
Recent Advances in the Molecular Design and Applications of Multispecific Biotherapeutics

Abstract
1. Introduction
2. Major Classes of Multispecific Biotherapeutic Drugs

{kind=link}
{kind=link}
| Binding Modules | Binding Modules | Molecular Weight | Molecular Properties | References |
|---|---|---|---|---|
| Fragments of antibody (Fabs) |  | ~50 kDa | Medium half-life/less aggregated | [9,10] |
| Single-chain variable fragment (scFv) |  | ~25 kDa | Short half-life/less aggregated | [9,10] |
| Diabodies (Db) |  | ~25 kDa | Short half-life/less aggregated | [9,10] |
| Single-domain antibodies (VHH from llama or camel, VNAR from shark) |  | ~15 kDa | Short half-life/less aggregated | [9,10] |
| Synthetic peptides |  | ~3 kDa | Extremely short half-life/less aggregated | [41,42,43,44] |
| TCR domains |  | ~50 kDa | Medium half-life/less aggregated | [45,46,47,48] |
| Enzyme domains |  | ~50 kDa | Medium half-life/less aggregated | [49] |
| Small scaffolds (Affibody, Fynomers, Monobodies, DARPins, Knottins, VLRs, Nanoantibodies) |  | ~5–12 kDa | Short half-life/less aggregated | [21,50,51,52,53,54,55,56] |
| Chemical molecules |  | ~3 kDa | Short half-life/aggregated | [6,17] |
| Oligonucleotides |  | ~10 kDa | Short half-life/less aggregated | [19,57] |
| Technology Name | Mutations in First Chain | Mutations in Second Chain | References |
|---|---|---|---|
| Knobs-into-holes (Genentech) | HC1: S354C, T366W | HC2: Y349C, T366S, L368A, Y407V | [58,59] |
| Electrostatic steering (Amgen) | HC1:K409D, K392D | HC2: D399K, E356K | [60] |
| Electrostatic steering (Pfizer) | IgG1 HC1: D221E, P228E, L368E IgG2 HC1: C223E, P228E, L368E | IgG1 HC2: D221R, P228R, K409R IgG2 HC2: C223R, E225R, P228R, K409R | [61] |
| Electrostatic Steering (Merus) | HC1: L351D, L368E | HC2: L351K, T366K | [62] |
| Fab-arm exchange (Genmab) | HC1: K409R | HC2: F405L | [63,64] |
| SEED (EMD Serono) | HC1: IgG/IgA chimera | HC2: IgG/IgA chimera | [65] |
| LUZ-Y (Genentech) | HC1: cleavable leucine zipper | HC2: cleavable leucine zipper | [66] |
| HA-TF (Xencor) | HC1: S364H, F405A | HC2: Y349T, T394F | [67] |
| EW-RVT (EW-RVTs-s) (Ajou University) | HC1: K360E, K409W (Y349C) | HC2: Q347R, D399V, F405T (S354C) | [68,69] |
| ZW1 (VYAV-VLLW) (Zymeworks) | HC1: T350V, L351Y, F405A, Y407V | HC2: T350V, T366L, K392L, T394W | [70] |
| DMA-RRVV (SYMV-GDQA) (UNC/Eli Lily) | HC1:K360D, D399M, Y407A (Y349S, K370Y, T366M, K409V) | HC2: E345R, Q347R, T366V, K409V (E356G, E357D, S364Q, Y407A) | [71] |
| Protein A affinity (Regeneron) | HC1: H435R | None | [72] |
| Protein A and Protein G Avidity (Glenmark) | HC1: IgG3Fc, N82aS | HC2: M428G/N434A/K213V | [73] |
| CrossMab (Roche) | HC1:CL-VH | LC1:CH1-VL | [74] |
| Fab-Interface engineering (Eli Lily) | HC1: Q39K, R62E, H172A, F174G LC1: D1R, Q38D, L135Y, S176W | HC2: Q39Y LC2: Q38R | [75] |
| Fab-Interface electrostatic steering (Amgen) | HC1: Q39K, Q105K, S183D LC1: Q38D, A43D, S176K | HC2: Q39D, Q105D, S183K LC2: Q38K, A43K, S176D | [76] |
| Κλ-bodies (Novimmune SA) | LC1:κ | LC2:λ | [77] |
| Common Light Chain (Genentech & Merck KGaA) | Shared LC | Shared LC | [78,79] |
| Tetravalent IgG-like Charged CR3 mutant (Biomunex) | Mab1 CH1: T192E | Mab1 CL: N137K, S114A | [80] |
| Tetravalent IgG-like Hydrophobicity-polarity swap MUT4 mutant (Biomunex) | Mab1 CH1: L143Q, S188V | Mab1 CL: V133T, S176V | [80] |
| Fabs-in-Tandem (FIT-Ig) (EpimAb) | Long chain: VLA-CL-VHB-CH1-CH2-CH3 | Short Chain A: VHA-CH1, Short Chain B:VLB-CL | [81] |
| DuetMab (AstraZeneca) | CH1: F126C | CL: S121C | [82,83] |
| BEAT (Glenmark) | HC1-CH3: Residues from TCR α interface | HC2-CH3: Residues from TCR β interface | [84] |
| TCR CαCβ (Eli Lily) | HC1-CH1: TCR Cα | LC1:CL: TCR Cβ | [47] |
| WuXiBody (WuXi Biologics) | HC1-CH1: TCR Cβ | LC1-CL: TCR Cα | [48] |
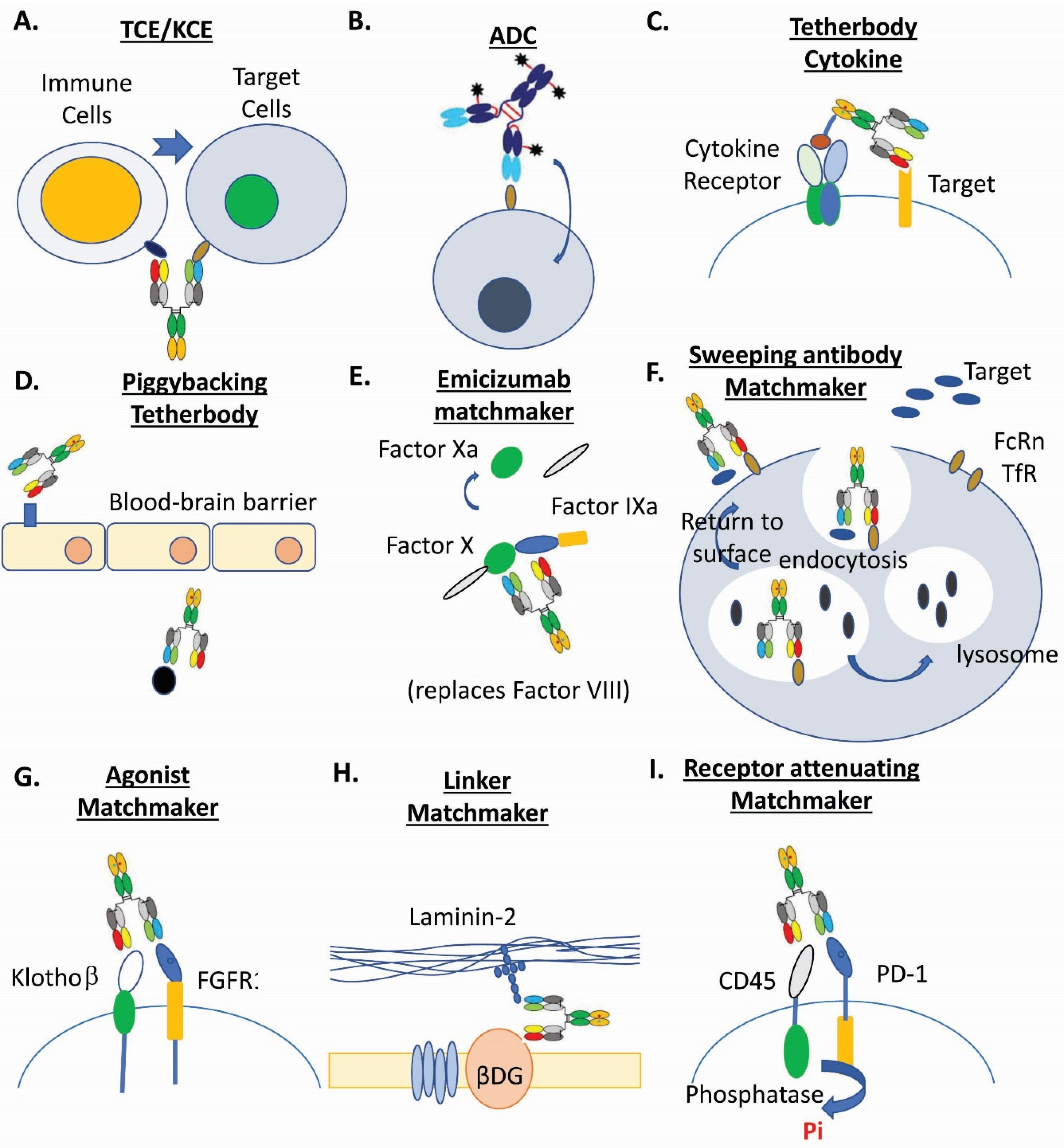
2.1. Immune-Cell Engagers

2.1.1. Multispecific T-Cell Engagers
2.1.2. Multispecific NK-Cell Engagers
2.2. Antibody Drug Conjugates
2.3. Tetherbodies
2.4. Biologic Matchmaker Drugs
2.5. Small-Scaffold Multispecific Modalities
3. Challenges and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hodgson, J. Refreshing the biologic pipeline 2020. Nat. Biotechnol. 2021, 39, 135–143. [Google Scholar] [CrossRef]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef]
- Deshaies, R.J. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 2020, 580, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Goebeler, M.E.; Bargou, R.C. T cell-engaging therapies—BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Nisonoff, A.; Rivers, M.M. Recombination of a mixture of univalent antibody fragments of different specificity. Arch. Biochem. Biophys. 1961, 93, 460–462. [Google Scholar] [CrossRef]
- Aalberse, R.C.; van der Gaag, R.; van Leeuwen, J. Serologic aspects of IgG4 antibodies. I. Prolonged immunization results in an IgG4-restricted response. J. Immunol. 1983, 130, 722–726. [Google Scholar]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Thakur, A.; Huang, M.; Lum, L.G. Bispecific antibody based therapeutics: Strengths and challenges. Blood Rev. 2018, 32, 339–347. [Google Scholar] [CrossRef]
- Staerz, U.D.; Kanagawa, O.; Bevan, M.J. Hybrid antibodies can target sites for attack by T cells. Nature 1985, 314, 628–631. [Google Scholar] [CrossRef]
- Perez, P.; Hoffman, R.W.; Shaw, S.; Bluestone, J.A.; Segal, D.M. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature 1985, 316, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Nagorsen, D.; Kufer, P.; Baeuerle, P.A.; Bargou, R. Blinatumomab: A historical perspective. Pharmacol. Ther. 2012, 136, 334–342. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Shim, H. Bispecific Antibodies and Antibody-Drug Conjugates for Cancer Therapy: Technological Considerations. Biomolecules 2020, 10, 360. [Google Scholar] [CrossRef]
- Abdollahpour-Alitappeh, M.; Lotfinia, M.; Gharibi, T.; Mardaneh, J.; Farhadihosseinabadi, B.; Larki, P.; Faghfourian, B.; Sepehr, K.S.; Abbaszadeh-Goudarzi, K.; Abbaszadeh-Goudarzi, G.; et al. Antibody-drug conjugates (ADCs) for cancer therapy: Strategies, challenges, and successes. J. Cell. Physiol. 2019, 234, 5628–5642. [Google Scholar] [CrossRef]
- Gauzy-Lazo, L.; Sassoon, I.; Brun, M.P. Advances in Antibody-Drug Conjugate Design: Current Clinical Landscape and Future Innovations. SLAS Discov. 2020, 25, 843–868. [Google Scholar] [CrossRef]
- Leung, D.; Wurst, J.M.; Liu, T.; Martinez, R.M.; Datta-Mannan, A.; Feng, Y. Antibody Conjugates-Recent Advances and Future Innovations. Antibodies 2020, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Voynov, V.; Adam, P.J.; Nixon, A.E.; Scheer, J.M. Discovery Strategies to Maximize the Clinical Potential of T-Cell Engaging Antibodies for the Treatment of Solid Tumors. Antibodies 2020, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Jost, C.; Pluckthun, A. Engineered proteins with desired specificity: DARPins, other alternative scaffolds and bispecific IgGs. Curr. Opin. Struct. Biol. 2014, 27, 102–112. [Google Scholar] [CrossRef]
- Caputi, A.P.; Navarra, P. Beyond antibodies: Ankyrins and DARPins. From basic research to drug approval. Curr. Opin. Pharmacol. 2020, 51, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R.; Naso, M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.A.; Desjarlais, J.R. Redirected T Cell Cytotoxicity in Cancer Therapy. Annu. Rev. Med. 2019, 70, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Ellerman, D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 2019, 154, 102–117. [Google Scholar] [CrossRef]
- Wu, Z.; Cheung, N.V. T cell engaging bispecific antibody (T-BsAb): From technology to therapeutics. Pharmacol. Ther. 2018, 182, 161–175. [Google Scholar] [CrossRef]
- Middelburg, J.; Kemper, K.; Engelberts, P.; Labrijn, A.F.; Schuurman, J.; van Hall, T. Overcoming Challenges for CD3-Bispecific Antibody Therapy in Solid Tumors. Cancers 2021, 13, 287. [Google Scholar] [CrossRef]
- Nie, S.; Wang, Z.; Moscoso-Castro, M.; D’Souza, P.; Lei, C.; Xu, J.; Gu, J. Biology drives the discovery of bispecific antibodies as innovative therapeutics. Antib. Ther. 2020, 3, 18–62. [Google Scholar] [CrossRef]
- Fan, G.; Wang, Z.; Hao, M.; Li, J. Bispecific antibodies and their applications. J. Hematol. Oncol. 2015, 8, 130. [Google Scholar] [CrossRef]
- Krah, S.; Sellmann, C.; Rhiel, L.; Schröter, C.; Dickgiesser, S.; Beck, J.; Zielonka, S.; Toleikis, L.; Hock, B.; Kolmar, H.; et al. Engineering bispecific antibodies with defined chain pairing. New Biotechnol. 2017, 39, 167–173. [Google Scholar] [CrossRef]
- Lambert, J.M.; Berkenblit, A. Antibody-Drug Conjugates for Cancer Treatment. Annu. Rev. Med. 2018, 69, 191–207. [Google Scholar] [CrossRef]
- Sawant, M.S.; Streu, C.N.; Wu, L.; Tessier, P.M. Toward Drug-Like Multispecific Antibodies by Design. Int. J. Mol. Sci. 2020, 21, 7496. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef]
- Gilbreth, R.N.; Koide, S. Structural insights for engineering binding proteins based on non-antibody scaffolds. Curr. Opin. Struct. Biol. 2012, 22, 413–420. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Engineered CH2 domains (nanoantibodies). MAbs 2009, 1, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Bonagura, V.R.; Morrison, S.L.; Bjorkman, P.J. Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry 1995, 34, 14649–14657. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Bjorkman, P.J. Fc receptors and their interactions with immunoglobulins. Annu. Rev. Cell Dev. Biol. 1996, 12, 181–220. [Google Scholar] [CrossRef]
- Krah, S.; Kolmar, H.; Becker, S.; Zielonka, S. Engineering IgG-Like Bispecific Antibodies-An Overview. Antibodies 2018, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Santich, B.H.; Park, J.A.; Tran, H.; Guo, H.F.; Huse, M.; Cheung, N.K. Interdomain spacing and spatial configuration drive the potency of IgG-[L]-scFv T cell bispecific antibodies. Sci. Transl. Med. 2020, 12, eaax1315. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Khare, P.; Wang, X.; Challa, D.K.; Greenberg, B.M.; Ober, R.J.; Ward, E.S. Selective Depletion of Antigen-Specific Antibodies for the Treatment of Demyelinating Disease. Mol. Ther. 2021, 29, 1312–1323. [Google Scholar] [CrossRef]
- Zhong, X.; Kieras, E.; Sousa, E.; D’Antona, A.; Baber, J.C.; He, T.; Desharnais, J.; Wood, L.; Luxenberg, D.; Stahl, M.; et al. Pyroglutamate and O-Linked Glycan Determine Functional Production of Anti-IL17A and Anti-IL22 Peptide-Antibody Bispecific Genetic Fusions. J. Biol. Chem. 2013, 288, 1409–1419. [Google Scholar] [CrossRef] [PubMed]
- Kanakaraj, P.; Puffer, B.A.; Yao, X.T.; Kankanala, S.; Boyd, E.; Shah, R.R.; Wang, G.; Patel, D.; Krishnamurthy, R.; Kaithamana, S.; et al. Simultaneous targeting of TNF and Ang2 with a novel bispecific antibody enhances efficacy in an in vivo model of arthritis. MAbs 2012, 4, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Suresh, M.R. Bispecific antibodies as novel bioconjugates. Bioconjug. Chem. 1998, 9, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Trier, N.; Hansen, P.; Houen, G. Peptides, Antibodies, Peptide Antibodies and More. Int. J. Mol. Sci. 2019, 20, 6289. [Google Scholar] [CrossRef] [PubMed]
- Lowe, K.L.; Cole, D.; Kenefeck, R.; OKelly, I.; Lepore, M.; Jakobsen, B.K. Novel TCR-based biologics: Mobilising T cells to warm ‘cold’ tumours. Cancer Treat. Rev. 2019, 77, 35–43. [Google Scholar] [CrossRef]
- Oates, J.; Hassan, N.J.; Jakobsen, B.K. ImmTACs for targeted cancer therapy: Why, what, how, and which. Mol. Immunol. 2015, 67, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Sereno, A.J.; Huang, F.; Zhang, K.; Batt, M.; Fitchett, J.R.; He, D.; Rick, H.L.; Conner, E.M.; Demarest, S.J. Protein design of IgG/TCR chimeras for the co-expression of Fab-like moieties within bispecific antibodies. MAbs 2015, 7, 364–376. [Google Scholar] [CrossRef]
- Guo, G.; Han, J.; Wang, Y.; Li, Y. A potential downstream platform approach for WuXiBody-based IgG-like bispecific antibodies. Protein Expr. Purif. 2020, 173, 105647. [Google Scholar] [CrossRef]
- Dai, Z.; Zhang, X.N.; Nasertorabi, F.; Cheng, Q.; Li, J.; Katz, B.B.; Smbatyan, G.; Pei, H.; Louie, S.G.; Lenz, H.J.; et al. Synthesis of site-specific antibody-drug conjugates by ADP-ribosyl cyclases. Sci. Adv. 2020, 6, eaba6752. [Google Scholar] [CrossRef] [PubMed]
- Sha, F.; Salzman, G.; Gupta, A.; Koide, S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, S.; Gräslund, T.; Karlström, A.E.; Frejd, F.Y.; Nygren, P.Å.; Löfblom, J. Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef]
- Lipovšek, D.; Carvajal, I.; Allentoff, A.J.; Barros, A., Jr.; Brailsford, J.; Cong, Q.; Cotter, P.; Gangwar, S.; Hollander, C.; Lafont, V.; et al. Adnectin-drug conjugates for Glypican-3-specific delivery of a cytotoxic payload to tumors. Protein Eng. Des. Sel. 2018, 31, 159–171. [Google Scholar] [CrossRef]
- Alder, M.N.; Rogozin, I.B.; Iyer, L.M.; Glazko, G.V.; Cooper, M.D.; Pancer, Z. Diversity and function of adaptive immune receptors in a jawless vertebrate. Science 2005, 310, 1970–1973. [Google Scholar] [CrossRef] [PubMed]
- Kintzing, J.R.; Cochran, J.R. Engineered knottin peptides as diagnostics, therapeutics, and drug delivery vehicles. Curr. Opin. Chem. Biol. 2016, 34, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Brack, S.; Attinger-Toller, I.; Schade, B.; Mourlane, F.; Klupsch, K.; Woods, R.; Hachemi, H.; von der Bey, U.; Koenig-Friedrich, S.; Bertschinger, J.; et al. A bispecific HER2-targeting FynomAb with superior antitumor activity and novel mode of action. Mol. Cancer Ther. 2014, 13, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Gong, R.; Wang, Y.; Ying, T.; Dimitrov, D.S. Bispecific engineered antibody domains (nanoantibodies) that interact noncompetitively with an HIV-1 neutralizing epitope and FcRn. PLoS ONE 2012, 7, e42288. [Google Scholar] [CrossRef] [PubMed]
- Benizri, S.; Gissot, A.; Martin, A.; Vialet, B.; Grinstaff, M.W.; Barthélémy, P. Bioconjugated Oligonucleotides: Recent Developments and Therapeutic Applications. Bioconjug. Chem. 2019, 30, 366–383. [Google Scholar] [CrossRef]
- Atwell, S.; Ridgway, J.B.; Wells, J.A.; Carter, P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J. Mol. Biol. 1997, 270, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Ng, S.B.; Born, T.; Retter, M.; Manchulenko, K.; et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 2010, 285, 19637–19646. [Google Scholar] [CrossRef]
- Strop, P.; Ho, W.H.; Boustany, L.M.; Abdiche, Y.N.; Lindquist, K.C.; Farias, S.E.; Rickert, M.; Appah, C.T.; Pascua, E.; Radcliffe, T.; et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J. Mol. Biol. 2012, 420, 204–219. [Google Scholar] [CrossRef] [PubMed]
- De Nardis, C.; Hendriks, L.J.; Poirier, E.; Arvinte, T.; Gros, P.; Bakker, A.B.; de Kruif, J. A new approach for generating bispecific antibodies based on a common light chain format and the stable architecture of human immunoglobulin G1. J. Biol. Chem. 2017, 292, 14706–14717. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Rispens, T.; Meesters, J.; Rose, R.J.; den Bleker, T.H.; Loverix, S.; van den Bremer, E.T.; Neijssen, J.; Vink, T.; Lasters, I.; et al. Species-specific determinants in the IgG CH3 domain enable Fab-arm exchange by affecting the noncovalent CH3-CH3 interaction strength. J. Immunol. 2011, 187, 3238–3246. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Meesters, J.I.; de Goeij, B.E.; van den Bremer, E.T.; Neijssen, J.; van Kampen, M.D.; Strumane, K.; Verploegen, S.; Kundu, A.; Gramer, M.J.; et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc. Natl. Acad. Sci. USA 2013, 110, 5145–5150. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H.; Aperlo, C.; Li, Y.; Kurosawa, E.; Lan, Y.; Lo, K.M.; Huston, J.S. SEEDbodies: Fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng. Des. Sel. 2010, 23, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Wranik, B.J.; Christensen, E.L.; Schaefer, G.; Jackman, J.K.; Vendel, A.C.; Eaton, D. LUZ-Y, a novel platform for the mammalian cell production of full-length IgG-bispecific antibodies. J. Biol. Chem. 2012, 287, 43331–43339. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.L.; Bautista, C.; Pong, E.; Nguyen, D.H.; Jacinto, J.; Eivazi, A.; Muchhal, U.S.; Karki, S.; Chu, S.Y.; Lazar, G.A. A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. MAbs 2011, 3, 546–557. [Google Scholar] [CrossRef]
- Choi, H.J.; Kim, Y.J.; Lee, S.; Kim, Y.S. A heterodimeric Fc-based bispecific antibody simultaneously targeting VEGFR-2 and Met exhibits potent antitumor activity. Mol. Cancer Ther. 2013, 12, 2748–2759. [Google Scholar] [CrossRef]
- Choi, H.J.; Seok, S.H.; Kim, Y.J.; Seo, M.D.; Kim, Y.S. Crystal structures of immunoglobulin Fc heterodimers reveal the molecular basis for heterodimer formation. Mol. Immunol. 2015, 65, 377–383. [Google Scholar] [CrossRef]
- Von Kreudenstein, T.S.; Escobar-Carbrera, E.; Lario, P.I.; D’Angelo, I.; Brault, K.; Kelly, J.F.; Durocher, Y.; Baardsnes, J.; Woods, R.J.; Xie, M.H.; et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: Quality by molecular design. MAbs 2013, 5, 646–654. [Google Scholar] [CrossRef]
- Leaver-Fay, A.; Froning, K.J.; Atwell, S.; Aldaz, H.; Pustilnik, A.; Lu, F.; Huang, F.; Yuan, R.; Hassanali, S.; Chamberlain, A.K.; et al. Computationally Designed Bispecific Antibodies using Negative State Repertoires. Structure 2016, 24, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.J.; Olson, K.; Haber, L.J.; Varghese, B.; Duramad, P.; Tustian, A.D.; Oyejide, A.; Kirshner, J.R.; Canova, L.; Menon, J.; et al. A novel, native-format bispecific antibody triggering T-cell killing of B-cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci. Rep. 2015, 5, 17943. [Google Scholar] [CrossRef] [PubMed]
- Ollier, R.; Wassmann, P.; Monney, T.; Ries Fecourt, C.; Gn, S.; CA, V.; Ayoub, D.; Stutz, C.; Gudi, G.S.; Blein, S. Single-step Protein A and Protein G avidity purification methods to support bispecific antibody discovery and development. MAbs 2019, 11, 1464–1478. [Google Scholar] [CrossRef]
- Schaefer, W.; Regula, J.T.; Bähner, M.; Schanzer, J.; Croasdale, R.; Dürr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef]
- Lewis, S.M.; Wu, X.; Pustilnik, A.; Sereno, A.; Huang, F.; Rick, H.L.; Guntas, G.; Leaver-Fay, A.; Smith, E.M.; Ho, C.; et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat. Biotechnol. 2014, 32, 191–198. [Google Scholar] [CrossRef]
- Liu, Z.; Leng, E.C.; Gunasekaran, K.; Pentony, M.; Shen, M.; Howard, M.; Stoops, J.; Manchulenko, K.; Razinkov, V.; Liu, H.; et al. A novel antibody engineering strategy for making monovalent bispecific heterodimeric IgG antibodies by electrostatic steering mechanism. J. Biol. Chem. 2015, 290, 7535–7562. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.; Elson, G.; Magistrelli, G.; Dheilly, E.; Fouque, N.; Laurendon, A.; Gueneau, F.; Ravn, U.; Depoisier, J.F.; Moine, V.; et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat. Commun. 2015, 6, 6113. [Google Scholar] [CrossRef]
- Jackman, J.; Chen, Y.; Huang, A.; Moffat, B.; Scheer, J.M.; Leong, S.R.; Lee, W.P.; Zhang, J.; Sharma, N.; Lu, Y.; et al. Development of a two-part strategy to identify a therapeutic human bispecific antibody that inhibits IgE receptor signaling. J. Biol. Chem. 2010, 285, 20850–20859. [Google Scholar] [CrossRef]
- Krah, S.; Schröter, C.; Eller, C.; Rhiel, L.; Rasche, N.; Beck, J.; Sellmann, C.; Günther, R.; Toleikis, L.; Hock, B.; et al. Generation of human bispecific common light chain antibodies by combining animal immunization and yeast display. Protein Eng. Des. Sel. 2017, 30, 291–301. [Google Scholar] [CrossRef]
- Golay, J.; Choblet, S.; Iwaszkiewicz, J.; Cérutti, P.; Ozil, A.; Loisel, S.; Pugnière, M.; Ubiali, G.; Zoete, V.; Michielin, O.; et al. Design and Validation of a Novel Generic Platform for the Production of Tetravalent IgG1-like Bispecific Antibodies. J. Immunol. 2016, 196, 3199–3211. [Google Scholar] [CrossRef]
- Gong, S.; Ren, F.; Wu, D.; Wu, X.; Wu, C. Fabs-in-tandem immunoglobulin is a novel and versatile bispecific design for engaging multiple therapeutic targets. MAbs 2017, 9, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Mazor, Y.; Oganesyan, V.; Yang, C.; Hansen, A.; Wang, J.; Liu, H.; Sachsenmeier, K.; Carlson, M.; Gadre, D.V.; Borrok, M.J.; et al. Improving target cell specificity using a novel monovalent bispecific IgG design. MAbs 2015, 7, 377–389. [Google Scholar] [CrossRef]
- Mazor, Y.; Hansen, A.; Yang, C.; Chowdhury, P.S.; Wang, J.; Stephens, G.; Wu, H.; Dall’Acqua, W.F. Insights into the molecular basis of a bispecific antibody’s target selectivity. MAbs 2015, 7, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Moretti, P.; Skegro, D.; Ollier, R.; Wassmann, P.; Aebischer, C.; Laurent, T.; Schmid-Printz, M.; Giovannini, R.; Blein, S.; Bertschinger, M. BEAT the bispecific challenge: A novel and efficient platform for the expression of bispecific IgGs. BMC Proc. 2013, 7 (Suppl. 6), O9. [Google Scholar] [CrossRef]
- Seimetz, D.; Lindhofer, H.; Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat. Rev. 2010, 36, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Root, A.R.; Cao, W.; Li, B.; LaPan, P.; Meade, C.; Sanford, J.; Jin, M.; O’Sullivan, C.; Cummins, E.; Lambert, M.; et al. Development of PF-06671008, a Highly Potent Anti-P-cadherin/Anti-CD3 Bispecific DART Molecule with Extended Half-Life for the Treatment of Cancer. Antibodies 2016, 5, 6. [Google Scholar] [CrossRef]
- Mathur, D.; Root, A.R.; Bugaj-Gaweda, B.; Bisulco, S.; Tan, X.; Fang, W.; Kearney, J.C.; Lucas, J.; Guffroy, M.; Golas, J.; et al. A Novel GUCY2C-CD3 T-Cell Engaging Bispecific Construct (PF-07062119) for the Treatment of Gastrointestinal Cancers. Clin. Cancer Res. 2020, 26, 2188–2202. [Google Scholar] [CrossRef]
- Root, A.R.; Guntas, G.; Katragadda, M.; Apgar, J.R.; Narula, J.; Chang, C.S.; Hanscom, S.; McKenna, M.; Wade, J.; Meade, C.; et al. Discovery and optimization of a novel anti-GUCY2c x CD3 bispecific antibody for the treatment of solid tumors. MAbs 2021, 13, 1850395. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Foletti, D.; Liu, X.; Ding, S.; Witt, J.M.; Hasa-Moreno, A.; Rickert, M.; Holz, C.; Aschenbrenner, L.; Yang, A.H.; et al. Targeting CLDN18.2 by CD3 Bispecific and ADC Modalities for the Treatments of Gastric and Pancreatic Cancer. Sci. Rep. 2019, 9, 8420. [Google Scholar] [CrossRef]
- Yeung, Y.A.; Krishnamoorthy, V.; Dettling, D.; Sommer, C.; Poulsen, K.; Ni, I.; Pham, A.; Chen, W.; Liao-Chan, S.; Lindquist, K.; et al. An Optimized Full-Length FLT3/CD3 Bispecific Antibody Demonstrates Potent Anti-leukemia Activity and Reversible Hematological Toxicity. Mol. Ther. 2020, 28, 889–900. [Google Scholar] [CrossRef]
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007, 25, 1290–1297. [Google Scholar] [CrossRef]
- Bardwell, P.D.; Staron, M.M.; Liu, J.; Tao, Q.; Scesney, S.; Bukofzer, G.; Rodriguez, L.E.; Choi, C.H.; Wang, J.; Chang, Q.; et al. Potent and conditional redirected T cell killing of tumor cells using Half DVD-Ig. Protein Cell 2018, 9, 121–129. [Google Scholar] [CrossRef]
- Correnti, C.E.; Laszlo, G.S.; de van der Schueren, W.J.; Godwin, C.D.; Bandaranayake, A.; Busch, M.A.; Gudgeon, C.J.; Bates, O.M.; Olson, J.M.; Mehlin, C.; et al. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia 2018, 32, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, A.; Vallée, F.; Beil, C.; Lange, C.; Baurin, N.; Beninga, J.; Capdevila, C.; Corvey, C.; Dupuy, A.; Ferrari, P.; et al. CODV-Ig, a universal bispecific tetravalent and multifunctional immunoglobulin format for medical applications. MAbs 2016, 8, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Neri, D. Antibody-Cytokine Fusions: Versatile Products for the Modulation of Anticancer Immunity. Cancer Immunol. Res. 2019, 7, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Delivery of Biologics across the Blood-Brain Barrier with Molecular Trojan Horse Technology. BioDrugs 2017, 31, 503–519. [Google Scholar] [CrossRef]
- Yu, Y.J.; Atwal, J.K.; Zhang, Y.; Tong, R.K.; Wildsmith, K.R.; Tan, C.; Bien-Ly, N.; Hersom, M.; Maloney, J.A.; Meilandt, W.J.; et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci. Transl. Med. 2014, 6, 261ra154. [Google Scholar] [CrossRef]
- Yu, Y.J.; Zhang, Y.; Kenrick, M.; Hoyte, K.; Luk, W.; Lu, Y.; Atwal, J.; Elliott, J.M.; Prabhu, S.; Watts, R.J.; et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 2011, 3, 84ra44. [Google Scholar] [CrossRef]
- Sampei, Z.; Igawa, T.; Soeda, T.; Okuyama-Nishida, Y.; Moriyama, C.; Wakabayashi, T.; Tanaka, E.; Muto, A.; Kojima, T.; Kitazawa, T.; et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS ONE 2013, 8, e57479. [Google Scholar]
- Kitazawa, T.; Shima, M. Emicizumab, a humanized bispecific antibody to coagulation factors IXa and X with a factor VIIIa-cofactor activity. Int. J. Hematol. 2020, 111, 20–30. [Google Scholar] [CrossRef]
- Neiveyans, M.; Melhem, R.; Arnoult, C.; Bourquard, T.; Jarlier, M.; Busson, M.; Laroche, A.; Cerutti, M.; Pugnière, M.; Ternant, D.; et al. A recycling anti-transferrin receptor-1 monoclonal antibody as an efficient therapy for erythroleukemia through target up-regulation and antibody-dependent cytotoxic effector functions. MAbs 2019, 11, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Baruch, A.; Wong, C.; Chinn, L.W.; Vaze, A.; Sonoda, J.; Gelzleichter, T.; Chen, S.; Lewin-Koh, N.; Morrow, L.; Dheerendra, S.; et al. Antibody-mediated activation of the FGFR1/Klothobeta complex corrects metabolic dysfunction and alters food preference in obese humans. Proc. Natl. Acad. Sci. USA 2020, 117, 28992–29000. [Google Scholar] [CrossRef]
- Shi, S.Y.; Lu, Y.W.; Liu, Z.; Stevens, J.; Murawsky, C.M.; Wilson, V.; Hu, Z.; Richards, W.G.; Michaels, M.L.; Zhang, J.; et al. A biparatopic agonistic antibody that mimics fibroblast growth factor 21 ligand activity. J. Biol. Chem. 2018, 293, 5909–5919. [Google Scholar] [CrossRef]
- Gumlaw, N.; Sevigny, L.M.; Zhao, H.; Luo, Z.; Bangari, D.S.; Masterjohn, E.; Chen, Y.; McDonald, B.; Magnay, M.; Travaline, T.; et al. biAb Mediated Restoration of the Linkage between Dystroglycan and Laminin-211 as a Therapeutic Approach for alpha-Dystroglycanopathies. Mol. Ther. 2020, 28, 664–676. [Google Scholar] [CrossRef]
- Fernandes, R.A.; Su, L.; Nishiga, Y.; Ren, J.; Bhuiyan, A.M.; Cheng, N.; Kuo, C.J.; Picton, L.K.; Ohtsuki, S.; Majzner, R.G.; et al. Immune receptor inhibition through enforced phosphatase recruitment. Nature 2020, 586, 779–784. [Google Scholar] [CrossRef]
- Kontermann, R.E. Half-life extended biotherapeutics. Expert Opin. Biol. Ther. 2016, 16, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, V.; Wang, C.W.; Geething, N.C.; Spink, B.J.; Campbell, A.; To, W.; Scholle, M.D.; Yin, Y.; Yao, Y.; Bogin, O.; et al. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat. Biotechnol. 2009, 27, 1186–1190. [Google Scholar] [CrossRef]
- Hutt, M.; Färber-Schwarz, A.; Unverdorben, F.; Richter, F.; Kontermann, R.E. Plasma half-life extension of small recombinant antibodies by fusion to immunoglobulin-binding domains. J. Biol. Chem. 2012, 287, 4462–4469. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Pagès, F.; Galon, J. Tumor immunosurveillance in human cancers. Cancer Metastasis Rev. 2011, 30, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Aptsiauri, N.; Ruiz-Cabello, F.; Garrido, F. The transition from HLA-I positive to HLA-I negative primary tumors: The road to escape from T-cell responses. Curr. Opin. Immunol. 2018, 51, 123–132. [Google Scholar] [CrossRef] [PubMed]
- de la Roche, M.; Asano, Y.; Griffiths, G.M. Origins of the cytolytic synapse. Nat. Rev. Immunol. 2016, 16, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Isaaz, S.; Baetz, K.; Olsen, K.; Podack, E.; Griffiths, G.M. Serial killing by cytotoxic T lymphocytes: T cell receptor triggers degranulation, re-filling of the lytic granules and secretion of lytic proteins via a non-granule pathway. Eur. J. Immunol. 1995, 25, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.R.; Sukumaran, S.; Hristopoulos, M.; Totpal, K.; Stainton, S.; Lu, E.; Wong, A.; Tam, L.; Newman, R.; Vuillemenot, B.R.; et al. An anti-CD3/anti-CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood 2017, 129, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Bortoletto, N.; Scotet, E.; Myamoto, Y.; D’Oro, U.; Lanzavecchia, A. Optimizing anti-CD3 affinity for effective T cell targeting against tumor cells. Eur. J. Immunol. 2002, 32, 3102–3107. [Google Scholar] [CrossRef]
- List, T.; Neri, D. Biodistribution studies with tumor-targeting bispecific antibodies reveal selective accumulation at the tumor site. MAbs 2012, 4, 775–783. [Google Scholar] [CrossRef]
- Mandikian, D.; Takahashi, N.; Lo, A.A.; Li, J.; Eastham-Anderson, J.; Slaga, D.; Ho, J.; Hristopoulos, M.; Clark, R.; Totpal, K.; et al. Relative Target Affinities of T-Cell-Dependent Bispecific Antibodies Determine Biodistribution in a Solid Tumor Mouse Model. Mol. Cancer Ther. 2018, 17, 776–785. [Google Scholar] [CrossRef]
- Reusch, U.; Harrington, K.H.; Gudgeon, C.J.; Fucek, I.; Ellwanger, K.; Weichel, M.; Knackmuss, S.H.; Zhukovsky, E.A.; Fox, J.A.; Kunkel, L.A.; et al. Characterization of CD33/CD3 Tetravalent Bispecific Tandem Diabodies (TandAbs) for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2016, 22, 5829–5838. [Google Scholar] [CrossRef]
- Reusch, U.; Duell, J.; Ellwanger, K.; Herbrecht, C.; Knackmuss, S.H.; Fucek, I.; Eser, M.; McAleese, F.; Molkenthin, V.; Le Gall, F.; et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. MAbs 2015, 7, 584–604. [Google Scholar] [CrossRef] [PubMed]
- Liddy, N.; Bossi, G.; Adams, K.J.; Lissina, A.; Mahon, T.M.; Hassan, N.J.; Gavarret, J.; Bianchi, F.C.; Pumphrey, N.J.; Ladell, K.; et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med. 2012, 18, 980–987. [Google Scholar] [CrossRef]
- Hsiue, E.H.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef]
- Weidanz, J. Targeting cancer with bispecific antibodies. Science 2021, 371, 996–997. [Google Scholar] [CrossRef] [PubMed]
- Pfosser, A.; Brandl, M.; Salih, H.; Grosse-Hovest, L.; Jung, G. Role of target antigen in bispecific-antibody-mediated killing of human glioblastoma cells: A pre-clinical study. Int. J. Cancer 1999, 80, 612–616. [Google Scholar] [CrossRef]
- Bluemel, C.; Hausmann, S.; Fluhr, P.; Sriskandarajah, M.; Stallcup, W.B.; Baeuerle, P.A.; Kufer, P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol. Immunother. 2010, 59, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, K.; Wiseman, D.; Brown, M.H.; Gould, K.; van der Merwe, P.A. T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature 2005, 436, 578–582. [Google Scholar] [CrossRef]
- Davis, S.J.; van der Merwe, P.A. The kinetic-segregation model: TCR triggering and beyond. Nat. Immunol. 2006, 7, 803–809. [Google Scholar] [CrossRef]
- James, J.R.; Vale, R.D. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature 2012, 487, 64–69. [Google Scholar] [CrossRef]
- Chen, W.; Yang, F.; Wang, C.; Narula, J.; Pascua, E.; Ni, I.; Ding, S.; Deng, X.; Chu, M.L.; Pham, A.; et al. One size does not fit all: Navigating the multi-dimensional space to optimize T-cell engaging protein therapeutics. MAbs 2021, 13, 1871171. [Google Scholar] [CrossRef]
- Braig, F.; Brandt, A.; Goebeler, M.; Tony, H.P.; Kurze, A.K.; Nollau, P.; Bumm, T.; Böttcher, S.; Bargou, R.C.; Binder, M. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood 2017, 129, 100–104. [Google Scholar] [CrossRef]
- Dimasi, N.; Fleming, R.; Hay, C.; Woods, R.; Xu, L.; Wu, H.; Gao, C. Development of a Trispecific Antibody Designed to Simultaneously and Efficiently Target Three Different Antigens on Tumor Cells. Mol. Pharm. 2015, 12, 3490–3501. [Google Scholar] [CrossRef]
- Skokos, D.; Waite, J.C.; Haber, L.; Crawford, A.; Hermann, A.; Ullman, E.; Slim, R.; Godin, S.; Ajithdoss, D.; Ye, X.; et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci. Transl. Med. 2020, 12, eaaw7888. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; June, C.H. Trispecific antibodies offer a third way forward for anticancer immunotherapy. Nature 2019, 575, 450–451. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Waite, J.C.; Wang, B.; Haber, L.; Hermann, A.; Ullman, E.; Ye, X.; Dudgeon, D.; Slim, R.; Ajithdoss, D.K.; Godin, S.J.; et al. Tumor-targeted CD28 bispecific antibodies enhance the antitumor efficacy of PD-1 immunotherapy. Sci. Transl. Med. 2020, 12, eaba2325. [Google Scholar] [CrossRef]
- Herrmann, M.; Krupka, C.; Deiser, K.; Brauchle, B.; Marcinek, A.; Ogrinc Wagner, A.; Rataj, F.; Mocikat, R.; Metzeler, K.H.; Spiekermann, K.; et al. Bifunctional PD-1 × alphaCD3 × alphaCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood 2018, 132, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.A.; Ciavattone, N.G.; Atkinson, R.; Woldergerima, N.; Wolf, J.; Clements, V.K.; Sinha, P.; Poudel, M.; Ostrand-Rosenberg, S. CD3xPDL1 bi-specific T cell engager (BiTE) simultaneously activates T cells and NKT cells, kills PDL1(+) tumor cells, and extends the survival of tumor-bearing humanized mice. Oncotarget 2017, 8, 57964–57980. [Google Scholar] [CrossRef] [PubMed]
- Herberman, R.B.; Nunn, M.E.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int. J. Cancer 1975, 16, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Davis, Z.B.; Vallera, D.A.; Miller, J.S.; Felices, M. Natural killer cells unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin. Immunol. 2017, 31, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Saw, P.E.; Song, E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol. Immunol. 2020, 17, 451–461. [Google Scholar] [CrossRef]
- Fang, F.; Xiao, W.; Tian, Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017, 31, 37–54. [Google Scholar] [CrossRef]
- Hartmann, F.; Renner, C.; Jung, W.; Deisting, C.; Juwana, M.; Eichentopf, B.; Kloft, M.; Pfreundschuh, M. Treatment of refractory Hodgkin’s disease with an anti-CD16/CD30 bispecific antibody. Blood 1997, 89, 2042–2047. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, F.; Renner, C.; Jung, W.; da Costa, L.; Tembrink, S.; Held, G.; Sek, A.; König, J.; Bauer, S.; Kloft, M.; et al. Anti-CD16/CD30 bispecific antibody treatment for Hodgkin’s disease: Role of infusion schedule and costimulation with cytokines. Clin. Cancer Res. 2001, 7, 1873–1881. [Google Scholar]
- Chan, W.K.; Kang, S.; Youssef, Y.; Glankler, E.N.; Barrett, E.R.; Carter, A.M.; Ahmed, E.H.; Prasad, A.; Chen, L.; Zhang, J.; et al. A CS1-NKG2D Bispecific Antibody Collectively Activates Cytolytic Immune Cells against Multiple Myeloma. Cancer Immunol. Res. 2018, 6, 776–787. [Google Scholar] [CrossRef]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed]
- Hodgins, J.J.; Khan, S.T.; Park, M.M.; Auer, R.C.; Ardolino, M. Killers 2.0: NK cell therapies at the forefront of cancer control. J. Clin. Investig. 2019, 129, 3499–3510. [Google Scholar] [CrossRef] [PubMed]
- Cytovia, Cytovia Is Developing NKp46 Multifunctional Engagers with the Potential for Better Disease Control without Hurting the Healthy Cells. 2021. Available online: https://www.cytoviatx.com/multi-specific-antibodies (accessed on 29 March 2021).
- Pahl, J.H.; Koch, J.; Götz, J.J.; Arnold, A.; Reusch, U.; Gantke, T.; Rajkovic, E.; Treder, M.; Cerwenka, A. CD16A Activation of NK Cells Promotes NK Cell Proliferation and Memory-Like Cytotoxicity against Cancer Cells. Cancer Immunol. Res. 2018, 6, 517–527. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Glasner, A.; Levi, A.; Enk, J.; Isaacson, B.; Viukov, S.; Orlanski, S.; Scope, A.; Neuman, T.; Enk, C.D.; Hanna, J.H.; et al. NKp46 Receptor-Mediated Interferon-gamma Production by Natural Killer Cells Increases Fibronectin 1 to Alter Tumor Architecture and Control Metastasis. Immunity 2018, 48, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef]
- Affimed, Actualizing the Untapped Potential of the Innate Immune System-Affimed’s Approach to Advancing Immuno-Oncology. 2021. Available online: https://www.affimed.com/ (accessed on 29 March 2021).
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. Engl. 2014, 53, 3796–3827. [Google Scholar] [CrossRef]
- Sellmann, C.; Doerner, A.; Knuehl, C.; Rasche, N.; Sood, V.; Krah, S.; Rhiel, L.; Messemer, A.; Wesolowski, J.; Schuette, M.; et al. Balancing Selectivity and Efficacy of Bispecific Epidermal Growth Factor Receptor (EGFR) x c-MET Antibodies and Antibody-Drug Conjugates. J. Biol. Chem. 2016, 291, 25106–25119. [Google Scholar] [CrossRef]
- Wagner-Rousset, E.; Janin-Bussat, M.C.; Colas, O.; Excoffier, M.; Ayoub, D.; Haeuw, J.F.; Rilatt, I.; Perez, M.; Corvaïa, N.; Beck, A. Antibody-drug conjugate model fast characterization by LC-MS following IdeS proteolytic digestion. MAbs 2014, 6, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.A. Treating Disease at the RNA Level with Oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70. [Google Scholar] [CrossRef]
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release 2016, 237, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Satake, N.; Duong, C.; Yoshida, S.; Oestergaard, M.; Chen, C.; Peralta, R.; Guo, S.; Seth, P.P.; Li, Y.; Beckett, L.; et al. Novel Targeted Therapy for Precursor B Cell Acute Lymphoblastic Leukemia: Anti-CD22 Antibody-MXD3 Antisense Oligonucleotide Conjugate. Mol. Med. 2016, 22, 632–642. [Google Scholar] [CrossRef]
- Arnold, A.E.; Malek-Adamian, E.; Le, P.U.; Meng, A.; Martínez-Montero, S.; Petrecca, K.; Damha, M.J.; Shoichet, M.S. Antibody-Antisense Oligonucleotide Conjugate Downregulates a Key Gene in Glioblastoma Stem Cells. Mol. Ther. Nucleic Acids 2018, 11, 518–527. [Google Scholar] [CrossRef]
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blättler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef] [PubMed]
- Acchione, M.; Kwon, H.; Jochheim, C.M.; Atkins, W.M. Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody-drug conjugates. MAbs 2012, 4, 362–372. [Google Scholar] [CrossRef]
- Sun, M.M.; Beam, K.S.; Cerveny, C.G.; Hamblett, K.J.; Blackmore, R.S.; Torgov, M.Y.; Handley, F.G.; Ihle, N.C.; Senter, P.D.; Alley, S.C. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug. Chem. 2005, 16, 1282–1290. [Google Scholar] [CrossRef]
- Junutula, J.R.; Gerber, H.P. Next-Generation Antibody-Drug Conjugates (ADCs) for Cancer Therapy. ACS Med. Chem. Lett. 2016, 7, 972–973. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Shen, B.Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Kung Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. SGN-CD33A: A novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; He, T.; Prashad, A.S.; Wang, W.; Cohen, J.; Ferguson, D.; Tam, A.S.; Sousa, E.; Lin, L.; Tchistiakova, L.; et al. Mechanistic understanding of the cysteine capping modifications of antibodies enables selective chemical engineering in live mammalian cells. J. Biotechnol. 2017, 248, 48–58. [Google Scholar] [CrossRef]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef]
- Strop, P.; Liu, S.H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.T.; Ho, W.H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef]
- Lhospice, F.; Brégeon, D.; Belmant, C.; Dennler, P.; Chiotellis, A.; Fischer, E.; Gauthier, L.; Boëdec, A.; Rispaud, H.; Savard-Chambard, S.; et al. Site-Specific Conjugation of Monomethyl Auristatin E to Anti-CD30 Antibodies Improves Their Pharmacokinetics and Therapeutic Index in Rodent Models. Mol. Pharm. 2015, 12, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Hell, T.; Merkel, A.S.; Grawunder, U. Sortase Enzyme-Mediated Generation of Site-Specifically Conjugated Antibody Drug Conjugates with High In Vitro and In Vivo Potency. PLoS ONE 2015, 10, e0131177. [Google Scholar] [CrossRef] [PubMed]
- van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody-Drug Conjugates. Bioconjug. Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Rabuka, D.; Rush, J.S.; Gregory, W.D.; Wu, P.; Bertozzi, C.R. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 2012, 7, 1052–1067. [Google Scholar] [CrossRef]
- Lin, S.; Yang, X.; Jia, S.; Weeks, A.M.; Hornsby, M.; Lee, P.S.; Nichiporuk, R.V.; Iavarone, A.T.; Wells, J.A.; Toste, F.D.; et al. Redox-based reagents for chemoselective methionine bioconjugation. Science 2017, 355, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Elledge, S.K.; Tran, H.L.; Christian, A.H.; Steri, V.; Hann, B.; Toste, F.D.; Chang, C.J.; Wells, J.A. Systematic identification of engineered methionines and oxaziridines for efficient, stable, and site-specific antibody bioconjugation. Proc. Natl. Acad. Sci. USA 2020, 117, 5733–5740. [Google Scholar] [CrossRef]
- Mäger, I.; Meyer, A.H.; Li, J.; Lenter, M.; Hildebrandt, T.; Leparc, G.; Wood, M.J. Targeting blood-brain-barrier transcytosis—Perspectives for drug delivery. Neuropharmacology 2017, 120, 4–7. [Google Scholar] [CrossRef]
- Niewoehner, J.; Bohrmann, B.; Collin, L.; Urich, E.; Sade, H.; Maier, P.; Rueger, P.; Stracke, J.O.; Lau, W.; Tissot, A.C.; et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 2014, 81, 49–60. [Google Scholar] [CrossRef]
- Bezabeh, B.; Fleming, R.; Fazenbaker, C.; Zhong, H.; Coffman, K.; Yu, X.Q.; Leow, C.C.; Gibson, N.; Wilson, S.; Stover, C.K.; et al. Insertion of scFv into the hinge domain of full-length IgG1 monoclonal antibody results in tetravalent bispecific molecule with robust properties. MAbs 2017, 9, 240–256. [Google Scholar] [CrossRef]
- Wec, A.Z.; Nyakatura, E.K.; Herbert, A.S.; Howell, K.A.; Holtsberg, F.W.; Bakken, R.R.; Mittler, E.; Christin, J.R.; Shulenin, S.; Jangra, R.K.; et al. A “Trojan horse” bispecific-antibody strategy for broad protection against ebolaviruses. Science 2016, 354, 350–354. [Google Scholar] [CrossRef]
- Desnoyers, L.R.; Vasiljeva, O.; Richardson, J.H.; Yang, A.; Menendez, E.E.; Liang, T.W.; Wong, C.; Bessette, P.H.; Kamath, K.; Moore, S.J.; et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci. Transl. Med. 2013, 5, 207ra144. [Google Scholar] [CrossRef]
- Mimoto, F.; Tatsumi, K.; Shimizu, S.; Kadono, S.; Haraya, K.; Nagayasu, M.; Suzuki, Y.; Fujii, E.; Kamimura, M.; Hayasaka, A.; et al. Exploitation of Elevated Extracellular ATP to Specifically Direct Antibody to Tumor Microenvironment. Cell Rep. 2020, 33, 108542. [Google Scholar] [CrossRef]
- Kamata-Sakurai, M.; Narita, Y.; Hori, Y.; Nemoto, T.; Uchikawa, R.; Honda, M.; Hironiwa, N.; Taniguchi, K.; Shida-Kawazoe, M.; Metsugi, S.; et al. Antibody to CD137 Activated by Extracellular Adenosine Triphosphate Is Tumor Selective and Broadly Effective In Vivo without Systemic Immune Activation. Cancer Discov. 2021, 11, 158–175. [Google Scholar] [CrossRef]
- Polu, K.R.; Lowman, H.B. Probody therapeutics for targeting antibodies to diseased tissue. Expert Opin. Biol. Ther. 2014, 14, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Maniaci, C.; Ciulli, A. Bifunctional chemical probes inducing protein-protein interactions. Curr. Opin. Chem. Biol. 2019, 52, 145–156. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef]
- Verma, R.; Mohl, D.; Deshaies, R.J. Harnessing the Power of Proteolysis for Targeted Protein Inactivation. Mol. Cell. 2020, 77, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Kanner, S.A.; Morgenstern, T.; Colecraft, H.M. Sculpting ion channel functional expression with engineered ubiquitin ligases. Elife 2017, 6, e29744. [Google Scholar] [CrossRef]
- Kanner, S.A.; Shuja, Z.; Choudhury, P.; Jain, A.; Colecraft, H.M. Targeted deubiquitination rescues distinct trafficking-deficient ion channelopathies. Nat. Methods 2020, 17, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Grabulovski, D.; Kaspar, M.; Neri, D. A novel, non-immunogenic Fyn SH3-derived binding protein with tumor vascular targeting properties. J. Biol. Chem. 2007, 282, 3196–3204. [Google Scholar] [CrossRef] [PubMed]
- Silacci, M.; Lembke, W.; Woods, R.; Attinger-Toller, I.; Baenziger-Tobler, N.; Batey, S.; Santimaria, R.; von der Bey, U.; Koenig-Friedrich, S.; Zha, W.; et al. Discovery and characterization of COVA322, a clinical-stage bispecific TNF/IL-17A inhibitor for the treatment of inflammatory diseases. MAbs 2016, 8, 141–149. [Google Scholar] [CrossRef]
- Bloom, L.; Calabro, V. FN3: A new protein scaffold reaches the clinic. Drug Discov. Today 2009, 14, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Wojcik, J.; Gilbreth, R.N.; Hoey, R.J.; Koide, S. Teaching an old scaffold new tricks: Monobodies constructed using alternative surfaces of the FN3 scaffold. J. Mol. Biol. 2012, 415, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Bailey, C.W.; Huang, X.; Koide, S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 1998, 284, 1141–1151. [Google Scholar] [CrossRef]
- Diem, M.D.; Hyun, L.; Yi, F.; Hippensteel, R.; Kuhar, E.; Lowenstein, C.; Swift, E.J.; O’Neil, K.T.; Jacobs, S.A. Selection of high-affinity Centyrin FN3 domains from a simple library diversified at a combination of strand and loop positions. Protein Eng. Des. Sel. 2014, 27, 419–429. [Google Scholar] [CrossRef]
- Wojcik, J.; Hantschel, O.; Grebien, F.; Kaupe, I.; Bennett, K.L.; Barkinge, J.; Jones, R.B.; Koide, A.; Superti-Furga, G.; Koide, S. A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat. Struct. Mol. Biol. 2010, 17, 519–527. [Google Scholar] [CrossRef]
- Ludwicki, M.B.; Li, J.; Stephens, E.A.; Roberts, R.W.; Koide, S.; Hammond, P.T.; DeLisa, M.P. Broad-Spectrum Proteome Editing with an Engineered Bacterial Ubiquitin Ligase Mimic. ACS Cent. Sci. 2019, 5, 852–866. [Google Scholar] [CrossRef]
- Goldberg, S.D.; Cardoso, R.M.; Lin, T.; Spinka-Doms, T.; Klein, D.; Jacobs, S.A.; Dudkin, V.; Gilliland, G.; O’Neil, K.T. Engineering a targeted delivery platform using Centyrins. Protein Eng. Des. Sel. 2016, 29, 563–572. [Google Scholar] [CrossRef]
- Feldwisch, J.; Tolmachev, V. Engineering of affibody molecules for therapy and diagnostics. Methods Mol. Biol. 2012, 899, 103–126. [Google Scholar]
- Löfblom, J.; Feldwisch, J.; Tolmachev, V.; Carlsson, J.; Ståhl, S.; Frejd, F.Y. Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010, 584, 2670–2680. [Google Scholar] [CrossRef]
- Frejd, F.Y.; Kim, K.T. Affibody molecules as engineered protein drugs. Exp. Mol. Med. 2017, 49, e306. [Google Scholar] [CrossRef]
- Akhtari, J.; Rezayat, S.M.; Teymouri, M.; Alavizadeh, S.H.; Gheybi, F.; Badiee, A.; Jaafari, M.R. Targeting, bio distributive and tumor growth inhibiting characterization of anti-HER2 affibody coupling to liposomal doxorubicin using BALB/c mice bearing TUBO tumors. Int. J. Pharm. 2016, 505, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Meister, S.W.; Hjelm, L.C.; Dannemeyer, M.; Tegel, H.; Lindberg, H.; Ståhl, S.; Löfblom, J. An Affibody Molecule Is Actively Transported into the Cerebrospinal Fluid via Binding to the Transferrin Receptor. Int. J. Mol. Sci. 2020, 21, 2999. [Google Scholar] [CrossRef] [PubMed]
- Pluckthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef]
- Simon, M.; Stefan, N.; Borsig, L.; Plückthun, A.; Zangemeister-Wittke, U. Increasing the antitumor effect of an EpCAM-targeting fusion toxin by facile click PEGylation. Mol. Cancer Ther. 2014, 13, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, E.; Proshkina, G.; Kutova, O.; Shilova, O.; Ryabova, A.; Schulga, A.; Stremovskiy, O.; Zdobnova, T.; Balalaeva, I.; Deyev, S. Recombinant targeted toxin based on HER2-specific DARPin possesses a strong selective cytotoxic effect in vitro and a potent antitumor activity in vivo. J. Control. Release 2016, 233, 48–56. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Chen, D.; Nabuurs, A.G.; Quax, W.J.; Demaria, M.; Boersma, Y.L. A Bispecific Inhibitor of the EGFR/ADAM17 Axis Decreases Cell Proliferation and Migration of EGFR-Dependent Cancer Cells. Cancers 2020, 12, 411. [Google Scholar] [CrossRef] [PubMed]
- Baird, R.; Linossi, C.; Middleton, M.; Lord, S.; Harris, A.; Rodón, J.; Zitt, C.; Fiedler, U.; Dawson, K.M.; Leupin, N.; et al. First-in-Human Phase I Study of MP0250, a First-in-Class DARPin Drug Candidate Targeting VEGF and HGF, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, JCO2000596. [Google Scholar] [CrossRef]
- Babaee, N.; Garoosi, Y.T.; Karimipoor, M.; Davami, F.; Bayat, E.; Safarpour, H.; Mahboudi, F.; Barkhordari, F. DARPin Ec1-LMWP protein scaffold in targeted delivery of siRNA molecules through EpCAM cancer stem cell marker. Mol. Biol. Rep. 2020, 47, 7323–7331. [Google Scholar] [CrossRef] [PubMed]
- Cox, N.; Kintzing, J.R.; Smith, M.; Grant, G.A.; Cochran, J.R. Integrin-Targeting Knottin Peptide-Drug Conjugates Are Potent Inhibitors of Tumor Cell Proliferation. Angew. Chem. Int. Ed. Engl. 2016, 55, 9894–9897. [Google Scholar] [CrossRef]
- Lee, S.C.; Park, K.; Han, J.; Lee, J.J.; Kim, H.J.; Hong, S.; Heu, W.; Kim, Y.J.; Ha, J.S.; Lee, S.G.; et al. Design of a binding scaffold based on variable lymphocyte receptors of jawless vertebrates by module engineering. Proc. Natl. Acad. Sci. USA 2012, 109, 3299–3304. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Eggenstein, E.; Skerra, A. Anticalins: Exploiting a non-Ig scaffold with hypervariable loops for the engineering of binding proteins. FEBS Lett. 2014, 588, 213–218. [Google Scholar] [CrossRef]
- Rothe, C.; Skerra, A. Anticalin((R)) Proteins as Therapeutic Agents in Human Diseases. BioDrugs 2018, 32, 233–243. [Google Scholar] [CrossRef]
- Thakur, A.; Lum, L.G. Cancer therapy with bispecific antibodies: Clinical experience. Curr. Opin. Mol. Ther. 2010, 12, 340–349. [Google Scholar] [PubMed]
- Doppalapudi, V.R.; Huang, J.; Liu, D.; Jin, P.; Liu, B.; Li, L.; Desharnais, J.; Hagen, C.; Levin, N.J.; Shields, M.J.; et al. Chemical generation of bispecific antibodies. Proc. Natl. Acad. Sci. USA 2010, 107, 22611–22616. [Google Scholar] [CrossRef] [PubMed]
- White, B.H.; Whalen, K.; Kriksciukaite, K.; Alargova, R.; Au Yeung, T.; Bazinet, P.; Brockman, A.; DuPont, M.; Oller, H.; Lemelin, C.A.; et al. Discovery of an SSTR2-Targeting Maytansinoid Conjugate (PEN-221) with Potent Activity in Vitro and in Vivo. J. Med. Chem. 2019, 62, 2708–2719. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, J.; Kong, Y.; Liang, R.; Li, W.; Li, J.; Lu, J.; Dimitrov, D.S.; Yu, F.; Wu, Y.; et al. Engineering a Novel Antibody-Peptide Bispecific Fusion Protein against MERS-CoV. Antibodies 2019, 8, 53. [Google Scholar] [CrossRef]
- Kang, J.C.; Sun, W.; Khare, P.; Karimi, M.; Wang, X.; Shen, Y.; Ober, R.J.; Ward, E.S. Engineering a HER2-specific antibody-drug conjugate to increase lysosomal delivery and therapeutic efficacy. Nat. Biotechnol. 2019, 37, 523–526. [Google Scholar] [CrossRef]
- Andreev, J.; Thambi, N.; Bay, A.E.; Delfino, F.; Martin, J.; Kelly, M.P.; Kirshner, J.R.; Rafique, A.; Kunz, A.; Nittoli, T.; et al. Bispecific Antibodies and Antibody-Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol. Cancer Ther. 2017, 16, 681–693. [Google Scholar] [CrossRef]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Morisaki, J.H.; et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Liu, R.; Wang, R.E.; Wang, F. Antibody-drug conjugates for non-oncological indications. Expert Opin. Biol. Ther. 2016, 16, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Kobold, S.; Pantelyushin, S.; Rataj, F.; Vom Berg, J. Rationale for Combining Bispecific T Cell Activating Antibodies with Checkpoint Blockade for Cancer Therapy. Front. Oncol. 2018, 8, 285. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, M.; Poggi, A. Checkpoint Inhibitors and Engineered Cells: New Weapons for Natural Killer Cell Arsenal against Hematological Malignancies. Cells 2020, 9, 1578. [Google Scholar] [CrossRef]
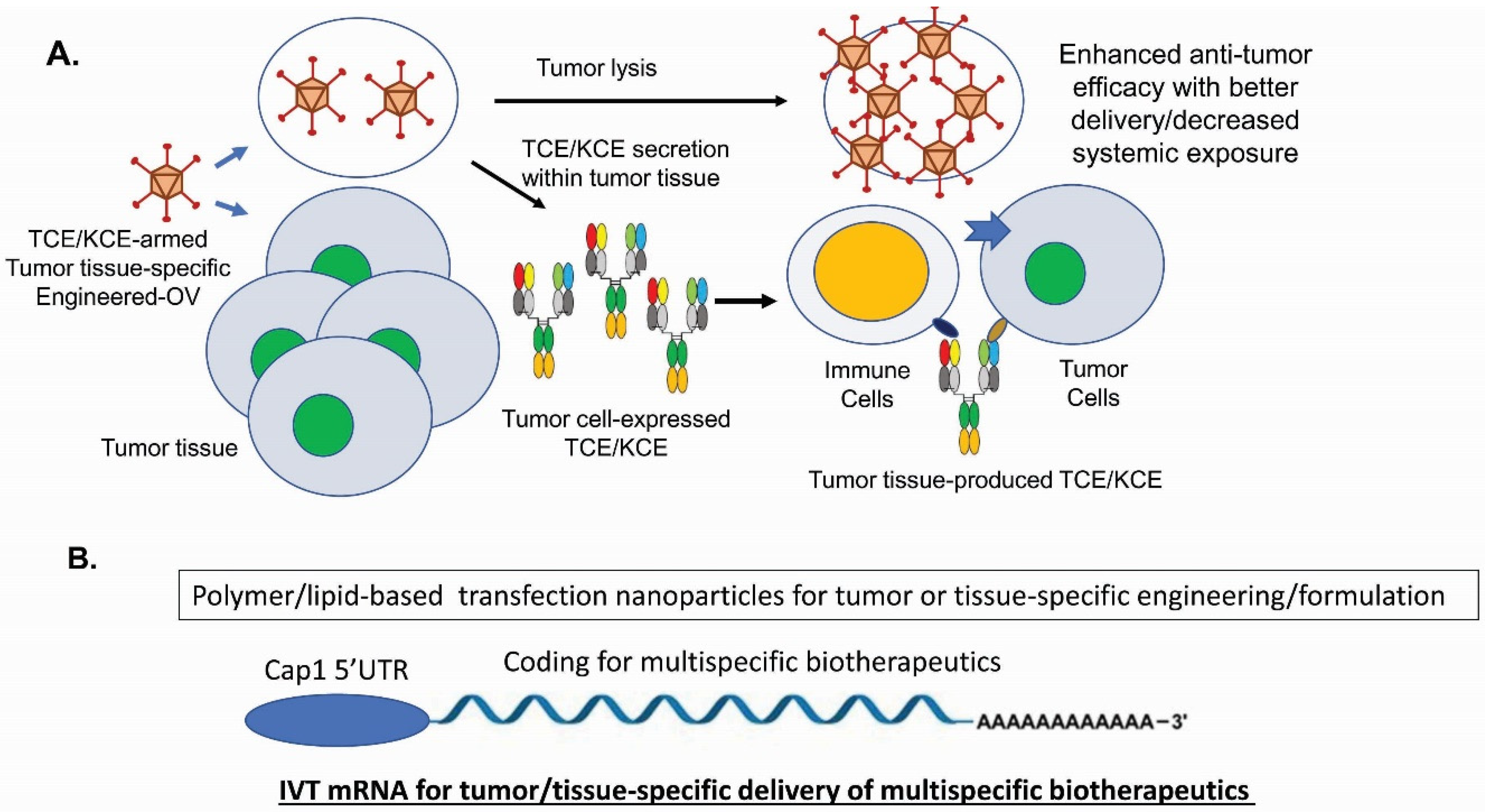
- Guo, Z.S.; Lotze, M.T.; Zhu, Z.; Storkus, W.J.; Song, X.T. Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy. Biomedicines 2020, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Stadler, C.R.; Bähr-Mahmud, H.; Celik, L.; Hebich, B.; Roth, A.S.; Roth, R.P.; Karikó, K.; Türeci, Ö.; Sahin, U. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat. Med. 2017, 23, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Rehman, H.; Silk, A.W.; Kane, M.P.; Kaufman, H.L. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J. Immunother. Cancer 2016, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.M.; Jacobus, E.J.; Lyons, B.; Frost, S.; Freedman, J.D.; Dyer, A.; Khalique, H.; Taverner, W.K.; Carr, A.; Champion, B.R.; et al. Bi- and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J. Immunother. Cancer 2019, 7, 320. [Google Scholar] [CrossRef]
- Speck, T.; Heidbuechel, J.P.; Veinalde, R.; Jaeger, D.; Von Kalle, C.; Ball, C.R.; Ungerechts, G.; Engeland, C.E. Targeted BiTE Expression by an Oncolytic Vector Augments Therapeutic Efficacy Against Solid Tumors. Clin. Cancer Res. 2018, 24, 2128–2137. [Google Scholar] [CrossRef]
- Fajardo, C.A.; Guedan, S.; Rojas, L.A.; Moreno, R.; Arias-Badia, M.; de Sostoa, J.; June, C.H.; Alemany, R. Oncolytic Adenoviral Delivery of an EGFR-Targeting T-cell Engager Improves Antitumor Efficacy. Cancer Res. 2017, 77, 2052–2063. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.; Pol, J.G.; Kroemer, G. Heating it up: Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. Oncoimmunology 2018, 7, e1442169. [Google Scholar] [CrossRef]
- Scott, E.M.; Duffy, M.R.; Freedman, J.D.; Fisher, K.D.; Seymour, L.W. Solid Tumor Immunotherapy with T Cell Engager-Armed Oncolytic Viruses. Macromol. Biosci. 2018, 18, 1700187. [Google Scholar] [CrossRef] [PubMed]
- Trinklein, N.D.; Pham, D.; Schellenberger, U.; Buelow, B.; Boudreau, A.; Choudhry, P.; Clarke, S.C.; Dang, K.; Harris, K.E.; Iyer, S.; et al. Efficient tumor killing and minimal cytokine release with novel T-cell agonist bispecific antibodies. MAbs 2019, 11, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; DiGiandomenico, A.; Keller, A.E.; Smith, T.R.; Park, D.H.; Ramos, S.; Schultheis, K.; Elliott, S.T.; Mendoza, J.; Broderick, K.E.; et al. An engineered bispecific DNA-encoded IgG antibody protects against Pseudomonas aeruginosa in a pneumonia challenge model. Nat. Commun. 2017, 8, 637. [Google Scholar] [CrossRef]

| Approved Product Name | Year of Approval | Indication | Modality |
|---|---|---|---|
| catumaxomab (RemovabTM) | 2009 and withdrawal in 2017 for commercial reasons | Solid malignancies (malignant ascites owing to epithelial carcinomas) | bsTCE |
| blinatumomab (Blincyto®) | 2014 | Hematological malignancies [acute lymphoblastic leukemia (ALL) and B-ALL] | bsTCE |
| emicizumab (Hemlibra®) | 2017 | Routine prophylaxis of Hemophilia A with and without FVIII inhibitors | Matchmaker |
| moxetumomab pasudotox (LumoxitiTM) | 2018 | Relapsed or refractory hairy cell leukemia | Tetherbody |
| gemtuzumab ozogamicin (Mylotarg®) | First-approved in 2000, withdrawal in 2010, and re-approved in 2017 | Acute myeloid leukemia | ADC |
| brentuximab vedotin (Adcetris®) | 2011 | Hodgkin lymphoma, anaplastic large cell lymphoma, CD30-expressing mycosis fungoides | ADC |
| ado-trastuzumab emtansine (Kadcyla®) | 2013 | HER2+ metastatic breast cancer | ADC |
| inotuzumab ozogamicin (Besponsa®) | 2017 | Relapsed or refractory B-cell precursor acute lymphoblastic leukemia | ADC |
| enfortumab vedotin (PadcevTM) | 2019 | Locally advanced or metastatic urothelial cancer | ADC |
| Fam-trastuzumab deruxtecan-nxki (Enhertu®) | 2019 | HER2+ unresectable or metastatic breast cancer | ADC |
| Polatuzumab vedotin-piiq (PolivyTM) | 2019 | Relapsed or refractory diffuse large B-cell lymphoma | ADC |
| Belantamab mafodotin-blmf (BlenrepTM) | 2020 | Relapsed or refractory multiple myeloma | ADC |
| Sacituzumab govitecan (TrodelvyTM) | 2020 | Metastatic triple-negative breast cancer | ADC |
| ADC Products | Target Antigen/Antibody | Conjugation Methods (Lys/Cys) Random/Site-Specific | Linker Payload | Average Drug Antibody Ratio |
|---|---|---|---|---|
| gemtuzumab ozogamicin (Pfizer) | CD33/humanized IgG4κ | Lys/random | N-acetyl-γ calicheamicin 1,2 dimethyl hydrazine dichloride | ~1.5 |
| brentuximab vedotin (Seattle Genetics) | CD30/chimeric IgG1 | Interchain Cys/random | mc-vc-PABC-MMAE | ~4 |
| ado-trastuzumab emtansine (Roche) | HER2/humanized IgG1 | Lys/random | SMCC-DM1 | ~3.5 |
| inotuzumab ozogamicin (Pfizer) | CD22/humanized IgG4 | Lys/random | N-acetyl-γ calicheamicin 1,2 dimethyl hydrazine dichloride | ~5–7 |
| enfortumab vedotin (Astellas) | Nectin-4/human IgG1κ | Interchain Cys/random | mc-vc-PABC-MMAE | ~4 |
| fam-trastuzumab deruxtecan-nxki (Daiichi Sankyo) | HER2/humanized IgG1κ | Interchain Cys/site-specific | mc-GGFG-DX-8951 derivative | ~7.7 |
| polatuzumab vedotin-piiq (Roche) | CD79b/humanized IgG1κ | Interchain Cys/random | mc-vc-PABC-MMAE | ~4 |
| belantamab mafodotin-blmf (GlaxoSmithKline) | BCMA/Afucosylated humanized IgG1 | Interchain Cys/random | mc-MMAF | ~4 |
| sacituzumab govitecan (Immunomedics) | TROP2/humanized IgG1κ | Interchain Cys/random | Cl2A-SN38 | ~7.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, X.; D’Antona, A.M. Recent Advances in the Molecular Design and Applications of Multispecific Biotherapeutics. Antibodies 2021, 10, 13. https://doi.org/10.3390/antib10020013
Zhong X, D’Antona AM. Recent Advances in the Molecular Design and Applications of Multispecific Biotherapeutics. Antibodies. 2021; 10(2):13. https://doi.org/10.3390/antib10020013
Chicago/Turabian StyleZhong, Xiaotian, and Aaron M. D’Antona. 2021. "Recent Advances in the Molecular Design and Applications of Multispecific Biotherapeutics" Antibodies 10, no. 2: 13. https://doi.org/10.3390/antib10020013
APA StyleZhong, X., & D’Antona, A. M. (2021). Recent Advances in the Molecular Design and Applications of Multispecific Biotherapeutics. Antibodies, 10(2), 13. https://doi.org/10.3390/antib10020013