
Structural Investigation of Therapeutic Antibodies Using Hydroxyl Radical Protein Footprinting Methods


Abstract
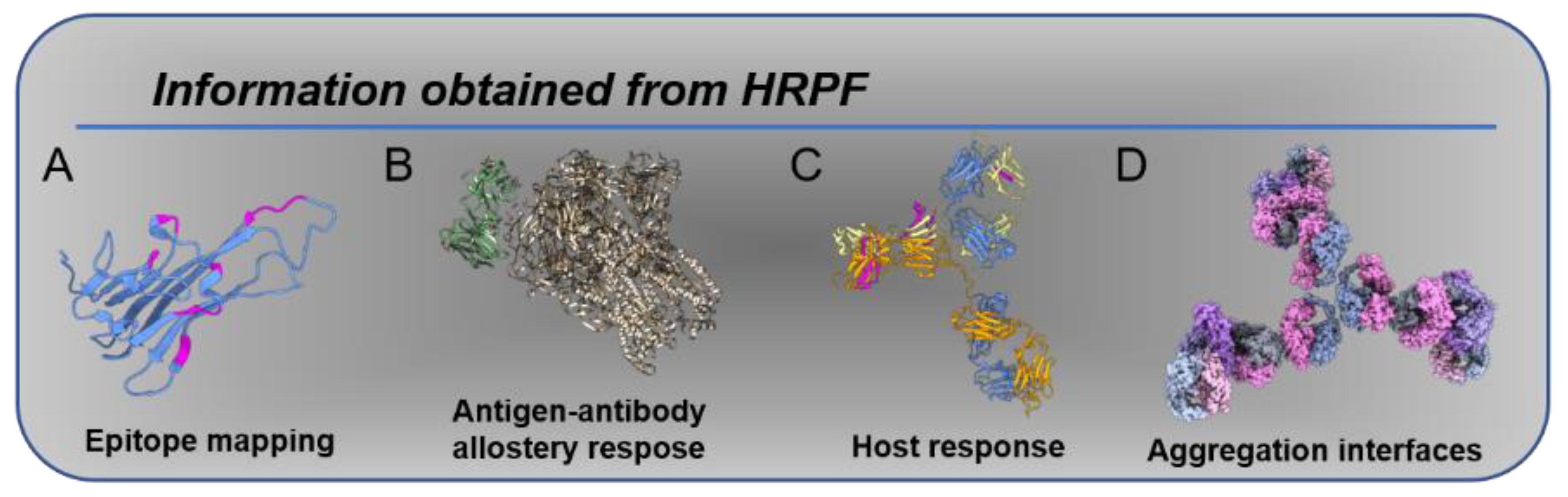
1. Introduction
2. A Brief History of Hydroxyl Radical Footprinting Methods
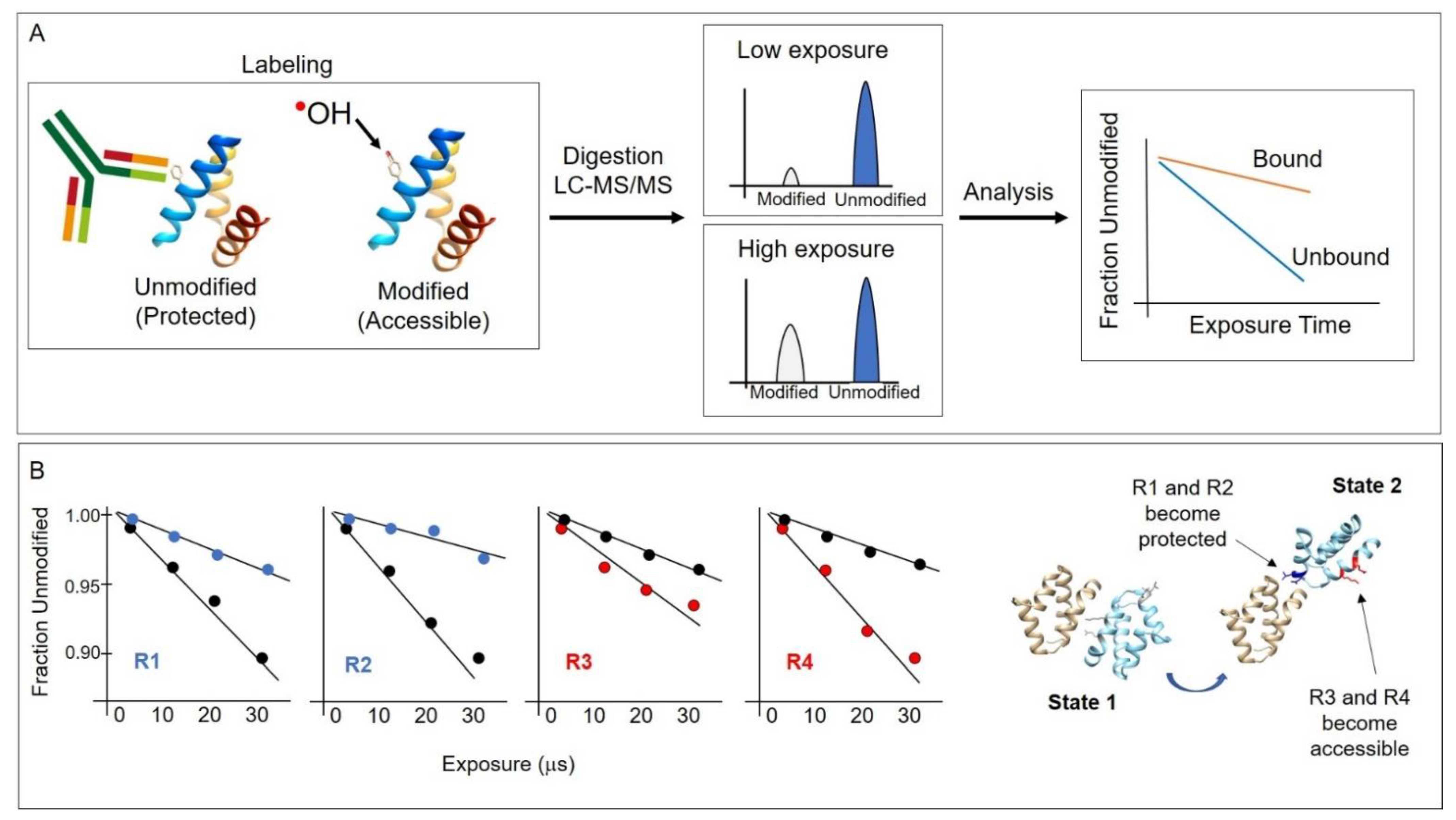
3. Practical Applications and Considerations
3.1. Chemical ˙OH Footprinting
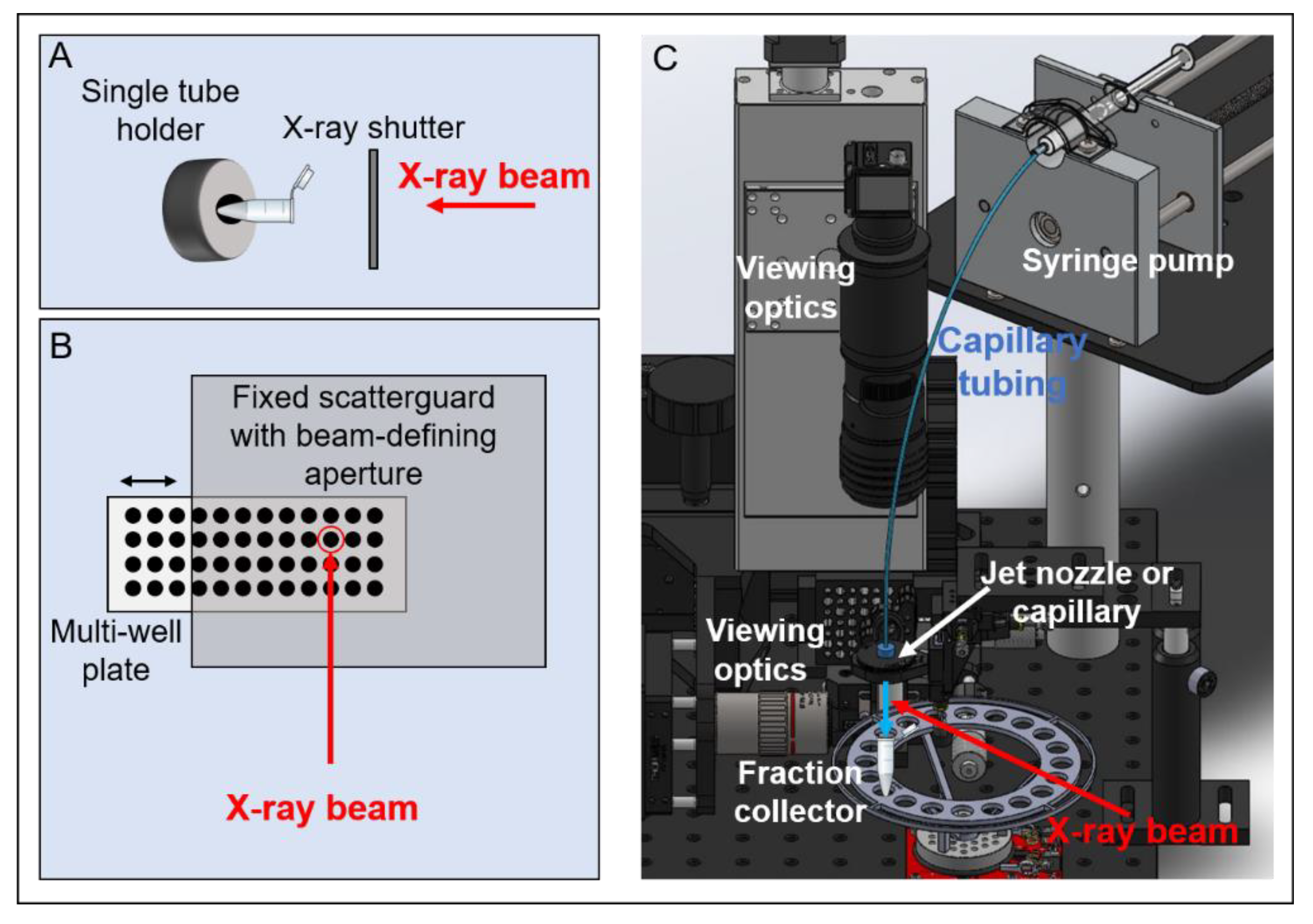
3.2. X-Ray-Based ˙OH Footprinting
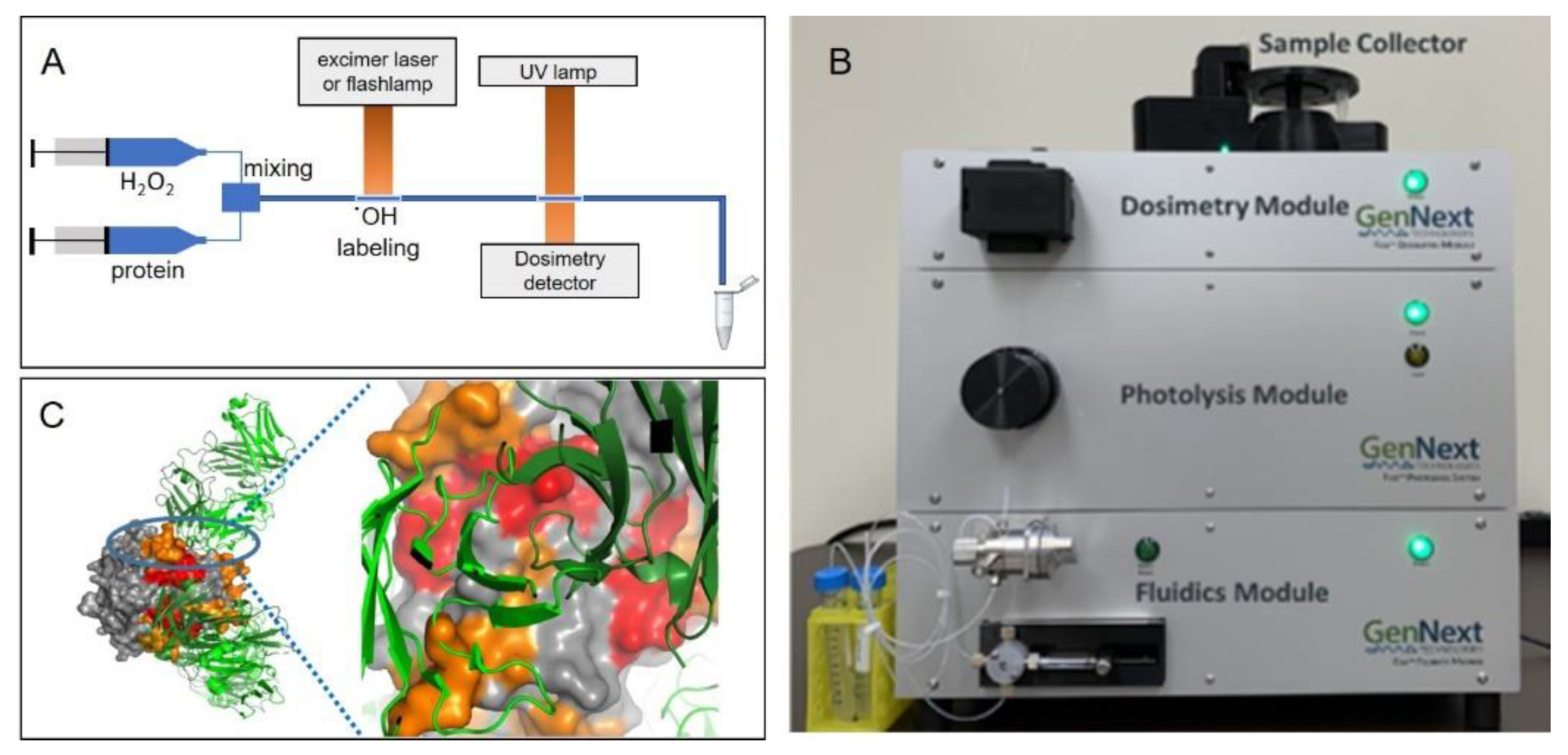
3.3. Laser-Photolysis-Based ˙OH Footprinting
4. Recent Advances
4.1. Automation
4.1.1. FOX System
4.1.2. HTP 96-Well Plate and Software
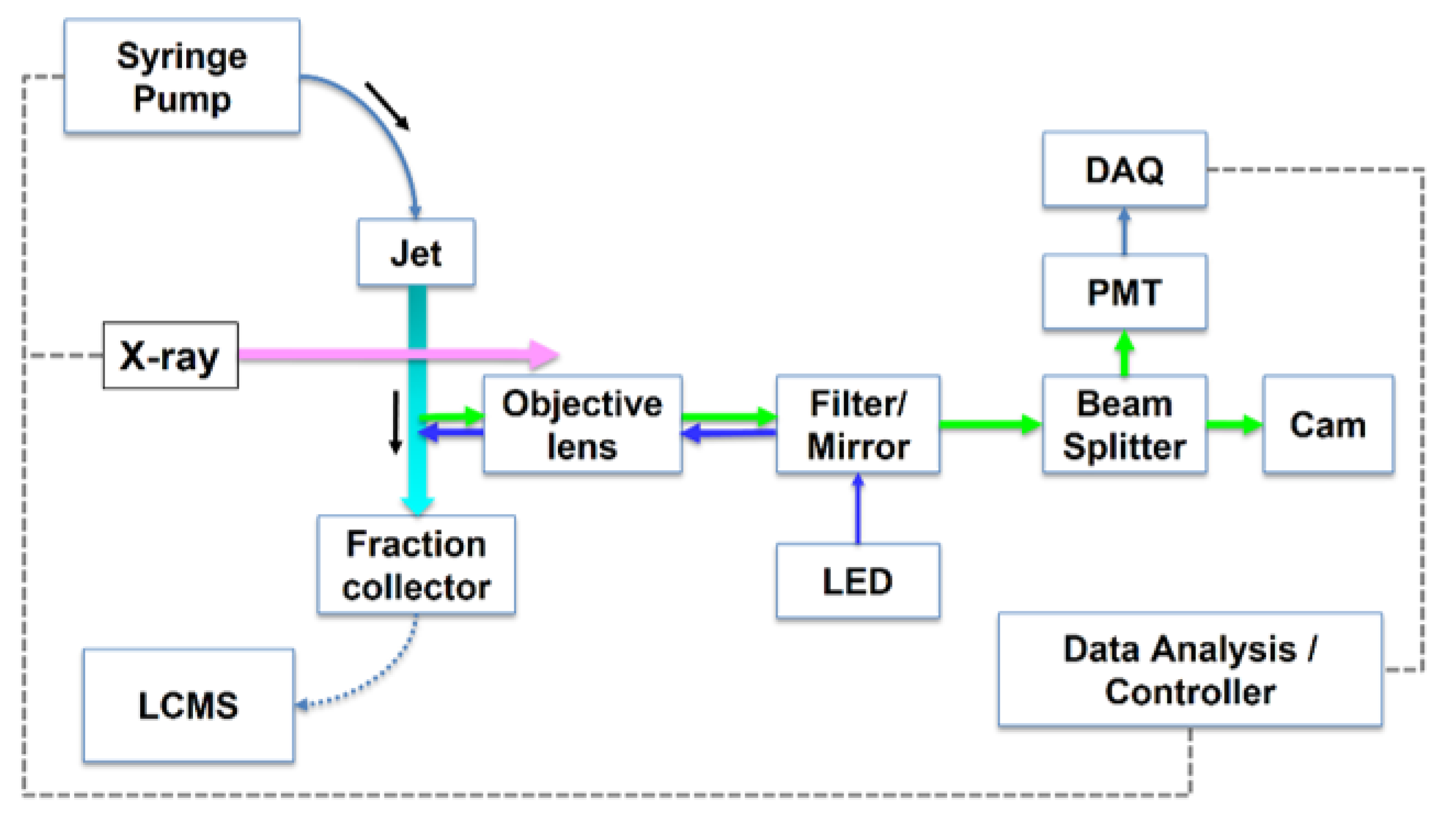
4.1.3. Jet Delivery System and Software
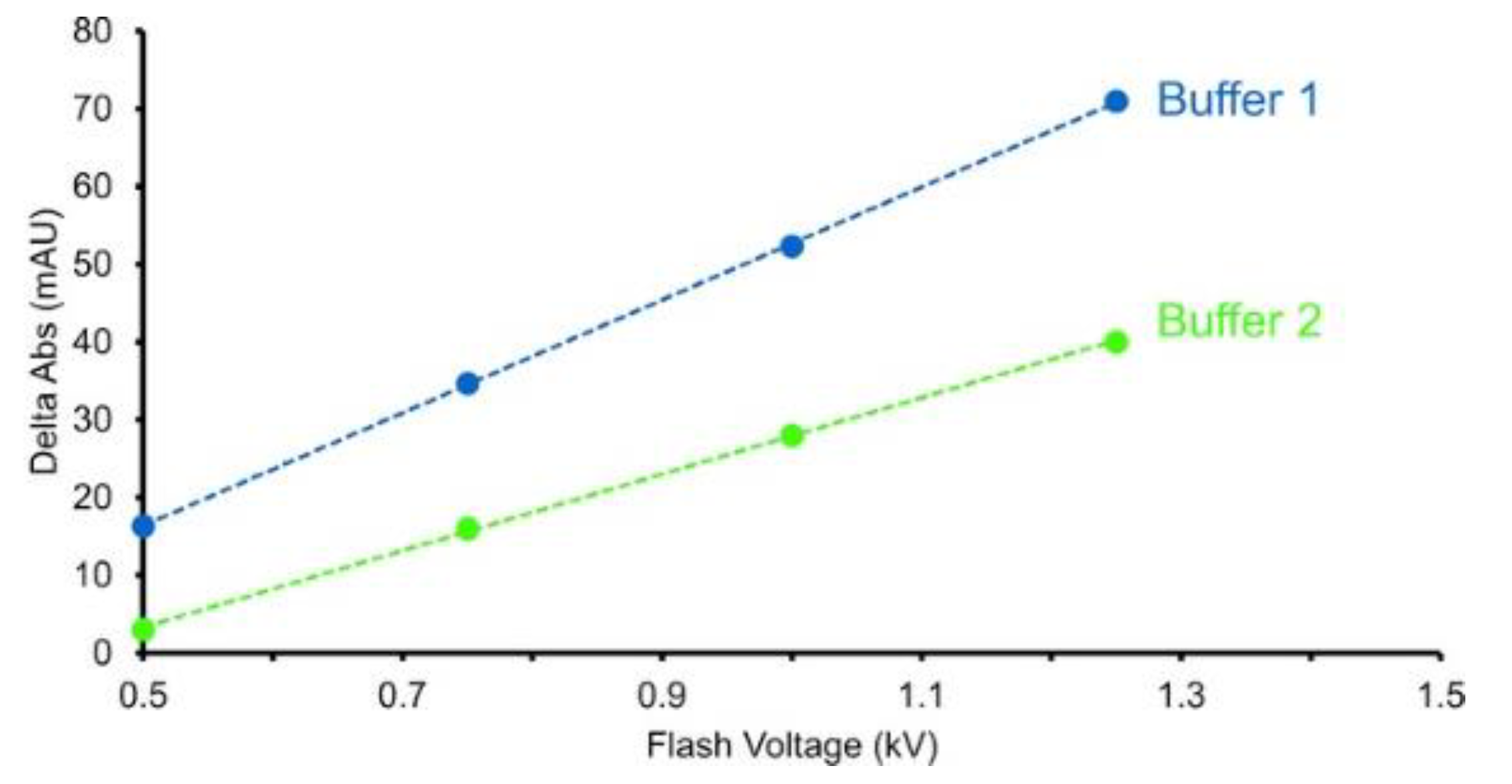
4.2. Inline Dosimetry
4.2.1. UV Absorbance Dosimeters (FPOP/FOX)
4.2.2. Inline Fluorescence Monitoring in XFMS Experiments
5. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rudra, A.; Li, J.; Shakur, R.; Bhagchandani, S.; Langer, R. Trends in Therapeutic Conjugates: Bench to Clinic. Bioconjug. Chem. 2020, 31, 462–473. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Safdari, Y.; Farajnia, S.; Asgharzadeh, M.; Khalili, M. Antibody humanization methods-a review and update. Biotechnol. Genet. Engy. Rev. 2013, 29, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.X.; Tong, X.; Li, C.; Giddens, J.P.; Li, T. Glycoengineering of Antibodies for Modulating Functions. Annu. Rev. Biochem. 2019, 88, 433–459. [Google Scholar] [CrossRef] [PubMed]
- Lerner, R.A. Combinatorial antibody libraries: New advances, new immunological insights. Nat. Rev. Immunol 2016, 16, 498–508. [Google Scholar] [CrossRef]
- Rouet, R.; Jackson, K.J.L.; Langley, D.B.; Christ, D. Next-Generation Sequencing of Antibody Display Repertoires. Front. Immunol. 2018, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to watch in 2022. MAbs 2022, 14, 2014296. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S. The cryo-EM revolution: Fueling the next phase. IUCrJ 2019, 6, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.C.; Yeates, T.O.; Rodriguez, J.A. Advances in methods for atomic resolution macromolecular structure determination. F1000Res 2020, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hilty, C.; Kurzbach, D.; Frydman, L. Hyperpolarized water as universal sensitivity booster in biomolecular NMR. Nat. Protoc. 2022, 17, 1621–1657. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Prakash, K.; Diederich, B.; Heintzmann, R.; Schermelleh, L. Super-resolution microscopy: A brief history and new avenues. Philos. Trans. A Math. Phys. Engy. Sci. 2022, 380, 20210110. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, Present, and Future of Rituximab-The World’s First Oncology Monoclonal Antibody Therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, H.; Zhong, C.; Peng, B.; Zhang, M.; Li, B.; Huo, S.; Guo, Y.; Ding, J. Structural basis for recognition of CD20 by therapeutic antibody Rituximab. J. Biol. Chem. 2007, 282, 15073–15080. [Google Scholar] [CrossRef] [PubMed]
- Puthenveetil, R.; Vinogradova, O. Solution NMR: A powerful tool for structural and functional studies of membrane proteins in reconstituted environments. J. Biol. Chem. 2019, 294, 15914–15931. [Google Scholar] [CrossRef] [PubMed]
- Stander, S.; Grauslund, L.R.; Scarselli, M.; Norais, N.; Rand, K. Epitope Mapping of Polyclonal Antibodies by Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS). Anal. Chem. 2021, 93, 11669–11678. [Google Scholar] [CrossRef] [PubMed]
- Sharp, J.S.; Chea, E.E.; Misra, S.K.; Orlando, R.; Popov, M.; Egan, R.W.; Holman, D.; Weinberger, S.R. Flash Oxidation (FOX) System: A Novel Laser-Free Fast Photochemical Oxidation Protein Footprinting Platform. J. Am. Soc. Mass Spectrom. 2021, 32, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Minkoff, B.B.; Blatz, J.M.; Choudhury, F.A.; Benjamin, D.; Shohet, J.L.; Sussman, M.R. Plasma-Generated OH Radical Production for Analyzing Three-Dimensional Structure in Protein Therapeutics. Sci. Rep. 2017, 7, 12946. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.K.; Orlando, R.; Weinberger, S.R.; Sharp, J.S. Compensated Hydroxyl Radical Protein Footprinting Measures Buffer and Excipient Effects on Conformation and Aggregation in an Adalimumab Biosimilar. AAPS J. 2019, 21, 87. [Google Scholar] [CrossRef] [PubMed]
- Schick, A.J., 3rd; Lundin, V.; Low, J.; Peng, K.; Vandlen, R.; Wecksler, A.T. Epitope mapping of anti-drug antibodies to a clinical candidate bispecific antibody. MAbs 2022, 14, 2028337. [Google Scholar] [CrossRef] [PubMed]
- Schoof, M.; Faust, B.; Saunders, R.A.; Sangwan, S.; Rezelj, V.; Hoppe, N.; Boone, M.; Billesbolle, C.B.; Puchades, C.; Azumaya, C.M.; et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive Spike. Science 2020, 370, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Sevillano, N.; Bohn, M.F.; Zimanyi, M.; Chen, Y.; Petzold, C.; Gupta, S.; Ralston, C.Y.; Craik, C.S. Structure of an affinity-matured inhibitory recombinant fab against urokinase plasminogen activator reveals basis of potency and specificity. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140562. [Google Scholar] [CrossRef]
- Deperalta, G.; Alvarez, M.; Bechtel, C.; Dong, K.; McDonald, R.; Ling, V. Structural analysis of a therapeutic monoclonal antibody dimer by hydroxyl radical footprinting. MAbs 2013, 5, 86–101. [Google Scholar] [CrossRef]
- Li, X.; Grant, O.C.; Ito, K.; Wallace, A.; Wang, S.; Zhao, P.; Wells, L.; Lu, S.; Woods, R.J.; Sharp, J.S. Structural Analysis of the Glycosylated Intact HIV-1 gp120-b12 Antibody Complex Using Hydroxyl Radical Protein Footprinting. Biochemistry 2017, 56, 957–970. [Google Scholar] [CrossRef]
- Li, J.; Wei, H.; Krystek, S.R., Jr.; Bond, D.; Brender, T.M.; Cohen, D.; Feiner, J.; Hamacher, N.; Harshman, J.; Huang, R.Y.; et al. Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Anal. Chem. 2017, 89, 2250–2258. [Google Scholar] [CrossRef]
- Zhang, Y.; Wecksler, A.T.; Molina, P.; Deperalta, G.; Gross, M.L. Mapping the Binding Interface of VEGF and a Monoclonal Antibody Fab-1 Fragment with Fast Photochemical Oxidation of Proteins (FPOP) and Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 850–858. [Google Scholar] [CrossRef]
- Zheng, B.; Yu, S.F.; Del Rosario, G.; Leong, S.R.; Lee, G.Y.; Vij, R.; Chiu, C.; Liang, W.C.; Wu, Y.; Chalouni, C.; et al. An Anti-CLL-1 Antibody-Drug Conjugate for the Treatment of Acute Myeloid Leukemia. Clin. Cancer. Res. 2019, 25, 1358–1368. [Google Scholar] [CrossRef]
- Kiselar, J.; Chance, M.R. High-Resolution Hydroxyl Radical Protein Footprinting: Biophysics Tool for Drug Discovery. Annu. Rev. Biophys. 2018, 47, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Galas, D.J.; Schmitz, A. DNAse footprinting: A simple method for the detection of protein-DNA binding specificity. Nucleic. Acids. Res. 1978, 5, 3157–3170. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, M.W.; Dervan, P.B. Chromomycin, mithramycin, and olivomycin binding sites on heterogeneous deoxyribonucleic acid. Footprinting with (methidiumpropyl-EDTA)iron(II). Biochemistry 1983, 22, 5555–5567. [Google Scholar] [CrossRef]
- Tullius, T.D. DNA footprinting with hydroxyl radical. Nature 1988, 332, 663–664. [Google Scholar] [CrossRef] [PubMed]
- Tullius, T.D.; Dombroski, B.A.; Churchill, M.E.; Kam, L. Hydroxyl radical footprinting: A high-resolution method for mapping protein-DNA contacts. Methods Enzym. 1987, 155, 537–558. [Google Scholar]
- Hayes, J.J.; Kam, L.; Tullius, T.D. Footprinting protein-DNA complexes with gamma-rays. Methods Enzym. 1990, 186, 545–549. [Google Scholar]
- Sclavi, B.; Sullivan, M.; Chance, M.R.; Brenowitz, M.; Woodson, S.A. RNA folding at millisecond intervals by synchrotron hydroxyl radical footprinting. Science 1998, 279, 1940–1943. [Google Scholar] [CrossRef]
- Hachimori, Y.; Horinishi, H.; Kurihara, K.; Shibata, K. States of Amino Acid Residues in Proteins. V. Different Reactivities with H2o2 of Tryptophan Residues in Lysozyme, Proteinases and Zymogens. Biochim. Biophys. Acta. 1964, 93, 346–360. [Google Scholar] [CrossRef]
- Sheshberadaran, H.; Payne, L.G. Protein antigen-monoclonal antibody contact sites investigated by limited proteolysis of monoclonal antibody-bound antigen: Protein "footprinting". Proc. Natl. Acad. Sci. USA 1988, 85, 1–5. [Google Scholar] [CrossRef]
- Sheshberadaran, H.; Payne, L.G. Protein footprinting method for studying antigen-antibody interactions and epitope mapping. Methods Enzym. 1989, 178, 746–764. [Google Scholar]
- Shcherbakova, I.; Mitra, S.; Beer, R.H.; Brenowitz, M. Fast Fenton footprinting: A laboratory-based method for the time-resolved analysis of DNA, RNA and proteins. Nucleic Acids Res. 2006, 34, 48–49. [Google Scholar] [CrossRef] [PubMed]
- Maleknia, S.D.; Brenowitz, M.; Chance, M.R. Millisecond radiolytic modification of peptides by synchrotron X-rays identified by mass spectrometry. Anal. Chem. 1999, 71, 3965–3973. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.; Janik, I.; Zhuang, T.; Charvatova, O.; Woods, R.J.; Sharp, J.S. Pulsed electron beam water radiolysis for submicrosecond hydroxyl radical protein footprinting. Anal. Chem. 2009, 81, 2496–2505. [Google Scholar] [CrossRef]
- Hambly, D.M.; Gross, M.L. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 2005, 16, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Sharp, J.S.; Becker, J.M.; Hettich, R.L. Analysis of protein solvent accessible surfaces by photochemical oxidation and mass spectrometry. Anal. Chem. 2004, 76, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R., 3rd. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, S.R.; Chea, E.E.; Sharp, J.S.; Misra, S.K. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-al Analysis of Proteins. J. Vis. Exp. 2021, 16, 2057–2063. [Google Scholar]
- Gupta, S.; Celestre, R.; Petzold, C.J.; Chance, M.R.; Ralston, C. Development of a microsecond X-ray protein footprinting facility at the Advanced Light Source. J. Synchrotron Radiat. 2014, 21, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Asuru, A.; Farquhar, E.R.; Sullivan, M.; Abel, D.; Toomey, J.; Chance, M.R.; Bohon, J. The XFP (17-BM) beamline for X-ray footprinting at NSLS-II. J. Synchrotron Radiat. 2019, 26, 1388–1399. [Google Scholar] [CrossRef]
- Misra, S.K.; Sharp, J.S. Enabling Real-Time Compensation in Fast Photochemical Oxidations of Proteins for the Determination of Protein Topography Changes. J. Vis. Exp. 2020, 163, e61580. [Google Scholar] [CrossRef] [PubMed]
- Roush, A.E.; Riaz, M.; Misra, S.K.; Weinberger, S.R.; Sharp, J.S. Intrinsic Buffer Hydroxyl Radical Dosimetry Using Tris(hydroxymethyl)aminomethane. J. Am. Soc. Mass Spectrom. 2020, 31, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Sharp, J.S.; Misra, S.K.; Persoff, J.J.; Egan, R.W.; Weinberger, S.R. Real Time Normalization of Fast Photochemical Oxidation of Proteins Experiments by Inline Adenine Radical Dosimetry. Anal. Chem. 2018, 90, 12625–12630. [Google Scholar] [CrossRef]
- Gupta, S.; Raskatov, J.A.; Ralston, C.Y. A Hybrid Structural Method for Investigating Low Molecular Weight Oligomeric Structures of Amyloid Beta. Chembiochem 2022, 13, e202200333. [Google Scholar] [CrossRef]
- Tadi, S.; Misra, S.K.; Sharp, J.S. Inline Liquid Chromatography-Fast Photochemical Oxidation of Proteins for Targeted Structural Analysis of Conformationally Heterogeneous Mixtures. Anal. Chem. 2021, 93, 3510–3516. [Google Scholar] [CrossRef] [PubMed]
- Buxton, G.V. Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH/O−) in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Garrison, W.M. Reaction Mechanisms in the Radiolysis of Peptides, Polypeptides, and Proteins. Chem. Rev. 1987, 87, 381–398. [Google Scholar] [CrossRef]
- Xu, G.; Chance, M.R. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev. 2007, 107, 3514–3543. [Google Scholar] [CrossRef]
- Sharp, J.S.; Tomer, K.B. Effects of anion proximity in peptide primary sequence on the rate and mechanism of leucine oxidation. Anal. Chem. 2006, 78, 4885–4893. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ravikumar, K.M.; Chance, M.R.; Yang, S. Quantitative mapping of protein structure by hydroxyl radical footprinting-mediated structural mass spectrometry: A protection factor analysis. Biophys. J. 2015, 108, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Sood, A.; Woods, R.J.; Sharp, J.S. Quantitative Protein Topography Measurements by High Resolution Hydroxyl Radical Protein Footprinting Enable Accurate Molecular Model Selection. Sci. Rep. 2017, 7, 4552. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Lousada, C.M.; Yang, M.; Nilsson, K.; Jonsson, M. Catalytic decomposition of hydrogen peroxide on transition metal and lanthanide oxides. J. Mol. Catal. A Chem. 2013, 379, 178–184. [Google Scholar] [CrossRef]
- Florence, T.M. The production of hydroxyl radical from hydrogen peroxide. J. Inorg. Biochem. 1984, 22, 221–230. [Google Scholar] [CrossRef]
- Novikov, A.S.; Kuznetsiv, M.; Pombeiro, A.J.L.; Bokach, N.A.; Shulpin, G.B. Generation of HO• Radical from Hydrogen Peroxide Catalyzed by Aqua Complexes of the Group III Metals [M(H2O)n]3+ (M = Ga, In, Sc, Y, or La): A Theoretical Study. ACS Catal. 2013, 3, 1195–1208. [Google Scholar] [CrossRef]
- Koppenol, W.H. The Haber-Weiss cycle--70 years later. Redox Rep. 2001, 6, 229–234. [Google Scholar] [CrossRef]
- Tullius, T.D.; Dombroski, B.A. Iron(II) EDTA used to measure the helical twist along any DNA molecule. Science 1985, 230, 679–681. [Google Scholar] [CrossRef]
- Tullius, T.D.; Dombroski, B.A. Hydroxyl radical “footprinting”: High-resolution information about DNA-protein contacts and application to lambda repressor and Cro protein. Proc. Natl. Acad. Sci. USA 1986, 83, 5469–5473. [Google Scholar] [CrossRef] [PubMed]
- Leser, M.; Chapman, J.R.; Khine, M.; Pegan, J.; Law, M.; Makkaoui, M.E.; Ueberheide, B.M.; Brenowitz, M. Chemical Generation of Hydroxyl Radical for Oxidative ‘Footprinting’. Protein Pept. Let. 2019, 26, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Sharp, J.S.; Becker, J.M.; Hettich, R.L. Protein surface mapping by chemical oxidation: Structural analysis by mass spectrometry. Anal. Biochem. 2003, 313, 216–225. [Google Scholar] [CrossRef]
- Gau, B.C.; Sharp, J.S.; Rempel, D.L.; Gross, M.L. Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal. Chem. 2009, 81, 6563–6571. [Google Scholar] [CrossRef]
- Xu, G.; Kiselar, J.; He, Q.; Chance, M.R. Secondary reactions and strategies to improve quantitative protein footprinting. Anal. Chem. 2005, 77, 3029–3037. [Google Scholar] [CrossRef] [PubMed]
- Roots, R.; Okada, S. Estimation of life times and diffusion distances of radicals involved in x-ray-induced DNA strand breaks of killing of mammalian cells. Radiat. Res. 1975, 64, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Abel, D.; Rakitin, M.; Sullivan, M.; Lodowski, D.T.; Chance, M.R.; Farquhar, E.R. New high-throughput endstation to accelerate the experimental optimization pipeline for synchrotron X-ray footprinting. J. Synchrotron Radiat. 2021, 28, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Asuru, A.; Kiselar, J.; Mathai, G.; Chance, M.R.; Gross, M.L. Fast Protein Footprinting by X-ray Mediated Radical Trifluoromethylation. J. Am. Soc. Mass Spectrom. 2020, 31, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Dhillon, N.S.; Farquhar, E.R.; Wang, B.; Li, X.; Kiselar, J.; Chance, M.R. Multiplex Chemical Labeling of Amino Acids for Protein Footprinting Structure Assessment. Anal. Chem. 2022, 94, 9819–9825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rempel, D.L.; Zhang, H.; Gross, M.L. An improved fast photochemical oxidation of proteins (FPOP) platform for protein therapeutics. J. Am. Soc. Mass Spectrom. 2015, 26, 526–529. [Google Scholar] [CrossRef]
- Liu, X.R.; Rempel, D.L.; Gross, M.L. Protein higher-order-structure determination by fast photochemical oxidation of proteins and mass spectrometry analysis. Nat. Protoc. 2020, 15, 3942–3970. [Google Scholar] [CrossRef] [PubMed]
- Espino, J.A.; Mali, V.S.; Jones, L.M. In Cell Footprinting Coupled with Mass Spectrometry for the Structural Analysis of Proteins in Live Cells. Anal. Chem. 2015, 87, 7971–7978. [Google Scholar] [CrossRef]
- Espino, J.A.; Jones, L.M. Illuminating Biological Interactions with in Vivo Protein Footprinting. Anal. Chem. 2019, 91, 6577–6584. [Google Scholar] [CrossRef] [PubMed]
- Espino, J.A.; Zhang, Z.; Jones, L.M. Chemical Penetration Enhancers Increase Hydrogen Peroxide Uptake in C. elegans for In Vivo Fast Photochemical Oxidation of Proteins. J. Proteome. Res. 2020, 19, 3708–3715. [Google Scholar] [CrossRef]
- Xie, B.; Sharp, J.S. Hydroxyl Radical Dosimetry for High Flux Hydroxyl Radical Protein Footprinting Applications Using a Simple Optical Detection Method. Anal. Chem. 2015, 87, 10719–10723. [Google Scholar] [CrossRef] [PubMed]
- Rosi, M.; Russell, B.; Kristensen, L.G.; Farquhar, E.R.; Jain, R.; Abel, D.; Sullivan, M.; Costello, S.M.; Dominguez-Martin, M.A.; Chen, Y. An automated liquid jet for fluorescence dosimetry and microsecond radiolytic labeling of proteins. Commun. Biol. 2022, 5, 866. [Google Scholar] [CrossRef] [PubMed]
- McKenzie-Coe, A.; Montes, N.S.; Jones, L.M. Hydroxyl Radical Protein Footprinting: A Mass Spectrometry-Based Structural Method for Studying the Higher Order Structure of Proteins. Chem. Rev. 2022, 122, 7532–7561. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Sullivan, M.; Toomey, J.; Kiselar, J.; Chance, M.R. The Beamline X28C of the Center for Synchrotron Biosciences: A national resource for biomolecular structure and dynamics experiments using synchrotron footprinting. J. Synchrotron. Radiat. 2007, 14, 233–243. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method of OH Radical Generation | Sample/Buffer Considerations | Access | Timescale Accessible |
|---|---|---|---|
| Fenton reaction | Fe-EDTA and H2O2 added | Lab-based approach | Seconds to minutes |
| X-ray | No exogenous chemicals added | Synchrotron facilities (user proposals required) | 10 microseconds to hours |
| UV | H2O2 added | Lab-based; UV laser required | 1 microsecond to minutes |
| Plasma | H2O2 added | Lab-based (flash-lamp required) or through commercial venue | 1 microsecond to minutes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ralston, C.Y.; Sharp, J.S. Structural Investigation of Therapeutic Antibodies Using Hydroxyl Radical Protein Footprinting Methods. Antibodies 2022, 11, 71. https://doi.org/10.3390/antib11040071
Ralston CY, Sharp JS. Structural Investigation of Therapeutic Antibodies Using Hydroxyl Radical Protein Footprinting Methods. Antibodies. 2022; 11(4):71. https://doi.org/10.3390/antib11040071
Chicago/Turabian StyleRalston, Corie Y., and Joshua S. Sharp. 2022. "Structural Investigation of Therapeutic Antibodies Using Hydroxyl Radical Protein Footprinting Methods" Antibodies 11, no. 4: 71. https://doi.org/10.3390/antib11040071
APA StyleRalston, C. Y., & Sharp, J. S. (2022). Structural Investigation of Therapeutic Antibodies Using Hydroxyl Radical Protein Footprinting Methods. Antibodies, 11(4), 71. https://doi.org/10.3390/antib11040071