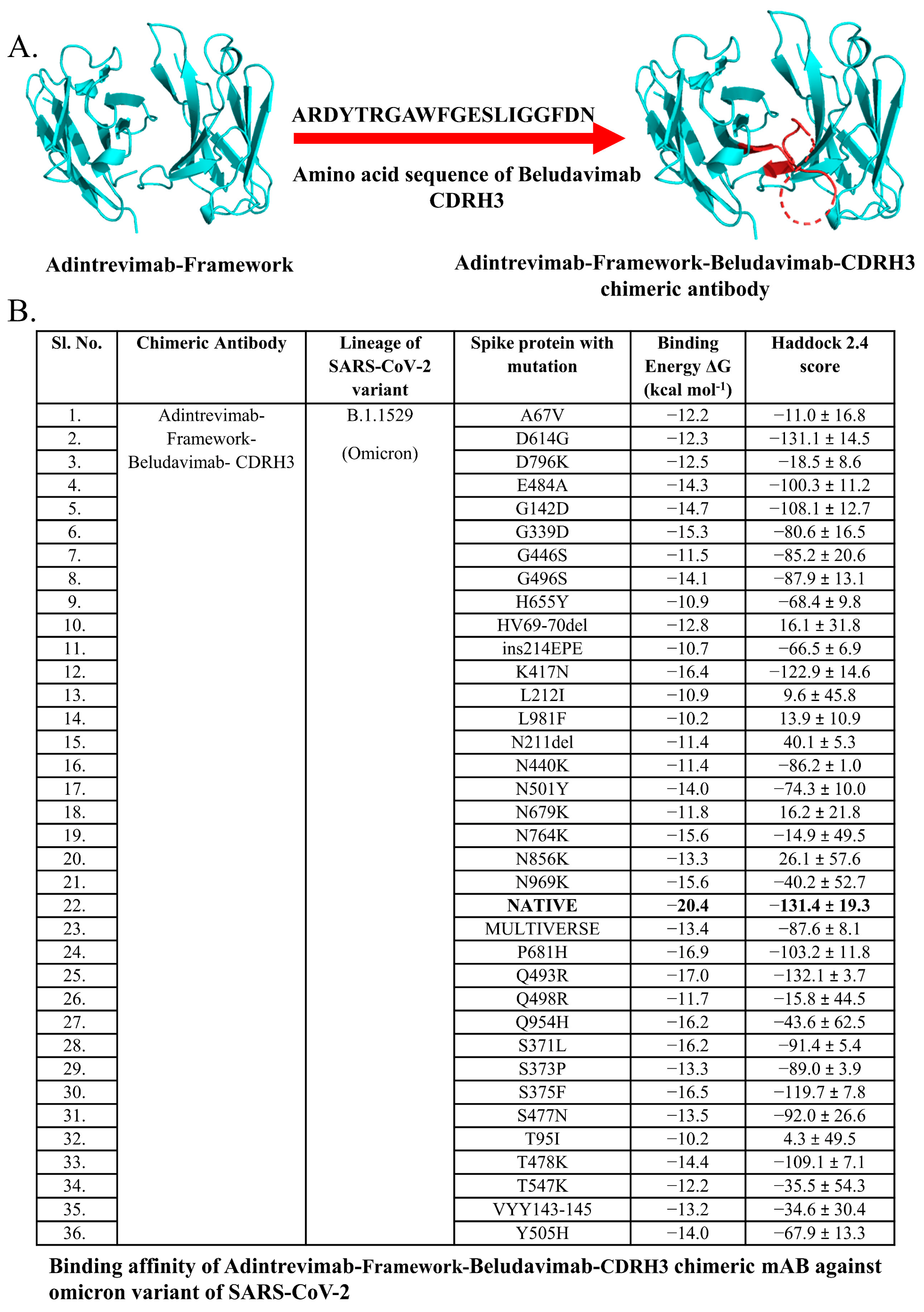
2.6. Designing and Characterization of Chimeric Antibody
3. Results
3.1. Homology Modelling
3.2. Screening of the Potential mAbs
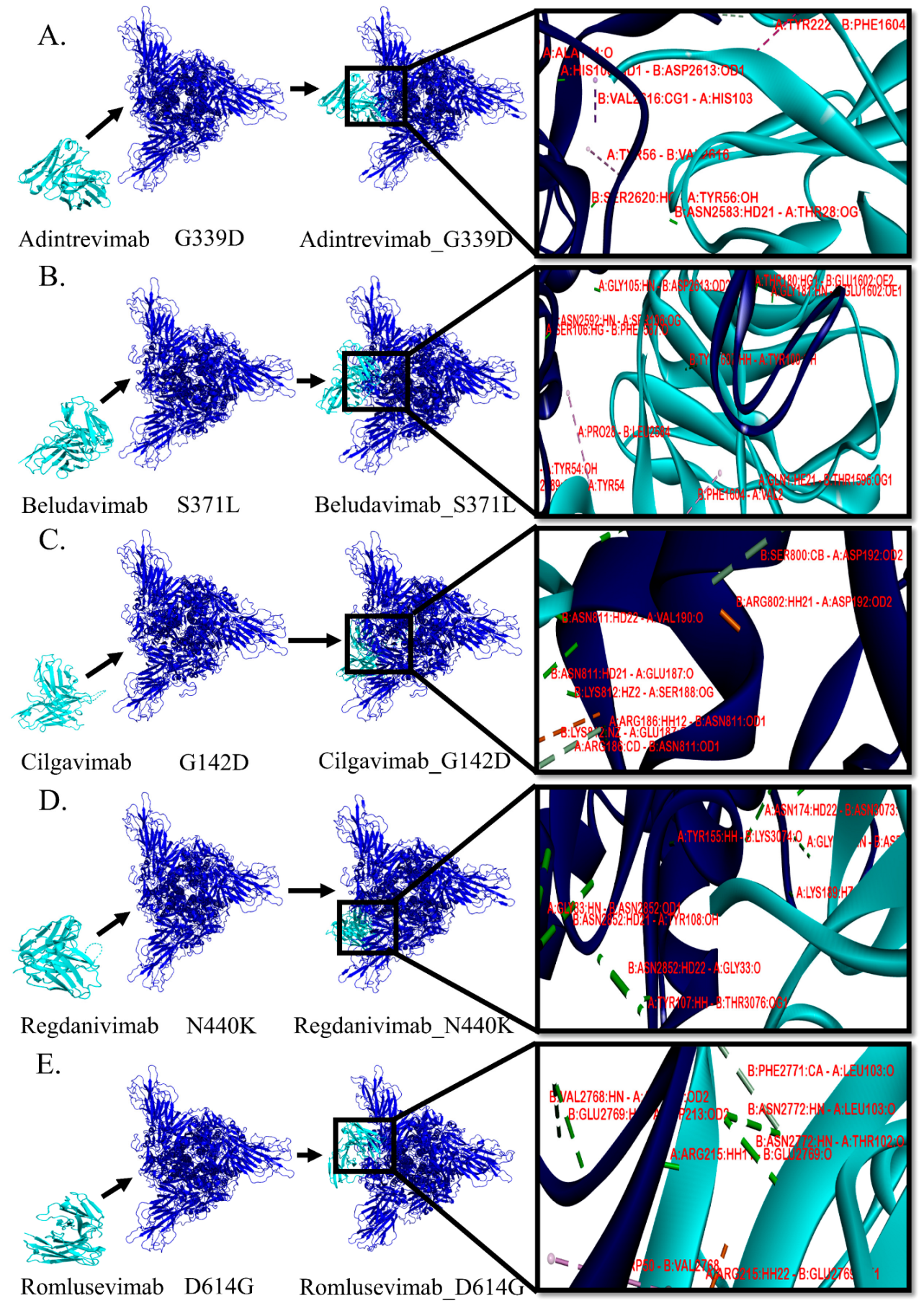
3.3. Protein–Protein Interactions between mAb and Mutant Spike Protein(s) of Omicron
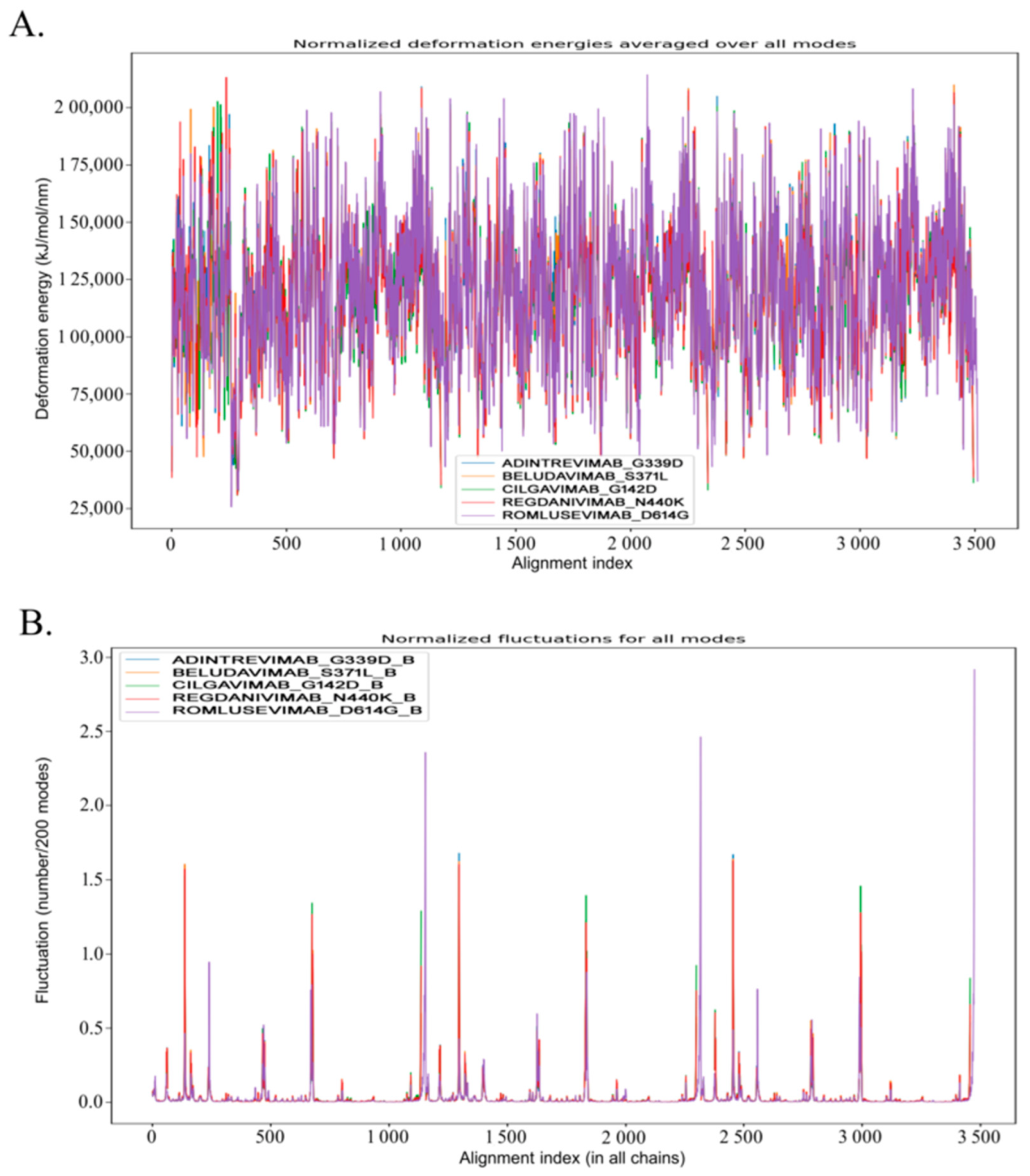
3.4. Molecular Dynamic Study to Verify the Stability
3.5. Chimeric mAbs and Their Potentiality as Immunotherapeutics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patra, R.; Chandra Das, N.; Mukherjee, S. Targeting human TLRs to combat COVID-19: A solution? J. Med. Virol. 2021, 93, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Gupta, P.S.S.; Panda, S.K.; Rana, M.K.; Mukherjee, S. Designing AbhiSCoVac-A single potential vaccine for all ‘corona culprits’: Immunoinformatics and immune simulation approaches. J. Mol. Liq. 2022, 351, 118633. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-C.; Shih, T.-P.; Ko, W.-C.; Tang, H.-J.; Hsueh, P.-R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Tiwari, S.; Deb, M.K.; Marty, J.L. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): A global pandemic and treatment strategies. Int. J. Antimicrob. Agents 2020, 56, 106054. [Google Scholar] [CrossRef]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020, 91, 157–160. [Google Scholar] [CrossRef]
- Kumar, S.; Karuppanan, K.; Subramaniam, G. Omicron (BA.1) and sub-variants (BA.1.1, BA.2, and BA.3) of SARS-CoV-2 spike infectivity and pathogenicity: A comparative sequence and structural-based computational assessment. J. Med. Virol. 2022, 94, 4780–4791. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Gilby, N.B.; Wei, G.-W. Omicron Variant (B.1.1.529): Infectivity, Vaccine Breakthrough, and Antibody Resistance. J. Chem. Inf. Model. 2022, 24, 412–422. [Google Scholar] [CrossRef]
- Halfmann, P.J.; Iida, S.; Iwatsuki-Horimoto, K.; Maemura, T.; Kiso, M.; Scheaffer, S.M.; Darling, T.L.; Joshi, A.; Loeber, S.; Singh, G.; et al. SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature 2022, 603, 687–692. [Google Scholar] [CrossRef]
- Guo, Y.; Han, J.; Zhang, Y.; He, J.; Yu, W.; Zhang, X.; Wu, J.; Zhang, S.; Kong, Y.; Guo, Y.; et al. SARS-CoV-2 Omicron Variant: Epidemiological Features, Biological Characteristics, and Clinical Significance. Front. Immunol. 2022, 13, 877101. [Google Scholar] [CrossRef]
- Medigeshi, G.R.; Batra, G.; Murugesan, D.R.; Thiruvengadam, R.; Chattopadhyay, S.; Das, B.; Gosain, M.; Ayushi; Singh, J.; Anbalagan, A.; et al. Sub-optimal neutralisation of omicron (B.1.1.529) variant by antibodies induced by vaccine alone or SARS-CoV-2 Infection plus vaccine (hybrid immunity) post 6-months. eBioMedicine 2022, 78, 103938. [Google Scholar] [CrossRef]
- Fan, Y.; Li, X.; Zhang, L.; Wan, S.; Zhang, L.; Zhou, F. SARS-CoV-2 Omicron variant: Recent progress and future perspectives. Signal Transduct. Target. Ther. 2022, 7, 141. [Google Scholar] [CrossRef]
- Vitiello, A.; Ferrara, F.; Auti, A.M.; Di Domenico, M.; Boccellino, M. Advances in the Omicron variant development. J. Intern. Med. 2022, 292, 81–90. [Google Scholar] [CrossRef]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef]
- Caputo, E.; Mandrich, L. Structural and Phylogenetic Analysis of SARS-CoV-2 Spike Glycoprotein from the Most Widespread Variants. Life 2022, 12, 1245. [Google Scholar] [CrossRef]
- Wu, C.; Yin, W.; Jiang, Y.; Xu, H.E. Structure genomics of SARS-CoV-2 and its Omicron variant: Drug design templates for COVID-19. Acta Pharmacol. Sin. 2022, 43, 3021–3033. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Tian, M.; Wu, N.; Yang, X.; Qi, J.; Ren, W.; Li, F.; Bian, H. SARS-CoV-2 and Emerging Variants: Unmasking Structure, Function, Infection, and Immune Escape Mechanisms. Front. Cell. Infect. Microbiol. 2022, 12, 869832. [Google Scholar] [CrossRef]
- Das, B.S.; Das, N.C.; Swain, S.S.; Mukherjee, S.; Bhattacharya, D. Andrographolide induces anti-SARS-CoV-2 response through host-directed mechanism: An in silico study. Future Virol. 2022, 17, 651–673. [Google Scholar] [CrossRef]
- Choudhury, A.; Chandra Das, N.; Patra, R.; Mukherjee, S. In silico analyses on the comparative sensing of SARS-CoV-2 mRNA by the intracellular TLRs of human. J. Med. Virol. 2021, 93, 2476–2486. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Hakami, A.R. Targeting the RBD of Omicron Variant (B.1.1.529) with Medicinal Phytocompounds to Abrogate the Binding of Spike Glycoprotein with the hACE2 Using Computational Molecular Search and Simulation Approach. Biology 2022, 11, 258. [Google Scholar] [CrossRef]
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.-D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Laffeber, C.; de Koning, K.; Kanaar, R.; Lebbink, J.H.G. Experimental Evidence for Enhanced Receptor Binding by Rapidly Spreading SARS-CoV-2 Variants. J. Mol. Biol. 2021, 433, 167058. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Mannar, D.; Srivastava, S.S.; Berezuk, A.M.; Demers, J.-P.; Saville, J.W.; Leopold, K.; Li, W.; Dimitrov, D.S.; Tuttle, K.S.; et al. Cryo-electron microscopy structures of the N501Y SARS-CoV-2 spike protein in complex with ACE2 and 2 potent neutralizing antibodies. PLoS Biol. 2021, 19, e3001237. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Quarleri, J.; Galvan, V.; Delpino, M.V. Omicron variant of the SARS-CoV-2: A quest to define the consequences of its high mutational load. GeroScience 2022, 44, 53–56. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e13. [Google Scholar] [CrossRef]
- Ching, W.-Y.; Adhikari, P.; Jawad, B.; Podgornik, R. Effect of Delta and Omicron Mutations on the RBD-SD1 Domain of the Spike Protein in SARS-CoV-2 and the Omicron Mutations on RBD-ACE2 Interface Complex. Int. J. Mol. Sci. 2022, 23, 10091. [Google Scholar] [CrossRef]
- Das, N.C.; Chakraborty, P.; Bayry, J.; Mukherjee, S. In silico analyses on the comparative potential of therapeutic human monoclonal antibodies against newly emerged SARS-CoV-2 variants bearing mutant spike protein. Front. Immunol. 2022, 12, 782506. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.; Schwede, T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009, 4, 1–13. [Google Scholar] [CrossRef]
- Dunbar, J.; Krawczyk, K.; Leem, J.; Marks, C.; Nowak, J.; Regep, C.; Georges, G.; Kelm, S.; Popovic, B.; Deane, C.M. SAbPred: A structure-based antibody prediction server. Nucleic Acids Res. 2016, 44, W474–W478. [Google Scholar] [CrossRef]
- Ruffolo, J.A.; Sulam, J.; Gray, J.J. Antibody structure prediction using interpretable deep learning. Patterns 2022, 3, 100406. [Google Scholar] [CrossRef]
- Robinson, S.A.; Raybould, M.I.J.; Schneider, C.; Wong, W.K.; Marks, C.; Deane, C.M. Epitope profiling using computational structural modelling demonstrated on coronavirus-binding antibodies. PLOS Comput. Biol. 2021, 17, e1009675. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Beg, M.A.; Shivangi; Thakur, S.C.; Meena, L.S. Structural Prediction and Mutational Analysis of Rv3906c Gene of Mycobacterium tuberculosis H37Rv to Determine Its Essentiality in Survival. Adv. Bioinform. 2018, 2018, 6152014. [Google Scholar] [CrossRef]
- Nadaradjane, A.A.; Guerois, R.; Andreani, J. Protein-Protein Docking Using Evolutionary Information. In Protein Complex Assembly: Methods and Protocols; Marsh, J.A., Ed.; Springer: New York, NY, USA, 2018; pp. 429–447. ISBN 978-1-4939-7759-8. [Google Scholar]
- Kangueane, P.; Nilofer, C. Protein-Protein Docking: Methods and Tools. In Protein-Protein and Domain-Domain Interactions; Kangueane, P., Nilofer, C., Eds.; Springer: Singapore, 2018; pp. 161–168. ISBN 978-981-10-7347-2. [Google Scholar]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; Van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Charitou, V.; van Keulen, S.C.; Bonvin, A.M.J.J. Cyclization and Docking Protocol for Cyclic Peptide–Protein Modeling Using HADDOCK2.4. J. Chem. Theory Comput. 2022, 18, 4027–4040. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Siebenmorgen, T.; Zacharias, M. Computational prediction of protein–protein binding affinities. WIREs Comput. Mol. Sci. 2020, 10, e1448. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef]
- Tiwari, S.P.; Fuglebakk, E.; Hollup, S.M.; Skjærven, L.; Cragnolini, T.; Grindhaug, S.H.; Tekle, K.M.; Reuter, N. WEBnm@ v2.0: Web server and services for comparing protein flexibility. BMC Bioinform. 2014, 15, 427. [Google Scholar] [CrossRef]
- Raybould, M.I.J.; Marks, C.; Krawczyk, K.; Taddese, B.; Nowak, J.; Lewis, A.P.; Bujotzek, A.; Shi, J.; Deane, C.M. Five computational developability guidelines for therapeutic antibody profiling. Proc. Natl. Acad. Sci. USA 2019, 116, 4025–4030. [Google Scholar] [CrossRef]
- Choudhury, A.; Das, N.C.; Patra, R.; Bhattacharya, M.; Ghosh, P.; Patra, B.C.; Mukherjee, S. Exploring the binding efficacy of ivermectin against the key proteins of SARS-CoV-2 pathogenesis: An in silico approach. Future Virol. 2021, 16, 277–291. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, G.; Mukherjee, S. Chemotherapy vs. Immunotherapy in combating nCOVID19: An update. Hum. Immunol. 2021, 82, 646–658. [Google Scholar] [CrossRef]
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; de la Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef]
- Hwang, Y.-C.; Lu, R.-M.; Su, S.-C.; Chiang, P.-Y.; Ko, S.-H.; Ke, F.-Y.; Liang, K.-H.; Hsieh, T.-Y.; Wu, H.-C. Monoclonal antibodies for COVID-19 therapy and SARS-CoV-2 detection. J. Biomed. Sci. 2022, 29, 1. [Google Scholar] [CrossRef]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug repurposing approach to fight COVID-19. Pharmacol. Rep. 2020, 72, 1479–1508. [Google Scholar] [CrossRef]
- WHO. Update on Omicron. Available online: https://www.who.int/news/item/28-11-2021-update-on-omicron (accessed on 7 October 2022).
- He, C.; He, X.; Yang, J.; Lei, H.; Hong, W.; Song, X.; Yang, L.; Li, J.; Wang, W.; Shen, G.; et al. Spike protein of SARS-CoV-2 Omicron (B.1.1.529) variant has a reduced ability to induce the immune response. Signal Transduct. Target. Ther. 2022, 7, 119. [Google Scholar] [CrossRef]
- Kumar, S.; Thambiraja, T.S.; Karuppanan, K.; Subramaniam, G. Omicron and Delta variant of SARS-CoV-2: A comparative computational study of spike protein. J. Med. Virol. 2022, 94, 1641–1649. [Google Scholar] [CrossRef]
- Chavda, V.P.; Apostolopoulos, V. Is Booster Dose Strategy Sufficient for Omicron Variant of SARS-CoV-2? Vaccines 2022, 10, 367. [Google Scholar] [CrossRef]
- Lusvarghi, S.; Pollett, S.D.; Neerukonda, S.N.; Wang, W.; Wang, R.; Vassell, R.; Epsi, N.J.; Fries, A.C.; Agan, B.K.; Lindholm, D.A.; et al. SARS-CoV-2 BA.1 variant is neutralized by vaccine booster–elicited serum but evades most convalescent serum and therapeutic antibodies. Sci. Transl. Med. 2022, 14, eabn8543. [Google Scholar] [CrossRef]
- Chauvin, C.; Levillayer, L.; Roumier, M.; Nielly, H.; Roth, C.; Karnam, A.; Bonam, S.R.; Bourgarit, A.; Dubost, C.; Bousquet, A.; et al. Tocilizumab treated convalescent COVID-19 patients retain the cross-neutralization potential against SARS-CoV-2 variants. iScience 2023, 26, 106124. [Google Scholar] [CrossRef]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature 2022, 602, 664–670. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Takashita, E.; Yamayoshi, S.; Simon, V.; van Bakel, H.; Sordillo, E.M.; Pekosz, A.; Fukushi, S.; Suzuki, T.; Maeda, K.; Halfmann, P.; et al. Efficacy of Antibodies and Antiviral Drugs against Omicron BA.2.12.1, BA.4, and BA.5 Subvariants. N. Engl. J. Med. 2022, 387, 468–470. [Google Scholar] [CrossRef]
- Hentzien, M.; Autran, B.; Piroth, L.; Yazdanpanah, Y.; Calmy, A. A monoclonal antibody stands out against omicron subvariants: A call to action for a wider access to bebtelovimab. Lancet Infect. Dis. 2022, 22, 1278. [Google Scholar] [CrossRef] [PubMed]
- U.S. Pauses Distribution Of Monoclonal Antibody Treatments That Proved Ineffective Against Omicron. Available online: https://www.forbes.com/sites/zacharysmith/2021/12/23/us-pauses-distribution-of-monoclonal-antibody-treatments-that-proved-ineffective-against-omicron/?sh=53d804314c62 (accessed on 28 September 2022).
- Most Monoclonal Antibody Treatments Fail against Omicron, Other in Short Supply. Available online: https://www.beckershospitalreview.com/supply-chain/most-monoclonal-antibody-treatments-fail-against-omicron-other-in-short-supply.html (accessed on 28 September 2022).
- Bakkari, M.A.; Moni, S.S.; Sultan, M.H.; Madkhali, O.A. Monoclonal Antibodies and their Target Specificity Against SARS-CoV-2 Infections: Perspectives and Challenges. Recent Pat. Biotechnol. 2022, 16, 64–78. [Google Scholar] [CrossRef] [PubMed]
- 2 Different COVID-19 Monoclonal Antibodies Effectively Neutralize Omicron. Available online: https://www.contagionlive.com/view/2-different-covid-19-monoclonal-antibodies-effectively-neutralize-omicron (accessed on 27 September 2022).
- Gottlieb, R.L.; Nirula, A.; Chen, P.; Boscia, J.; Heller, B.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. Effect of Bamlanivimab as Monotherapy or in Combination With Etesevimab on Viral Load in Patients With Mild to Moderate COVID-19: A Randomized Clinical Trial. JAMA 2021, 325, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferri, F.; Mirandola, M.; Savoldi, A.; De Nardo, P.; Morra, M.; Tebon, M.; Armellini, M.; De Luca, G.; Calandrino, L.; Sasset, L.; et al. Exploratory data on the clinical efficacy of monoclonal antibodies against SARS-CoV-2 Omicron Variant of Concern. Elife 2022, 11, e79639. [Google Scholar] [CrossRef] [PubMed]
- Anti-Coronavirus Spike Neutralizing Antibody ChimeraMAb, 40589-D003|Sino Biological. Available online: https://www.sinobiological.com/antibodies/cov-spike-40589-d003 (accessed on 27 September 2022).
- Kurella, V.B.; Gali, R. Antibody Design and Humanization via In Silico Modeling. Methods Mol. Biol. 2018, 1827, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Pelat, T.; Bedouelle, H.; Rees, A.R.; Crennell, S.J.; Lefranc, M.-P.; Thullier, P. Germline Humanization of a Non-human Primate Antibody that Neutralizes the Anthrax Toxin, by in Vitro and in Silico Engineering. J. Mol. Biol. 2008, 384, 1400–1407. [Google Scholar] [CrossRef]
- Khan, M.A.-A.-K.; Turjya, R.R.; Islam, A.B.M.M.K. Computational engineering the binding affinity of Adalimumab monoclonal antibody for designing potential biosimilar candidate. J. Mol. Graph. Model. 2021, 102, 107774. [Google Scholar] [CrossRef]
- Kuroda, D.; Tsumoto, K. Engineering Stability, Viscosity, and Immunogenicity of Antibodies by Computational Design. J. Pharm. Sci. 2020, 109, 1631–1651. [Google Scholar] [CrossRef]
- Wolf Pérez, A.-M.; Sormanni, P.; Andersen, J.S.; Sakhnini, L.I.; Rodriguez-Leon, I.; Bjelke, J.R.; Gajhede, A.J.; De Maria, L.; Otzen, D.E.; Vendruscolo, M.; et al. In vitro and in silico assessment of the developability of a designed monoclonal antibody library. MAbs 2019, 11, 388–400. [Google Scholar] [CrossRef]
- Breznik, M.; Ge, Y.; Bluck, J.P.; Briem, H.; Hahn, D.F.; Christ, C.D.; Mortier, J.; Mobley, D.L.; Meier, K. Prioritizing Small Sets of Molecules for Synthesis through in-silico Tools: A Comparison of Common Ranking Methods. ChemMedChem 2023, 18, e202200425. [Google Scholar] [CrossRef]
- Warren, G.L.; Andrews, C.W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No. | Lineage of SARS-CoV-2 Strain | Spike Protein with Mutation | Interacting Monoclonal Antibody | Binding Affinity ΔG (kcal mol−1) | Haddock 2.4 Score |
|---|---|---|---|---|---|
| 1. | B.1.1529 (Omicron) | G339D | Adintrevimab | −15.5 | −113.7 ± 5.7 |
| 2. | S371L | Beludavimab | −15.0 | −101.6 ± 4.9 | |
| 3. | N440K | Regdanivimab | −15.8 | −118.0 ± 17.7 | |
| 4. | G142D | Cilgavimab | −18.6 | −112.0 ± 14.4 | |
| 5. | D614G | Romlusevimab | −15.3 | −104.5 ± 3.7 |
| mAb/Spike Protein Complex Structure | Hydrogen Bond | Electrostatic Bond | Hydrophobic Bond | ||||||
|---|---|---|---|---|---|---|---|---|---|
| mAb Residue | Spike Protein Residue | Distance (Å) | mAb Residue | Spike Protein Residue | Distance (Å) | mAb Residue | Spike Protein Residue | Distance (Å) | |
| Adintrevimab/G339D | SER30 | GLU2589 | 1.73 | - | - | - | π-σ interaction | ||
| SER74 | ASP2588 | 1.67 | - | - | - | TYR214 | THR1596 | 3.49 | |
| HIS103 | ASP2613 | 1.64 | - | - | - | HIS103 | VAL2616 | 3.93 | |
| ALA104 | ASP2638 | 1.72 | - | - | - | π-π stacked | |||
| TYR154 | CYS1598 | 2.13 | - | - | - | TYR222 | PHE1604 | 4.35 | |
| THR28 | ASN2583 | 1.96 | - | - | - | π-alkyl interaction | |||
| TYR56 | SER2620 | 1.73 | - | - | - | TYR56 | VAL2616 | 5.14 | |
| ALA104 | THR2634 | 3.18 | - | - | - | - | - | - | |
| ALA104 | ASN2637 | 1.97 | - | - | - | - | - | - | |
| SER219 | GLY1603 | 3.77 | - | - | - | - | - | - | |
| Beludavimab/S371L | GLN1 | THR1596 | 1.97 | TYR54 | GLU2589 | 4.0984 | π-π shaped | ||
| GLY105 | ASP2613 | 1.70 | - | - | - | TRP104 | TYR2614 | 4.84 | |
| SER106 | PHE2587 | 1.76 | - | - | - | alkyl interaction | |||
| THR180 | GLU1602 | 1.95 | - | - | - | PRO28 | LEU2584 | 4.94 | |
| GLY181 | GLU1602 | 1.71 | - | - | - | π-alkyl interaction | |||
| TYR100 | TYR1607 | 2.37 | - | - | - | TYR54 | PRO2586 | 4.70 | |
| SER106 | ASN2592 | 2.30 | - | - | - | TRP104 | PRO2776 | 5.45 | |
| TYR54 | LYS2605 | 1.75 | - | - | - | VAL2 | PHE1604 | 5.26 | |
| TPR104 | PRO2776 | 3.02 | - | - | - | - | - | - | |
| Cilgavimab/G142D | LYS162 | ASP817 | 1.53 | alkyl interaction | |||||
| ASP106 | LYS291 | 2.42 | PRO108 | ILE1000 | 4.69 | ||||
| ASP106 | LYS291 | 1.62 | PRO108 | ILE2131 | 4.62 | ||||
| ASP192 | ARG802 | 1.84 | PRO108 | ILE3262 | 4.67 | ||||
| GLU187 | LYS812 | 4.52 | ARG193 | PRO799 | 4.82 | ||||
| ASP106 | LYS951 | 2.33 | ALA201 | ALA832 | 4.09 | ||||
| VAL107 | ARG1883 | 2.62 | LEU156 | ARG834 | 5.19 | ||||
| GLN153 | GLU268 | 2.31 | PRO108 | ALA2134 | 4.65 | ||||
| SER154 | GLU268 | 1.73 | PRO108 | ALA3265 | 5.34 | ||||
| TYR157 | THR294 | 2.48 | |||||||
| SER158 | SER33 | 1.83 | |||||||
| ASN161 | ASN947 | 2.83 | |||||||
| LYS162 | ASN940 | 2.56 | |||||||
| ARG186 | ASN811 | 2.37 | |||||||
| ASP192 | ASP807 | 2.66 | |||||||
| SER159 | THR294 | 2.56 | |||||||
| GLU187 | ASN811 | 2.90 | |||||||
| VAL190 | ASN811 | 1.80 | |||||||
| SER188 | LYS812 | 1.70 | |||||||
| LEU109 | LYS951 | 1.69 | |||||||
| ASP106 | ARG1883 | 1.96 | |||||||
| ASP106 | ARG1883 | 2.31 | |||||||
| VAL107 | THR1886 | 1.82 | |||||||
| PRO108 | ILE2131 | 2.41 | |||||||
| ASP106 | SER1876 | 2.85 | |||||||
| ARG186 | ASN811 | 2.99 | |||||||
| ASP192 | SESR800 | 3.39 | |||||||
| Regdanivimab/N440K | ASP56 | LYS2527 | 1.59 | alkyl interaction | |||||
| LYS66 | GLU2530 | 4.76 | LYS66 | ALA3094 | 4.95 | ||||
| LYS66 | ASP3092 | 5.10 | |||||||
| GLY33 | ASN2852 | 2.72 | |||||||
| LYS59 | ASN2529 | 1.80 | |||||||
| TYR107 | THR3076 | 1.84 | |||||||
| TYR155 | LYS3074 | 1.84 | |||||||
| ASN174 | ASN3073 | 2.83 | |||||||
| LYS189 | ASN3073 | 1.64 | |||||||
| GLY191 | ASP3069 | 2.39 | |||||||
| ASP56 | LYS2527 | 1.68 | |||||||
| ASP57 | THR2535 | 1.72 | |||||||
| TYR108 | ASN2852 | 3.06 | |||||||
| GLY33 | ASN2852 | 2.00 | |||||||
| SER190 | LYS3060 | 2.97 | |||||||
| LYS189 | ASN3073 | 2.89 | |||||||
| ASN153 | ALA3078 | 2.17 | |||||||
| ASN153 | LEU3077 | 1.91 | |||||||
| ASN106 | GLN3203 | 2.31 | |||||||
| TYR105 | THR3255 | 2.09 | |||||||
| LYS66 | GLU2530 | 3.57 | |||||||
| LYS176 | SER3189 | 3.18 | |||||||
| ASN175 | SER3188 | 3.24 | |||||||
| Romlusevimab/D614G | ARG215 | GLU2769 | 2.44 | TRP50 | GLU2769 | 4.38929 | π-π stacked | ||
| ASN52 | GLU2769 | 2.14 | TRP208 | PHE2771 | 4.33 | ||||
| SER210 | THR2763 | 2.83 | TRP208 | PHE2771 | 5.27 | ||||
| ARG215 | GLU2769 | 1.78 | π-alkyl interaction | ||||||
| TYR166 | ALA359 | 1.80 | TRP50 | VAL2768 | 4.87 | ||||
| SER75 | LYS2729 | 1.65 | TYR166 | ALA359 | 4.64 | ||||
| SER210 | ASN2766 | 1.65 | LEU103 | TYR2774 | 5.43 | ||||
| ASP213 | VAL2768 | 2.87 | |||||||
| ASP213 | GLU2769 | 2.67 | |||||||
| THR102 | ASN2772 | 2.23 | |||||||
| LEU103 | ASN2772 | 2.62 | |||||||
| LEU103 | PHE2771 | 3.02 | |||||||
| Sl. No. | Chimeric Antibody | mAb Framework | mAb CDR | CDR Sequence |
|---|---|---|---|---|
| 1. | Regdanivimab-Framework-Adintrevimab-CDRH3 | Regdanivimab | Adintrevimab | ARDFSGHTAWAGTGFEY |
| 2. | Adintrevimab-Framework-Regdanivimab-CDRH3 | Adintrevimab | Regdanivimab | ARIPGFLRYRNRYYYYGMDV |
| 3. | Regdanivimab-Framework-Beludavimab-CDRH3 | Regdanivimab | Beludavimab | ARDYTRGAWFGESLIGGFDN |
| 4. | Beludavimab-Framework-Regdanivimab-CDRH3 | Beludavimab | Regdanivimab | ARIPGFLRYRNRYYYYGMDV |
| 5. | Adintrevimab- Framework-Beludavimab-CDRH3 | Adintrevimab | Beludavimab | ARDYTRGAWFGESLIGGFDN |
| 6. | Beludavimab-Framework-Adintrevimab-CDRH3 | Beludavimab | Adintrevimab | ARDFSGHTAWAGTGFEY |
| 7. | Sotrovimab-Framework-Regdanivimab-CDRH3 | Sotrovimab | Regdanivimab | ARIPGFLRYRNRYYYYGMDV |
| Mutant Spike Proteins of B.1.1529 (Omicron) SARS-CoV-2 Strain | Chimeric Antibody | ||||||
|---|---|---|---|---|---|---|---|
| Regdanivimab-Framework-Adintrevimab-CDRH3 | Adintrevimab-Framework-Regdanivimab-CDRH3 | Regdanivimab-Framework-Beludavimab-CDRH3 | Beludavimab-Framework-Regdanivimab-CDRH3 | Adintrevimab-Framework-Beludavimab-CDRH3 | Beludavimab-Framework-Adintrevimab-CDRH3 | Sotrovimab-Framework-Regdanivimab-CDRH3 | |
| Binding Affinity ΔG (kcal mol−1) | |||||||
| A67V | −6.7 | −18.1 | −10.0 | −16.4 | −12.2 | −12.1 | −16.3 |
| D614G | −10.5 | −10.1 | −11.4 | −11.5 | −12.3 | −14.1 | −12.2 |
| D796K | −6.6 | −12.8 | −10.9 | −15.2 | −12.5 | −8.4 | −14.2 |
| E484A | −12.0 | −14.6 | −9.2 | −10.1 | −14.3 | −14.8 | −13.1 |
| G142D | −11.9 | −13.7 | −11.5 | −12.2 | −14.7 | −15.1 | −10.5 |
| G339D | −13.5 | −9.8 | −14.1 | −12.2 | −15.3 | −12.5 | −11.2 |
| G446S | −10.8 | −11.5 | −14.5 | −11.8 | −11.5 | −14.0 | −11.9 |
| G496S | −11.3 | −12.4 | −9.9 | −10.8 | −14.1 | −14.0 | −11.6 |
| H655Y | −8.5 | −15.3 | −10.8 | −16.4 | −10.9 | −10.1 | −16.8 |
| HV69-70del | −8.7 | −15.3 | −11.4 | −10.4 | −12.8 | −11.7 | −15.4 |
| ins214EPE | −11.1 | −15.6 | −10.1 | −16.7 | −10.7 | −12.9 | −12.3 |
| K417N | −11.1 | −12.6 | −12.2 | −11.6 | −16.4 | −12.1 | −11.5 |
| L212I | −8.5 | −13.2 | −9.1 | −13.6 | −10.9 | −10.9 | −17.8 |
| L981F | −11.7 | −13.9 | −10.6 | −14.7 | −10.2 | −10.7 | −17.0 |
| N211del | −7.9 | −15.6 | −10.8 | −14.6 | −11.4 | −13.6 | −12.5 |
| N440K | −12.2 | −10.5 | −11.7 | −12.4 | −11.4 | −11.7 | −13.7 |
| N501Y | −12.6 | −12.2 | −9.2 | −12.9 | −14.0 | −15.5 | −12.3 |
| N679K | −12.4 | −14.6 | −8.3 | −13.0 | −11.8 | −13.8 | −14.2 |
| N764K | −9.9 | −15.0 | −11.2 | −13.5 | −15.6 | −12.5 | −10.5 |
| N856K | −10.7 | −13.9 | −11.1 | −16.9 | −13.3 | −12.1 | −15.2 |
| N969K | −10.6 | −15.0 | −10.5 | −16.3 | −15.6 | −12.4 | −13.6 |
| NATIVE | −11.3 | −10.6 | −11.4 | −9.9 | −20.4 | −10.3 | −12.3 |
| MULTIVERSE | −13.8 | −11.7 | −11.4 | −11.8 | −13.4 | −12.4 | −12.9 |
| P681H | −13.7 | −10.3 | −14.8 | −11.1 | −16.9 | −11.7 | −11.4 |
| Q493R | −13.8 | −10.9 | −13.0 | −10.7 | −17.0 | −9.7 | −11.3 |
| Q498R | −10.9 | −13.7 | −12.3 | −10.8 | −11.7 | −14.5 | −12.6 |
| Q954H | −7.2 | −16.3 | −10.6 | −14.2 | −16.2 | −11.7 | −14.1 |
| S371L | −11.0 | −11.5 | −10.5 | −12.0 | −16.2 | −12.1 | −13.0 |
| S373P | −11.9 | −12.6 | −10.4 | −10.2 | −13.3 | −12.1 | −10.9 |
| S375F | −12.4 | −10.8 | −13.1 | −11.8 | −16.5 | −10.3 | −10.8 |
| S477N | −12.7 | −10.8 | −11.9 | −10.9 | −13.5 | −11.9 | −10.9 |
| T95I | −9.2 | −11.4 | −10.2 | −16.5 | −10.2 | −7.6 | −14.4 |
| T478K | −12.7 | −10.9 | −12.6 | −12.3 | −14.4 | −11.8 | −12.6 |
| T547K | −8.1 | −14.1 | −10.3 | −15.5 | −12.2 | −14.0 | −11.2 |
| VYY143-145 | −8.1 | −16.0 | −9.0 | −13.6 | −13.2 | −7.9 | −16.3 |
| Y505H | −10.2 | −14.0 | −10.9 | −10.3 | −14.0 | −9.5 | −15.1 |
| Sl. No. | Chimeric Antibody | BA.1 | BA.2 | BA.2.12.1 | BA.4/5 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Haddock 2.4 Score | Binding Affinity ΔG (kcal mol−1) | Haddock 2.4 Score | Binding Affinity ΔG (kcal mol−1) | Haddock 2.4 Score | Binding Affinity ΔG (kcal mol−1) | Haddock 2.4 Score | Binding Affinity ΔG (kcal mol−1) | ||
| 1. | Regdanivimab-Framework-Adintrevimab-CDRH3 | −51.7 ± 5.4 | - | −79.6 ± 14.1 | - | −98.0 ± 8.8 | - | −52.2 ± 2.6 | - |
| 2. | Adintrevimab-Framework-Regdanivimab-CDRH3 | −98.3 ± 8.5 | - | −115.1 ± 7.0 | −14.0 | −123.6 ± 5.9 | −12.6 | −92.5 ± 5.1 | - |
| 3. | Regdanivimab-Framework-Beludavimab-CDRH3 | −52.1 ± 6.0 | - | −51.4 ± 4.4 | - | −72.0 ± 5.7 | - | −62.9 ± 4.8 | - |
| 4. | Beludavimab-Framework-Regdanivimab-CDRH3 | −96.0 ± 16.0 | - | −145.4 ± 20.8 | −15.1 | −125.3 ± 21.1 | −14.4 | −102.0 ± 6.1 | −13.1 |
| 5. | Adintrevimab-Framework-Beludavimab-CDRH3 | −111.6 ± 4.6 | −15.0 | −131.6 ± 7.1 | −13.9 | −135.4 ± 9.3 | −17.4 | −109.1 ± 12.0 | −14.6 |
| 6. | Beludavimab -Framework-Adintrevimab-CDRH3 | −104.0 ± 7.1 | −14.0 | −118.2 ± 6.4 | −12.6 | −110.9 ± 2.8 | −11.9 | −77.7 ± 7.3 | - |
| 7. | Sotrovimab-Framework Regdanivimab-CDRH3 | −105.1 ± 15.1 | −15.8 | −119.0 ± 4.3 | −11.7 | −109.2 ± 8.4 | −12.0 | −78.5 ± 12.7 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, N.C.; Chakraborty, P.; Bayry, J.; Mukherjee, S. Comparative Binding Ability of Human Monoclonal Antibodies against Omicron Variants of SARS-CoV-2: An In Silico Investigation. Antibodies 2023, 12, 17. https://doi.org/10.3390/antib12010017
Das NC, Chakraborty P, Bayry J, Mukherjee S. Comparative Binding Ability of Human Monoclonal Antibodies against Omicron Variants of SARS-CoV-2: An In Silico Investigation. Antibodies. 2023; 12(1):17. https://doi.org/10.3390/antib12010017
Chicago/Turabian StyleDas, Nabarun Chandra, Pritha Chakraborty, Jagadeesh Bayry, and Suprabhat Mukherjee. 2023. "Comparative Binding Ability of Human Monoclonal Antibodies against Omicron Variants of SARS-CoV-2: An In Silico Investigation" Antibodies 12, no. 1: 17. https://doi.org/10.3390/antib12010017
APA StyleDas, N. C., Chakraborty, P., Bayry, J., & Mukherjee, S. (2023). Comparative Binding Ability of Human Monoclonal Antibodies against Omicron Variants of SARS-CoV-2: An In Silico Investigation. Antibodies, 12(1), 17. https://doi.org/10.3390/antib12010017