
The Role of Bispecific Antibodies in Non-Hodgkin’s Lymphoma: From Structure to Prospective Clinical Use


Abstract
:1. Introduction
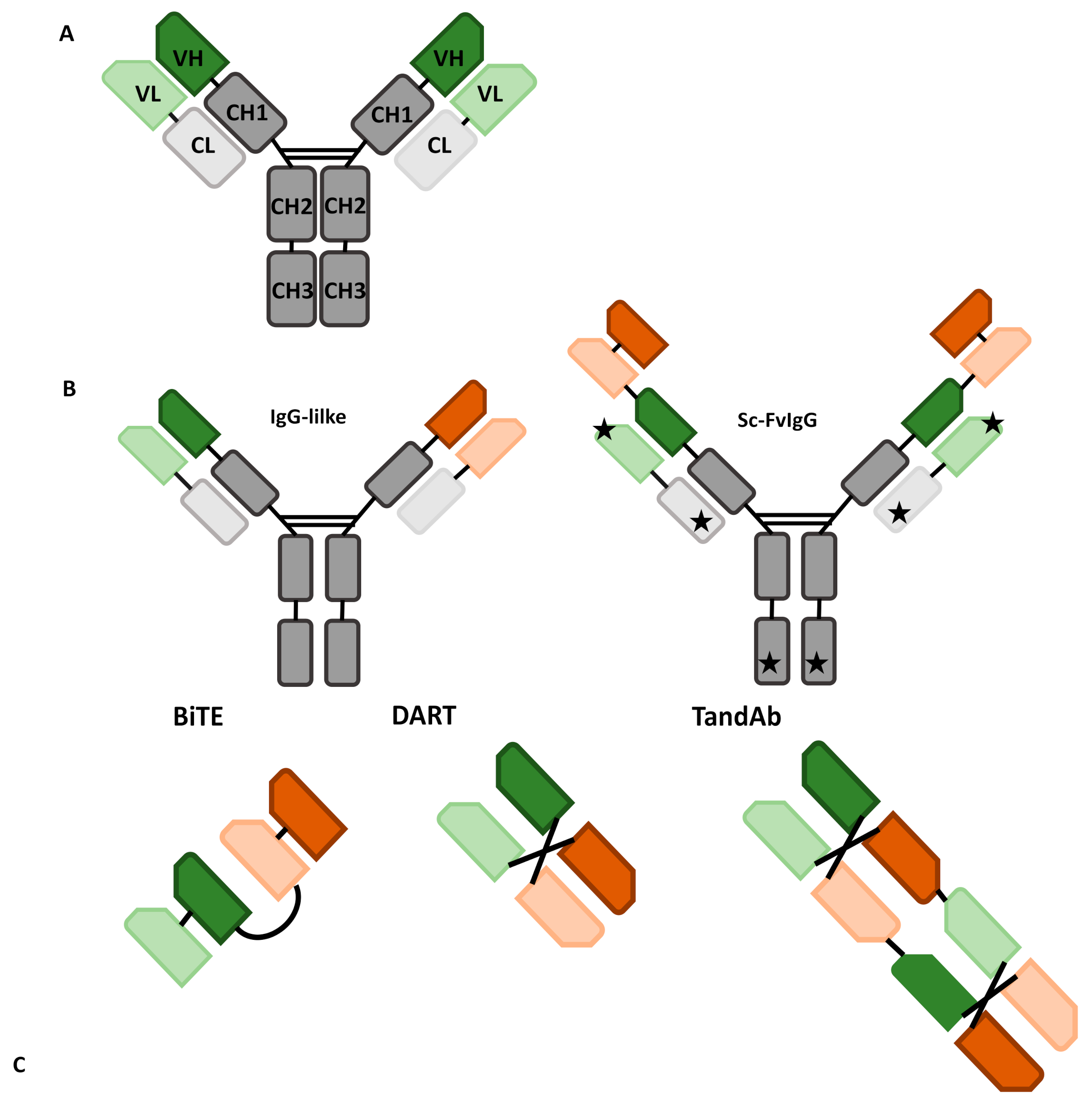
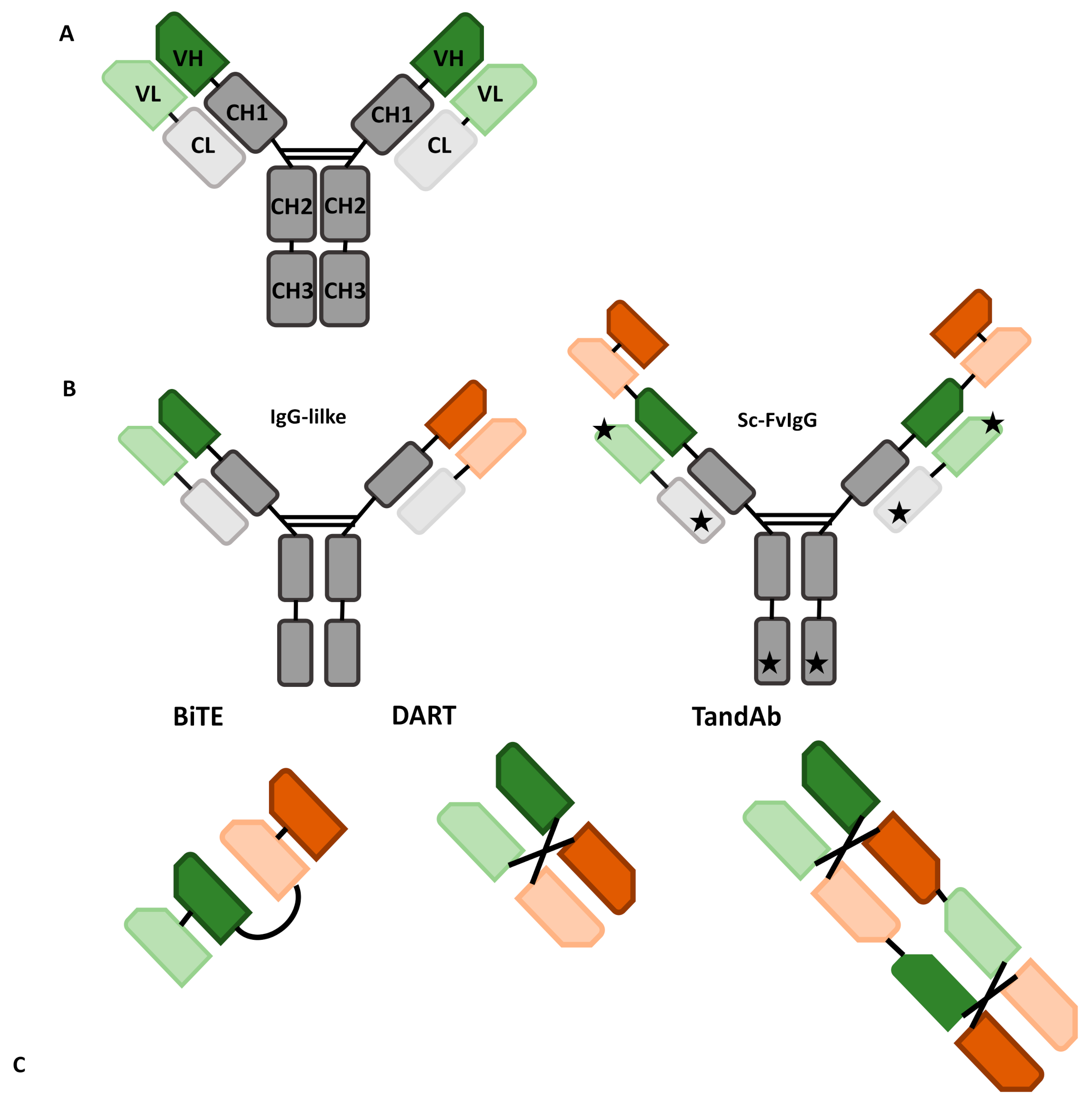
1.1. Formats of bsAbs: A Broad Overview
1.2. IgG-Like bsAbs
1.3. Non-IgG-Like bsAbs
2. Mechanism of Action
3. Bispecific Antibodies in Use and in Development in NHL
3.1. Blinatumomab
3.2. Glofitamab
3.3. Mosunetuzumab
3.4. Odronextamab
3.5. Epcoritamab
3.6. Novel Perspective and bsAbs in Clinical Development
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schuster, S.J. Bispecific antibodies for the treatment of lymphomas: Promises and challenges. Hematol. Oncol. 2021, 39, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Nisonoff, A.; Wissler, F.C.; Lipman, L.N. Properties of the major component of a peptic digest of rabbit antibody. Science 1960, 132, 1770–1771. [Google Scholar] [CrossRef] [PubMed]
- Nisonoff, A.; Rivers, M.M. Recombination of a mixture of univalent antibody fragments of different specificity. Arch. Biochem. Biophys. 1961, 93, 460–462. [Google Scholar] [CrossRef]
- Fudenberg, H.H.; Drews, G.; Nisonoff, A. Serologic demonstration of dual specificity of rabbit bivalent hybrid antibody. J. Exp. Med. 1964, 119, 151–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milstein, C.; Cuello, A.C. Hybrid hybridomas and their use in immunohistochemistry. Nature 1983, 305, 537–540. [Google Scholar] [CrossRef]
- Brennan, M.; Davison, P.F.; Paulus, H. Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin G1 fragments. Science 1985, 229, 81–83. [Google Scholar] [CrossRef]
- Renner, C.; Jung, W.; Sahin, U.; Denfeld, R.; Pohl, C.; Trümper, L.; Hartmann, F.; Diehl, V.; Van Lier, R.; Pfreundschuh, M. Cure of xenografted human tumors by bispecific monoclonal antibodies and human T cells. Science 1994, 264, 833–835. [Google Scholar] [CrossRef]
- De Jonge, J.; Heirman, C.; de Veerman, M.; Van Meirvenne, S.; Moser, M.; Leo, O.; Thielemans, K. In vivo retargeting of T cell effector function by recombinant bispecific single chain Fv (anti-CD3 x anti-idiotype) induces long-term survival in the murine BCL1 lymphoma model. J. Immunol. (Baltim. Md. 1950) 1998, 161, 1454–1461. [Google Scholar]
- Lu, D.; Zhang, H.; Ludwig, D.; Persaud, A.; Jimenez, X.; Burtrum, D.; Balderes, P.; Liu, M.; Bohlen, P.; Witte, L.; et al. Simultaneous Blockade of Both the Epidermal Growth Factor Receptor and the Insulin-like Growth Factor Receptor Signaling Pathways in Cancer Cells with a Fully Human Recombinant Bispecific Antibody. J. Biol. Chem. 2004, 279, 2856–2865. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, U.; Huhalov, A.; Harms, B.; Paragas, V.; Adams, S.; Gu, J.; Nguyen, S.; Luus, L.; Oyama, S.; Razlog, M.; et al. MM-111: A Novel Bispecific Antibody Targeting ErbB3 with Potent Anti-Tumor Activity in ErbB2 Over-Expressing Malignancies. Cancer Res. 2009, 69, 4166. [Google Scholar]
- Veri, M.C.; Burke, S.; Huang, L.; Li, H.; Gorlatov, S.; Tuaillon, N.; Rainey, G.J.; Ciccarone, V.; Zhang, T.; Shah, K.; et al. Therapeutic control of B cell activation via recruitment of Fcγ receptor IIb (CD32B) inhibitory function with a novel bispecific antibody scaffold. Arthritis Rheum. 2010, 62, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- De Bernardis, F.; Liu, H.; O’Mahony, R.; La Valle, R.; Bartollino, S.; Sandini, S.; Grant, S.; Brewis, N.; Tomlinson, I.; Basset, R.C.; et al. Human domain antibodies against virulence traits of Candida albicans inhibit fungus adherence to vaginal epithelium and protect against experimental vaginal candidiasis. J. Infect. Dis. 2007, 195, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves first bispecific. Nat. Rev. Drug Discov. 2015, 14, 7. [Google Scholar] [CrossRef]
- FDA Grants Roche Breakthrough Therapy Designation on Hemophilia Drug. Available online: https://www.biopharminternational.com/view/fda-grants-roche-breakthrough-therapy-designation-hemophilia-drug (accessed on 30 September 2021).
- Dolgin, E. Amivantamab OK’d for EGFR-Mutant NSCLC. Cancer Discov. 2021, 11, 1604. [Google Scholar]
- European Medicines Agency. Removab—Withdrawal of the marketing authorisation in the European Union. 2017. Available online: https://www.ema.europa.eu/en/documents/overview/removab-epar-summary-public_en.pdf (accessed on 14 October 2021).
- Duell, J.; Lammers, P.E.; Djuretic, I.; Chunyk, A.G.; Alekar, S.; Jacobs, I.; Gill, S. Bispecific Antibodies in the Treatment of Hematologic Malignancies. Clin. Pharmacol. Ther. 2019, 106, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Griffin, M.M.; Morley, N. Rituximab in the treatment of non-Hodgkin’s lymphoma-a critical evaluation of randomized controlled trials. Expert Opin. Biol. Ther. 2013, 13, 803–811. [Google Scholar] [CrossRef]
- Kallam, A.; Vose, J.M. Recent Advances in CAR-T Cell Therapy for Non-Hodgkin Lymphoma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 751–757. [Google Scholar] [CrossRef]
- Salvaris, R.; Ong, J.; Gregory, G.P. Bispecific antibodies: A review of development, clinical efficacy and toxicity in B-cell lymphomas. J. Pers. Med. 2021, 11, 355. [Google Scholar] [CrossRef]
- Kuo, T.T.; Aveson, V.G. Neonatal Fc receptor and IgG-based therapeutics. mAbs 2011, 3, 422–430. [Google Scholar] [CrossRef]
- Ying, T.; Ju, T.W.; Wang, Y.; Prabakaran, P.; Dimitrov, D.S. Interactions of IgG1 CH2 and CH3 domains with FcRn. Front. Immunol. 2014, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Giese, G.; Williams, A.; Rodriguez, M.; Persson, J. Bispecific antibody process development: Assembly and purification of knob and hole bispecific antibodies. Biotechnol. Prog. 2018, 34, 397–404. [Google Scholar] [CrossRef]
- Strop, P.; Ho, W.H.; Boustany, L.M.; Abdiche, Y.N.; Lindquist, K.C.; Farias, S.E.; Rickert, M.; Appah, C.T.; Pascua, E.; Radcliffe, T.; et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J. Mol. Biol. 2012, 420, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Spiess, C.; Bevers, J.; Jackman, J.; Chiang, N.; Nakamura, G.; Dillon, M.; Liu, H.; Molina, P.; Elliott, J.M.; Shatz, W.; et al. Development of a human IgG4 bispecific antibody for dual targeting of interleukin-4 (IL-4) and interleukin-13 (IL-13) cytokines. J. Biol. Chem. 2013, 288, 26583–26593. [Google Scholar] [CrossRef] [Green Version]
- Budde, E.; Gopal, A.K.; Flinn, I.W.; Nastoupil, L.J.; Gordon, M.S.; Pang, C.-F.; Keyt, B.; Carroll, S.; Leabman, M.; Hernandez, G.; et al. Preliminary Results of a Phase 1 Dose Escalation Study of the First-in-Class IgM Based Bispecific Antibody Igm-2323 (anti-CD20 x anti-CD3) in Patients with Advanced B-Cell Malignancies. Blood 2020, 136, 45–46. [Google Scholar] [CrossRef]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Sedykh, S.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Dev. Ther. 2018, 12, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Thakur, A.; Huang, M.; Lum, L.G. Bispecific antibody based therapeutics: Strengths and challenges. Blood Rev. 2018, 32, 339–347. [Google Scholar] [CrossRef]
- Pytlik, R.; Polgarova, K.; Karolova, J.; Klener, P. Current immunotherapy approaches in non-Hodgkin lymphomas. Vaccines 2020, 8, 708. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Tay, S.S.; Carol, H.; Biro, M. TriKEs and BiKEs join CARs on the cancer immunotherapy highway. Hum. Vaccines Immunother. 2016, 12, 2790–2796. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.; Burke, S.; Huang, L.; Gorlatov, S.; Li, H.; Wang, W.; Zhang, W.; Tuaillon, N.; Rainey, J.; Barat, B.; et al. Effector Cell Recruitment with Novel Fv-based Dual-affinity Re-targeting Protein Leads to Potent Tumor Cytolysis and in Vivo B-cell Depletion. J. Mol. Biol. 2010, 399, 436–449. [Google Scholar] [CrossRef]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef] [Green Version]
- Del Bano, J.; Chames, P.; Baty, D.; Kerfelec, B. Taking up Cancer Immunotherapy Challenges: Bispecific Antibodies, the Path Forward? Antibodies 2015, 5, 1. [Google Scholar] [CrossRef]
- Rothe, A.; Sasse, S.; Topp, M.S.; Eichenauer, D.A.; Hummel, H.; Reiners, K.S.; Dietlein, M.; Kuhnert, G.; Kessler, J.; Buerkle, C.; et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 2015, 125, 4024–4031. [Google Scholar] [CrossRef]
- Reusch, U.; Duell, J.; Ellwanger, K.; Herbrecht, C.; Knackmuss, S.H.J.; Fucek, I.; Eser, M.; McAleese, F.; Molkenthin, V.; Le Gall, F.; et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19+ tumor cells. mAbs 2015, 7, 584–604. [Google Scholar] [CrossRef] [Green Version]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Mimoto, F.; Katada, H.; Kadono, S.; Igawa, T.; Kuramochi, T.; Muraoka, M.; Wada, Y.; Haraya, K.; Miyazaki, T.; Hattori, K. Engineered antibody Fc variant with selectively enhanced FcγRIIb binding over both FcγRIIaR131 and FcγRIIaH131. Protein Eng. Des. Sel. 2013, 26, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Schlothauer, T.; Herter, S.; Koller, C.F.; Grau-Richards, S.; Steinhart, V.; Spick, C.; Kubbies, M.; Klein, C.; Umaña, P.; Mössner, E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 2016, 29, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Goebeler, M.E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-cell engager (BiTE) antibody construct Blinatumomab for the treatment of Patients with relapsed/refractory non-Hodgkin lymphoma: Final results from a phase I study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Viardot, A.; Goebeler, M.E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (bite) antibody Blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Hutchings, M.; Morschhauser, F.; Iacoboni, G.; Carlo-Stella, C.; Offner, F.C.; Sureda, A.; Salles, G.; Martinez-Lopez, J.; Crump, M.; Thomas, D.N.; et al. Glofitamab, a Novel, Bivalent CD20-targeting T-cell-engaging bispecific antibody, induces durable complete remissions in relapsed or refractory B-Cell Lymphoma: A phase i trial. J. Clin. Oncol. 2021, 39, 1959–1970. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bartlett, N.L.; Assouline, S.; Yoon, S.-S.; Bosch, F.; Sehn, L.H.; Cheah, C.Y.; Shadman, M.; Gregory, G.P.; Ku, M.; et al. Mosunetuzumab Induces Complete Remissions in Poor Prognosis Non-Hodgkin Lymphoma Patients, Including Those Who Are Resistant to or Relapsing After Chimeric Antigen Receptor T-Cell (CAR-T) Therapies, Is Active in Treatment through Multiple Lines. Blood 2019, 134, 6. [Google Scholar] [CrossRef]
- Bannerji, R.; Allan, J.N.; Arnason, J.E.; Brown, J.R.; Advani, R.; Ansell, S.M.; O’Brien, S.M.; Duell, J.; Martin, P.; Joyce, R.M.; et al. Odronextamab (REGN1979), a Human CD20 x CD3 Bispecific Antibody, Induces Durable, Complete Responses in Patients with Highly Refractory B-Cell Non-Hodgkin Lymphoma, Including Patients Refractory to CAR T Therapy. Blood 2020, 136, 42–43. [Google Scholar] [CrossRef]
- Hutchings, M.; Mous, R.; Clausen, M.R.; Johnson, P.; Linton, K.M.; Chamuleau, M.E.D.; Lewis, D.J.; Sureda Balari, A.; Cunningham, D.; Oliveri, R.S.; et al. Subcutaneous Epcoritamab Induces Complete Responses with an Encouraging Safety Profile across Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma Subtypes, Including Patients with Prior CAR-T Therapy: Updated Dose Escalation Data. Blood 2020, 136, 45–46. [Google Scholar] [CrossRef]
- Topp, M.S.; Gökbuget, N.; Stein, A.S.; Zugmaier, G.; O’Brien, S.; Bargou, R.C.; Dombret, H.; Fielding, A.K.; Heffner, L.; Larson, R.A.; et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: A multicentre, single-arm, phase 2 study. Lancet Oncol. 2015, 16, 57–66. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Coyle, L.; Morley, N.J.; Rambaldi, A.; Mason, K.D.; Verhoef, G.; Furness, C.L.; Zhang, A.; Jung, A.S.; Cohan, D.; Franklin, J.L. Open-Label, phase 2 study of blinatumomab as second salvage therapy in adults with relapsed/refractory aggressive B-cell non-Hodgkin lymphoma. Leuk. Lymphoma 2020, 61, 2103–2112. [Google Scholar] [CrossRef]
- Bacac, M.; Colombetti, S.; Herter, S.; Sam, J.; Perro, M.; Chen, S.; Bianchi, R.; Richard, M.; Schoenle, A.; Nicolini, V.; et al. CD20-TCB with obinutuzumab pretreatment as next-generation treatment of hematologic malignancies. Clin. Cancer Res. 2018, 24, 4785–4797. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, M.J.; Morschhauser, F.; Iacoboni, G.; Carlo-Stella, C.; Offner, F.C.; Sureda, A.; Salles, G.; Martinez-Lopez, J.; Crump, M.; Lundberg, L.; et al. CD20-TCB in relapsed or refractory non-Hodgkin lymphoma: Durable complete responses and manageable safety observed at clinically relevant doses in phase I dose escalation. EHA Libr. J. 2020, 290, S241. [Google Scholar]
- Assouline, S.E.; Kim, W.S.; Sehn, L.H.; Schuster, S.J.; Cheah, C.Y.; Nastoupil, L.J.; Shadman, M.; Yoon, S.-S.; Matasar, M.J.; Diefenbach, C.; et al. Mosunetuzumab Shows Promising Efficacy in Patients with Multiply Relapsed Follicular Lymphoma: Updated Clinical Experience from a Phase I Dose-Escalation Trial. Blood 2020, 136, 42–44. [Google Scholar] [CrossRef]
- Matasar, M.J.; Cheah, C.Y.; Yoon, D.H.; Assouline, S.E.; Bartlett, N.L.; Ku, M.; Giri, P.; Johnston, A.; Flinn, I.W.; Goy, A.H.; et al. Subcutaneous Mosunetuzumab in Relapsed or Refractory B-Cell Lymphoma: Promising Safety and Encouraging Efficacy in Dose Escalation Cohorts. Blood 2020, 136, 45–46. [Google Scholar] [CrossRef]
- Olszewski, A.J.; Avigdor, A.; Babu, S.; Levi, I.; Abadi, U.; Holmes, H.; McKinney, M.; McCord, R.; Xie, Y.; Chen, C.; et al. Single-Agent Mosunetuzumab Is a Promising Safe and Efficacious Chemotherapy-Free Regimen for Elderly/Unfit Patients with Previously Untreated Diffuse Large B-Cell Lymphoma. Blood 2020, 136, 43–45. [Google Scholar] [CrossRef]
- Phillips, T.J.; Olszewski, A.J.; Munoz, J.; Kim, T.M.; Yoon, D.H.; Greil, R.; Westin, J.; Jaeger, U.; Canales, M.; Chen, C.; et al. Mosunetuzumab, a Novel CD20/CD3 Bispecific Antibody, in Combination with CHOP Confers High Response Rates in Patients with Diffuse Large B-Cell Lymphoma. Blood 2020, 136, 37–38. [Google Scholar] [CrossRef]
- Qi, J.; Li, X.; Peng, H.; Cook, E.M.; Dadashian, E.L.; Wiestner, A.; Park, H.J.; Rader, C. Potent and selective antitumor activity of a T cell-engaging bispecific antibody targeting a membrane-proximal epitope of ROR1. Proc. Natl. Acad. Sci. USA 2018, 115, E5467–E5476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granger, D.; Gohil, S.; Barbarulo, A.; Baccaro, A.; Muczynski, V.; Chester, K.; Germaschewski, F.; Batten, T.; Brown, K.; Cook, S.; et al. NVG-111, a novel ROR1xCD3 bispecific antibody for non-Hodgkin lymphoma. J. Clin. Oncol. 2021, 39, 7549. [Google Scholar] [CrossRef]
- Shah, N.N.; Sokol, L. Targeting CD22 for the Treatment of B-Cell Malignancies. ImmunoTargets Ther. 2021, 10, 225–236. [Google Scholar] [CrossRef]
- Geuijen, C.; Tacken, P.; Wang, L.C.; Klooster, R.; van Loo, P.F.; Zhou, J.; Mondal, A.; Liu, Y.B.; Kramer, A.; Condamine, T.; et al. A human CD137×PD-L1 bispecific antibody promotes anti-tumor immunity via context-dependent T cell costimulation and checkpoint blockade. Nat. Commun. 2021, 12, 1–19. [Google Scholar] [CrossRef]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]

{kind=link}
| Trial | Enrolled Patients | ORR | PFS | CRS | ICANS-Like | |
|---|---|---|---|---|---|---|
| Blinatumomab | Phase 1 [42] | r/r NHL (N = 38); aNHL (N = 5) | 64% (CR 36%) | Median PFS 1.5 years (median follow-up 4.6 years) | 20% > G3 | 22% G 3 |
| Phase 2 [43] | r/r aNHL (N = 25) | 43% (CR19%) | Median PFS 3.7 years (median follow-up 15 months) | 13% > G3 | 22% G 3 | |
| Glofitamab | Phase 1 [44] | r/r NHL (N = 52); aNHL (N = 10) | 67% (CR 54%) iNHL/61% (CR 54%) aNHL | NR | 4% > G3 | NR |
| Mosunetuzumab | Phase 1/2b [45] | r/r NHL (N = 270); aNHL (N = 116) | 63% (CR 43%) iNHL/37% (CR 19%) aNHL | NR | 1% G3; no G4 | 1.1% G3 |
| Odronetoxomab | Phase 1 [46] | r/r NHL (N = 136); aNHL (N = 78) | 55% (CR 55%)/33% (CR 21%) in CAR T r/r | NR | 7% > G3 | 4% G3 |
| Epcoritamab | Phase 1 [47] | r/r NHL (N = 68); aNHL (N = 46) | 80% (CR 60%) iNHL/91% (CR 55%) aNHL (for maximum dose) | NR | no G3 | 3% G3 |
| Drug | Status | Study | Target Population | Treatment | Intervention Model | Number of Partecipants |
|---|---|---|---|---|---|---|
| Blinatumomab | Recruiting | NCT03114865 | Acute lymphoblastic leukemia (ALL) and B-cell non-Hodgkin lymphoma (NHLs) | Blinatumomab | Open label, phase 1b/2 study | 64 |
| Blinatumomab | Recruiting | NCT02568553 | Relapsed non-Hodgkin lymphoma | Lenalidomide and blinatumomab | Open label, phase 1 study | 44 |
| Blinatumomab | Active, not recruiting | NCT03072771 | DLBCL post-ASCT | Blinatumomab | Open label, phase 1 study | 14 |
| Blinatumomab | Active, not recruiting | NCT03340766 | Relapsed or refractory DLBCL | Blinatumomab in combination with pembrolizumab | Phase 1b open label study | 31 |
| Glofitamab | Recruiting | NCT04914741 | Younger, higher-risk patients with diffuse large B cell lymphoma | Combination of glofitamab and R-CHOP or pola-RCHP | Open label, multi-centre, phase 1b/2, parallel arm study | 80 |
| Glofitamab | Recruiting | NCT04408638 | Relapsed/refractory diffuse large B-cell lymphoma | Glofitamab in combination with gemcitabine + pxaliplatin | Phase III, open label, multicenter, randomized study | 270 |
| Glofitamab | Recruiting | NCT03467373 | Relapsed/refractory NHLs and untreated diffuse large B-cell lymphoma | Glofitamab in combination with rituximab or pbinutuzumab plus CHOP | Phase 1B, multi-center, dose-finding study | 172 |
| Glofitamab | Recruiting | NCT03075696 | Relapsed/refractory B-cell non-Hodgkin’s lymphoma | Glofitamab as a single agent and in combination with obinutuzumab | Phase 1b/2, multicenter, open label, dose-escalation study | 860 |
| Glofitamab | Recruiting | NCT04077723 | Relapsed/refractory B-cell non-Hodgkin’s lymphoma | Combination with obinutuzumab and glofitamab | Phase 1b/2, open label, dose-escalation study | 362 |
| Glofitamab | Recruiting | NCT03533283 | Relapsed/refractory B-cell non-Hodgkin’s lymphoma | Glofitamab and atezolizumab or polatuzumab vedotin | Open label, single arm, multicenter, dose finding, phase 1b study | 140 |
| Glofitamab | Recruiting | NCT04657302 | Relapsed/refractory diffuse large B-cell lymphoma | Glofitamab as single agent | Phase I, open label, multicenter study | 30 |
| Glofitamab | Recruiting | NCT04980222 | Untreated diffuse large B-cell lymphoma | Glofitamab in combination with rituximab plus CHOP | Phase II, open label, multicenter study | 40 |
| Glofitamab | Recruiting | NCT04889716 | Relapsed or refractory diffuse large B-cell or transformed follicular lymphomas | Glofitamab or mosunetuzumab after CAR T-cells | Open label, phase 2 study | 42 |
| Glofitamab | Recruiting | NCT04703686 | Relapse/refractory lymphomas | Glofitamab after CAR T-cell therapy | Open label, phase 2 study | 78 |
| Glofitamab | Active, not recruiting | NCT04313608 | Relapsed or refractory diffuse large B-cell lymphoma and high-grade large B-cell lymphoma | Glofitamab or mosunetuzumab in combination with gemcitabine plus oxaliplatin | Phase 1b, open label, multicenter study | 20 |
| Epcoritamab | Recruiting | NCT04628494 | Relapsed/refractory diffuse large B-cell lymphoma | Epcoritamab | Randomized, open label, phase 3 trial | 480 |
| Epcoritamab | Recruiting | NCT04663347 | B-cell non-Hodgkin lymphoma | Epcoritamab in combination with other standard of care | Phase 1b/2, open label trial | 270 |
| Epcoritamab | Recruiting | NCT03625037 | Relapsed, progressive, or refractory B-Cell lymphoma | Epcoritamab GEN3013 (DuoBody®-CD3xCD20) | Phase 1/2, open label safety trial | 486 |
| Epcoritamab | Recruiting | NCT04542824 | Relapsed, progressive, or refractory B-cell lymphoma (JAPANESE PATIENTS) | Epcoritamab | Phase 1/2, open label, dose-escalation trial | 73 |
| Odronextamab | Recruiting | NCT03888105 | Relapsed or refractory B-cell non-Hodgkin lymphoma | Odronextamab | Open label, phase 2 study | 512 |
| Odronextamab | Recruiting | NCT02290951 | B-cell non-Hodgkin lymphoma (NHLs) and chronic lymphocytic leukemia (CLL) | Odronextamab | Open label, multi-center phase 1 study | 256 |
| Mosunetuzumab | Recruiting | NCT03671018 | B-cell non-Hodgkin lymphoma | Mosunetuzumab in combination with polatuzumab vedotin | Open label, randomized, multicenter, phase 1b/2 study | 262 |
| Mosunetuzumab | Recruiting | NCT03677154 | Diffuse large B-cell lymphoma following first-line immunochemotherapy or untreated diffuse large B-cell lymphoma | Monotherapy or in combination with polatuzumab vedotin | Phase 1/2 study | 188 |
| Mosunetuzumab | Active, not recruiting | NCT04313608 | Relapsed or refractory diffuse large B-cell lymphoma, and high-grade large B-cell lymphoma | Mosunetuzumab or glofitamab in combination with gemcitabine plus oxaliplatin | Phase 1b, open label, multicenter Study | 20 |
| Mosunetuzumab | Active, not recruiting | NCT03677141 | Untreated diffuse large B-cell lymphoma | Mosunetuzumab in combination with CHOP or CHP-polatuzumab vedotin | Phase 1b/2, open label, multicenter, randomized, Controlled study | 160 |
| Mosunetuzumab | Not yet recruiting | NCT04792502 | Untreated FL or MZL | Mosunetuzumab with lenalidomide augmentation | Phase 2, open label study | 52 |
| Mosunetuzumab | Not yet recruiting | NCT04889716 | Relapsed or refractory diffuse large B-cell or transformed follicular lymphomas | Mosunetuzumab or glofitamab after CAR T-cells | Phase 2 study | 42 |
| Mosunetuzumab | Recruiting | NCT02500407 | Relapsed or refractory B-cell NHLs and CLL | Mosunetuzumab as a single agent and combined with atezolizumab | Open label, multicenter, phase 1/2 study | 836 |
| Name | Target | Population | Phase | Treatment | NR Patients | Clinicaltrials.Gov Identifier |
|---|---|---|---|---|---|---|
| MCLA-145 | PD-L1/CD137 | r/r B-cell lymphoma | 1 | Dose escalation | 118 | NCT03922204 |
| TG-1801 | CD47/CD19 | r/r B-cell lymphoma | 1b | Alone or in combination with ublituximab | 60 | NCT03804996 |
| TNB-486 | CD19/CD3 | r/r B-cell lymphoma | 1 | Dose escalation | 80 | NCT04594642 |
| MT103 | CD19/CD3 | r/r B-cell lymphoma | 1 | Dose escalation/single agent | 76 | NCT00274742 |
| IMM0306 | CD20/CD3 | r/r B-cell lymphoma | 1 | Dose escalation | 90 | NCT04746131 |
| AK104 | PD-1/CTLA-4 | r/r peripheral T-cell lymphoma | 1b/2 | Dose escalation/single agent | 80 | NCT04444141 |
| IBI318 | anti-PD1/PD-L1 | r/r extranodal NK/T-cell lymphoma | 1b/2 | Dose escalation/single agent | 129 | NCT04602065 |
| JNJ-75348780 | CD22/CD3 | r/r B-cell lymphoma | 1 | Dose escalation | 120 | NCT04540796 |
| NVG-111 | ROR1/CD3 | CLL/SLL and MCL | 1b/2 | Dose escalation | 90 | NCT04763083 |
| GB261 | CD20/CD3 | r/r B-cell lymphoma | 1b/2 | Dose escalation/single agent | 460 | NCT04923048 |
| REGN1979 | CD20/CD3 | r/r B-cell lymphoma | 1 | Dose escalation/single agent | 172 | NCT02651662 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tavarozzi, R.; Manzato, E. The Role of Bispecific Antibodies in Non-Hodgkin’s Lymphoma: From Structure to Prospective Clinical Use. Antibodies 2022, 11, 16. https://doi.org/10.3390/antib11010016
Tavarozzi R, Manzato E. The Role of Bispecific Antibodies in Non-Hodgkin’s Lymphoma: From Structure to Prospective Clinical Use. Antibodies. 2022; 11(1):16. https://doi.org/10.3390/antib11010016
Chicago/Turabian StyleTavarozzi, Rita, and Enrica Manzato. 2022. "The Role of Bispecific Antibodies in Non-Hodgkin’s Lymphoma: From Structure to Prospective Clinical Use" Antibodies 11, no. 1: 16. https://doi.org/10.3390/antib11010016
APA StyleTavarozzi, R., & Manzato, E. (2022). The Role of Bispecific Antibodies in Non-Hodgkin’s Lymphoma: From Structure to Prospective Clinical Use. Antibodies, 11(1), 16. https://doi.org/10.3390/antib11010016