Antibody Identification for Antigen Detection in Formalin-Fixed Paraffin-Embedded Tissue Using Phage Display and Naïve Libraries



Abstract
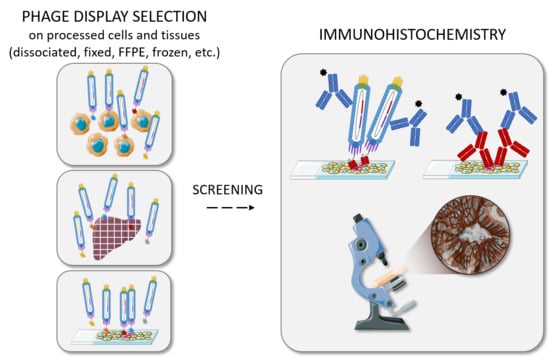
1. Introduction
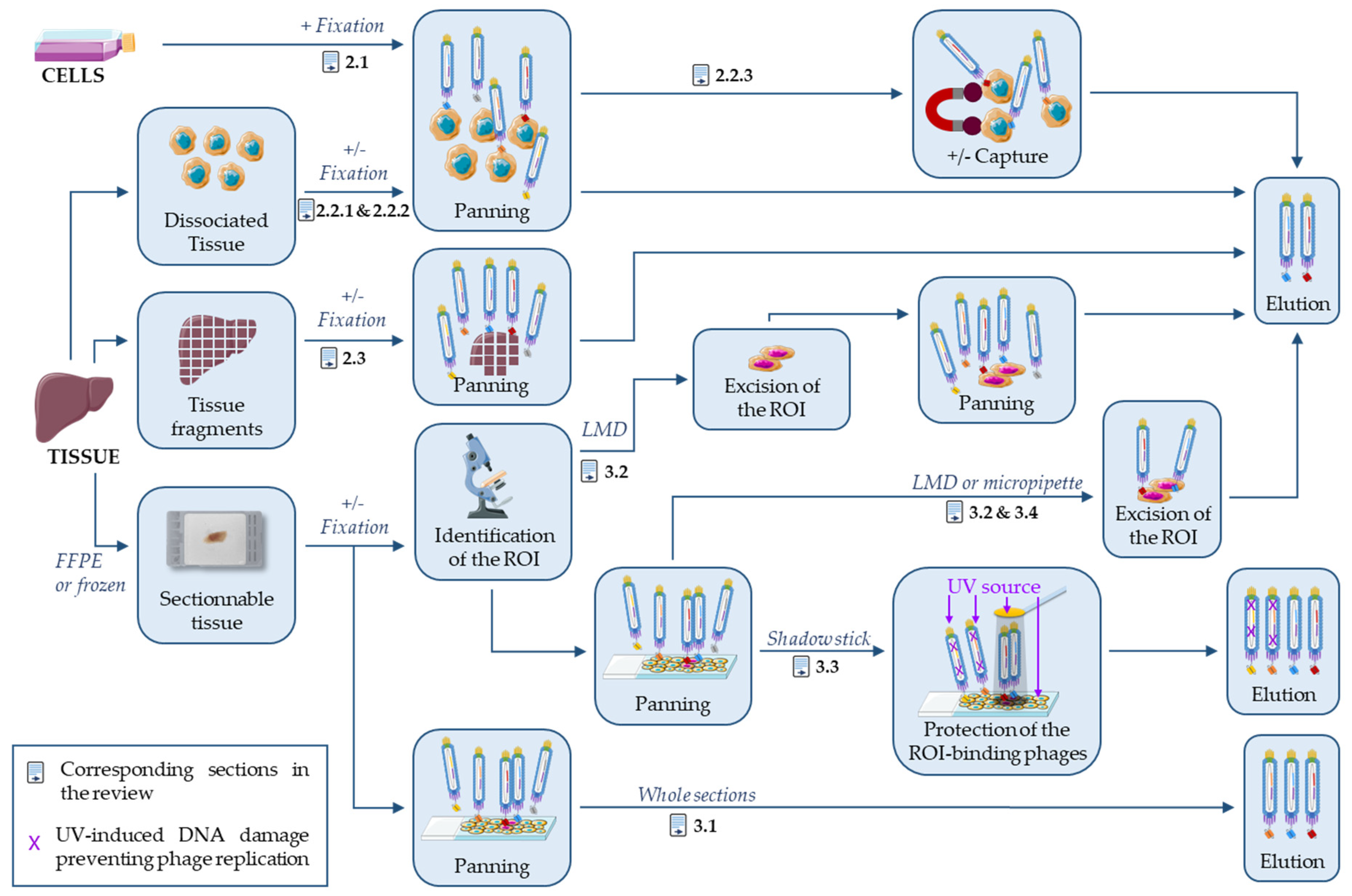
2. Antibody Selection on Processed Cells and Tissues
2.1. Antibody Selection on Fixed Cells
2.2. Antibody Selection on Dissociated Tissues
2.2.1. Antibody Selection on Freshly Dissociated Tissues
2.2.2. Antibody Selection on Fixed Dissociated Tissues
2.2.3. Antibody Selection against Specific Cell Subtypes
2.3. Antibody Selection on Tissue Fragments
2.3.1. Antibody Selection on Fresh Tissues
2.3.2. Antibody Selection on Fixed Tissues
3. On-Slide Antibody Selection
3.1. On Whole FFPE Sections
3.2. Laser-Assisted Microdissection Strategies

{kind=link}
{kind=link}
| Authors and References | Antibody Selection | Screening | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Selection | Depletion | First Screening | Immunohistochemical Staining | ||||||||
| Nb of Rounds | Support | Fixation | Before, during or after Selection | Support | Nb of Clones | Technique | Type | Antibody Format | Positive Clones | Note | |
| Selections on processed cells and tissues | |||||||||||
| Gur et al. [44] | 5 | Cell lines | 4% PFA | Before | Negatively-sorted cells | 171 | Phage-ICC on fixed cells | IHC-p/IHC-f | Phage-scFv | 2/2 | Both clones stain more intensely ALDH1+ than ALDH1- cells |
| Edwards et al. [47] | 3 | Tissue samples dissociated with collagenase | / | / | / | 2242 | Phage-ELISA on cell membranes | IHC-f | Phage-scFv | 82/109 | All cross-reacted with at least another cell type or structure |
| Jakobsen et al. [46] | 1–2 | Tissue samples dissociated with collagenase + hyaluronidase | / | / | / | Probably b between 83 and 98 | Phage-ELISA on fixed cells | IHC-p | Phage-scFv | 2/2 | Reactive with tumors of different histologic origins; no or weak binding to normal tissues |
| Roovers et al. [48] | 5 | Tissue samples dissociated with EDTA, EGTA, DTT | 0.25% PFA, 4 °C, 20 min | / | / | 42 clones with distinct fingerprint pattern tested | IHC | IHC-f | Fab | 3/40 | Only one clone can stain FFPE sections |
| Mutuberria et al. [49] | 3–4 | Tissue samples dissociated with trypsin, EDTA and cultured before selection | 1% PFA, RT, 30 min | During | Cells, magnetic sorting | 132 clones fingerprinted, 17 unique clones tested | Flow cytometry | IHC-f | Phage-scFv | 11/17 | / |
| Palmer et al. [45] | 6 | Tissue samples dissociated with collagenase | / | During | Cells, magnetic sorting | At least b 85 | Phage-IHC | IHC-f | Phage-scFv | 7 c | None stained exclusively all medullary epithelium |
| Edwards et al. [47] | 3 | Non-dissociated tissue fragments | / | / | / | 380 | Phage-ELISA on cell membranes | IHC-f | Phage-scFv | 82/109 | All cross-reacted with at least another cell type or structure |
| Dorfmueller et al. [50] | 3–4 | Non-dissociated tissue samples | / | Before and after | Cells (primary culture) | 1248 | On-cell ELISA | IF-f | scFv-alcaline phosphatase fusion proteins, then IgG | at least 4/20 | Number of non-specific clones not mentioned. |
| Jarutat et al. [53] | 6 | Free-floating FFPE sections | FFPE | Before (only from the 2nd round) | Healthy tissue sections | 240 | IHC | IHC-p | bacterial lysates containing Fab or mini-antibodies | 74/240 | Up to 6 clones tested per slide. |
| Van Ewijk et al. & Radošević et al. [51,52] | 3–4 | Non-dissociated tissue fragments | Glutaraldehyde a | Before and simultaneously | Cells (thymocytes and fixed spleen cells) | Probably b at least 28 | Phage-IHC | IHC-f | Phage-scFv then scFv | 3 c | / |
| On-slide selections | |||||||||||
| ten Haaf et al. [57,58] | 3 | FFPE sections on slides | FFPE | Before | Healthy tissue sections | 440 | Phage-ELISA on cell membranes | IHC-p | Fab | 3/3 | No or minimal cross-reactivity toward healthy tissues |
| Ruan et al. & Su et al. [60,61] | 2 | Cryosections on slides, with LMD | / | During | Rest of the slide | 192 | Flow cytometry | IHC-p/IHC-f | biotinylated-scFv | 1/1 | Clone can stain only cryosections; cross-reactive with some healthy tissues. |
| 2 | FFPE sections on slides, with LMD | FFPE | During | Rest of the slide | 760 | Flow cytometry | IHC-p/IHC-f | biotinylated-scFv | 1/1 | Clone can stain FFPE and cryosections; low cross-reactivity with healthy tissues. | |
| Tanaka et al. [59] | 1–2 | Cryosections on slides, with LMD | Acetone, 5 min | / | / | 409 PCR-controlled clones; 157 unique clones tested | Phage-IHC | IHC-f | Phage-scFv | 5/9 | / |
| Sun et al. [62] | 1 | Catapulted cryosections, with LMD | 2% PFA, RT, 15 min, or FFPE | / | / | 79, all unique | IF-f | IF-f | Phage-scFv | >14/79 | 14/79 bound to cancer cells more intensely than to tumor stroma |
| Sun et al. [63] | 1–3 | Cryosections on slides, with LMD | 2% PFA, RT, 15 min | / | / | 150 | IF-f | IF-f/ IHC-p | Phage-scFv | 31/150 and 6/150 | Selection of a patient-specific clone |
| Sørensen et al. [66,67] | 1 | Cytological preparations, with shadow stick | Methanol + PFA | During | Rest of the slide (male cells) | 1536 | On-cell phage-ELISA | / | / | / | / |
| Sørensen et al. [68] | 1 | Cytological preparations, with shadow stick | / | During | Rest of the slide (female cells) | 12 clones; 10 tested | ICC | IF-p | scFv | 5/10 | / |
| Larsen et al. [65] | 1 | FFPE sections, with shadow stick | FFPE | During | Rest of the slide | 40 | On-cell phage-ELISA | IHC-p | scFv | 2/3 | Clone 2E confirms the feasibility of shadow stick selections on tissue |
| Larsen et al. [69] | 1 | Cryosections, with shadow stick | PFA, 10 min | During | Rest of the slide | 315 | On-cell phage-ELISA | If-f | dAb | 1/11 d | Clone LH7, specific to some breast cancer cell subpopulation |
| Larsen et al. [70] | 1 | Cryosections, with shadow stick | PFA, 10 min | During | Rest of the slide | 315 | On-cell phage-ELISA | IF-f | dAb | 1/11 d | Clone LH8, no cross-reaction on healthy breast tissues |
| Sørensen et al. [71] | 1 | Cryosections, with shadow stick | Methanol, 5 min | During | Rest of the slide | 93 | Phage-ELISA on fixed cells | IF-f | dAb then dAb-rFc | 1/1 | Focus on only one clone |
| Lykkemark et al. [64] | 1 | Cryosections, with micropipette dissection | 4% PFA, RT, 12 min | During | Rest of the slide | 1150 clones; 192 tested | On-cell phage-ELISA | IF-f | dAb | 1/1 | / |
| Authors | Ref. | Target | Techniques |
|---|---|---|---|
| Jakobsen et al., 2007 | [28] | GRP78 | Yeast two-hybrid screening of a cDNA |
| Dorfmueller et al., 2016 | [32] | ALCAM | Immunoprecipitation + mass spectrometry |
| Jarutat et al., 2007 | [35] | Vimentin | Immunoprecipitation + mass spectrometry |
| Tanaka et al., 2002 | [41] | Actin, Tropomyosin, Actinin, Myosin | Mass spectrometry + cDNA expression library |
| Ruan et al., 2006 | [42] | ALCAM | Sequence similarity with a known anti-ALCAM antibody |
| Sørensen et al., 2017 | [53] | MRPS18A | Protein micro-array |
3.3. Shadow Stick-Based Antibody Selection
3.4. Micropipette-Assisted Microdissection Strategies
4. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- De Matos, L.L.; Trufelli, D.C.; de Matos, M.G.L.; da Silva Pinhal, M.A. Immunohistochemistry as an Important Tool in Biomarkers Detection and Clinical Practice. Biomark. Insights 2010, 5, 9–20. [Google Scholar] [CrossRef] [PubMed]
- O’Hurley, G.; Sjöstedt, E.; Rahman, A.; Li, B.; Kampf, C.; Pontén, F.; Gallagher, W.M.; Lindskog, C. Garbage in, garbage out: A critical evaluation of strategies used for validation of immunohistochemical biomarkers. Mol. Oncol. 2014, 8, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.-R.; Liu, C.; Pootrakul, L.; Tang, L.; Young, A.; Chen, R.; Cote, R.J.; Taylor, C.R. Evaluation of the Value of Frozen Tissue Section Used as “Gold Standard” for Immunohistochemistry. Am. J. Clin. Pathol. 2008, 129, 358–366. [Google Scholar] [CrossRef]
- Yamashita, S.; Okada, Y. Application of Heat-induced Antigen Retrieval to Aldehyde-fixed Fresh Frozen Sections. J. Histochem. Cytochem. 2005. [Google Scholar] [CrossRef]
- Grizzle, W.E.; Fredenburgh, J.L.; Myers, R.B. 4-Fixation of Tissues. In Theory and Practice of Histological Techniques, 6th ed.; Bancroft, J.D., Gamble, M., Eds.; Churchill Livingstone: Edinburgh, UK, 2008; pp. 53–74. ISBN 978-0-443-10279-0. [Google Scholar]
- Thavarajah, R.; Mudimbaimannar, V.K.; Elizabeth, J.; Rao, U.K.; Ranganathan, K. Chemical and physical basics of routine formaldehyde fixation. J. Oral Maxillofac. Pathol. JOMFP 2012, 16, 400–405. [Google Scholar] [CrossRef]
- Ramos-Vara, J.A.; Miller, M.A. When Tissue Antigens and Antibodies Get Along: Revisiting the Technical Aspects of Immunohistochemistry—The Red, Brown, and Blue Technique. Vet. Pathol. 2014, 51, 42–87. [Google Scholar] [CrossRef]
- Dunstan, R.W.; Wharton, K.A.; Quigley, C.; Lowe, A. The Use of Immunohistochemistry for Biomarker Assessment—Can It Compete with Other Technologies? Toxicol. Pathol. 2011, 39, 988–1002. [Google Scholar] [CrossRef]
- Shi, S.-R.; Shi, Y.; Taylor, C.R. Antigen Retrieval Immunohistochemistry. J. Histochem. Cytochem. 2011, 59, 13–32. [Google Scholar] [CrossRef]
- Fox, C.H.; Johnson, F.B.; Whiting, J.; Roller, P.P. Formaldehyde fixation. J. Histochem. Cytochem. 1985, 33, 845–853. [Google Scholar] [CrossRef]
- Shi, S.-R.; Gu, J.; Turrens, J.; Cote, R.J.; Taylor, C.R. Development of the antigen retrieval technique: Philosophical and theoretical bases. In Antigen Retrieval Techniques: Immunohistochemistry and Molecular Morphology; Shi, S.-R., Gu, J., Taylor, C.-R., Eds.; Eaton Publishing: Natick, MA, USA, 2000; pp. 17–40. ISBN 978-1-881299-43-1. [Google Scholar]
- Shi, S.R.; Key, M.E.; Kalra, K.L. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: An enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J. Histochem. Cytochem. 1991, 39, 741–748. [Google Scholar] [CrossRef]
- Gown, A.M. Unmasking the Mysteries of Antigen or Epitope Retrieval and Formalin Fixation. Am. J. Clin. Pathol. 2004, 121, 172–174. [Google Scholar] [CrossRef]
- Yamashita, S. Heat-induced antigen retrieval: Mechanisms and application to histochemistry. Prog. Histochem. Cytochem. 2007, 41, 141–200. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.B.; Evers, D.L.; O’Leary, T.J.; Mason, J.T. Antigen Retrieval Causes Protein Unfolding. J. Histochem. Cytochem. 2011, 59, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.-P.; Tachibana, K.; Hegen, M.; Scharpé, S.; Cho, D.; Schlossman, S.F.; Morimoto, C. Correlation of the epitopes defined by anti-CD26 mAbs and CD26 function. Mol. Immunol. 1998, 35, 13–21. [Google Scholar] [CrossRef]
- Hatano, R.; Yamada, T.; Madokoro, H.; Otsuka, H.; Komiya, E.; Itoh, T.; Narita, Y.; Iwata, S.; Yamazaki, H.; Matsuoka, S.; et al. Development of novel monoclonal antibodies with specific binding affinity for denatured human CD26 in formalin-fixed paraffin-embedded and decalcified specimens. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Pontén, F. Antibody-based proteomics for human tissue profiling. Mol. Cell. Proteom. MCP 2005, 4, 384–393. [Google Scholar] [CrossRef]
- Viegas Barroso, J.F.; Halder, M.E.; Whelan, M. EURL ECVAM Recommendation on Non-Animal-Derived Antibodies; EUR 30185 EN; Publications Office of the European Union: Luxembourg, 2020; ISBN 978-92-76-18346-4. [Google Scholar]
- Gray, A.C.; Bradbury, A.R.M.; Knappik, A.; Plückthun, A.; Borrebaeck, C.A.K.; Dübel, S. Animal-derived-antibody generation faces strict reform in accordance with European Union policy on animal use. Nat. Methods 2020, 17, 755–756. [Google Scholar] [CrossRef]
- Ledsgaard, L.; Kilstrup, M.; Karatt-Vellatt, A.; McCafferty, J.; Laustsen, A.H. Basics of Antibody Phage Display Technology. Toxins 2018, 10, 236. [Google Scholar] [CrossRef]
- Mimmi, S.; Maisano, D.; Quinto, I.; Iaccino, E. Phage Display: An Overview in Context to Drug Discovery. Trends Pharmacol. Sci. 2019, 40, 87–91. [Google Scholar] [CrossRef]
- Lloyd, C.; Lowe, D.; Edwards, B.; Welsh, F.; Dilks, T.; Hardman, C.; Vaughan, T. Modelling the human immune response: Performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng. Des. Sel. 2009, 22, 159–168. [Google Scholar] [CrossRef]
- Kurosawa, G.; Akahori, Y.; Morita, M.; Sumitomo, M.; Sato, N.; Muramatsu, C.; Eguchi, K.; Matsuda, K.; Takasaki, A.; Tanaka, M.; et al. Comprehensive screening for antigens overexpressed on carcinomas via isolation of human mAbs that may be therapeutic. Proc. Natl. Acad. Sci. USA 2008, 105, 7287–7292. [Google Scholar] [CrossRef] [PubMed]
- Torsvik, A.; Stieber, D.; Enger, P.Ø.; Golebiewska, A.; Molven, A.; Svendsen, A.; Westermark, B.; Niclou, S.P.; Olsen, T.K.; Chekenya Enger, M.; et al. U-251 revisited: Genetic drift and phenotypic consequences of long-term cultures of glioblastoma cells. Cancer Med. 2014, 3, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Gutbier, S.; May, P.; Berthelot, S.; Krishna, A.; Trefzer, T.; Behbehani, M.; Efremova, L.; Delp, J.; Gstraunthaler, G.; Waldmann, T.; et al. Major changes of cell function and toxicant sensitivity in cultured cells undergoing mild, quasi-natural genetic drift. Arch. Toxicol. 2018, 92, 3487–3503. [Google Scholar] [CrossRef] [PubMed]
- Uva, P.; Lahm, A.; Sbardellati, A.; Grigoriadis, A.; Tutt, A.; Rinaldis, E. de Comparative Membranome Expression Analysis in Primary Tumors and Derived Cell Lines. PLoS ONE 2010, 5, e11742. [Google Scholar] [CrossRef] [PubMed]
- Guadagni, F.; Roselli, M.; Schlom, J.; Greiner, J.W. In vitro and in vivo regulation of human tumor antigen expression by human recombinant interferons: A review. Int. J. Biol. Markers 1994, 9, 53–60. [Google Scholar] [CrossRef]
- Pietras, K.; Östman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Pimenidou, A.; Topping, K.; Hough, V.; Kirkpatrick, N.; Monson, J.; Greenman, J. Bacteriophage-Derived Antibodies in Cancer Research—Diagnosis, Imaging, and Treatment. Dis. Markers 2000, 16, 41–51. [Google Scholar] [CrossRef]
- Almagro, J.C.; Pedraza-Escalona, M.; Arrieta, H.I.; Pérez-Tapia, S.M. Phage Display Libraries for Antibody Therapeutic Discovery and Development. Antibodies 2019, 8, 44. [Google Scholar] [CrossRef]
- Ardelt, P.U.; Wood, C.G.; Chen, L.; Mintz, P.J.; Moya, C.; Arap, M.A.; Wright, K.C.; Pasqualini, R.; Arap, W. Targeting Urothelium: Ex Vivo Assay Standardization and Selection of Internalizing Ligands. J. Urol. 2003, 169, 1535–1540. [Google Scholar] [CrossRef]
- Koivistoinen, A.; Ilonen, I.I.K.; Punakivi, K.; Räsänen, J.V.; Helin, H.; Sihvo, E.I.; Bergman, M.; Salo, J.A. A novel peptide (Thx) homing to non-small cell lung cancer identified by ex vivo phage display. Clin. Transl. Oncol. 2013, 15, 492–498. [Google Scholar] [CrossRef]
- Maruta, F.; Parker, A.L.; Fisher, K.D.; Murray, P.G.; Kerr†, D.J.; Seymour, L.W. Use of a Phage Display Library to Identify Oligopeptides Binding to the Lumenal Surface of Polarized Endothelium by Ex Vivo Perfusion of Human Umbilical Veins. J. Drug Target. 2003, 11, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Maruta, F.; Akita, N.; Nakayama, J.; Miyagawa, S.; Ismail, T.; Rowlands, D.C.; Kerr, D.J.; Fisher, K.D.; Seymour, L.W.; Parker, A.L. Bacteriophage biopanning in human tumour biopsies to identify cancer-specific targeting ligands. J. Drug Target. 2007, 15, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-J.; Sui, Y.-X.; Budha, A.; Zheng, J.-B.; Sun, X.-J.; Hou, Y.-C.; Wang, T.D.; Lu, S.-Y. Affinity peptide developed by phage display selection for targeting gastric cancer. World J. Gastroenterol. WJG 2012, 18, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-F.; Shan, X.; Wang, Y.-X.; Wang, W.; Feng, S.-Y.; Cui, Y.-B. Screening and selection of peptides specific for esophageal cancer cells from a phage display peptide library. J. Cardiothorac. Surg. 2014, 9, 76. [Google Scholar] [CrossRef]
- Yao, V.J.; Ozawa, M.G.; Trepel, M.; Arap, W.; McDonald, D.M.; Pasqualini, R. Targeting Pancreatic Islets with Phage Display Assisted by Laser Pressure Catapult Microdissection. Am. J. Pathol. 2005, 166, 625–636. [Google Scholar] [CrossRef]
- Lu, H.; Jin, D.; Kapila, Y.L. Application of laser capture microdissection to phage display peptide library screening. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2004, 98, 692–697. [Google Scholar] [CrossRef]
- Kubo, N.; Akita, N.; Shimizu, A.; Kitahara, H.; Parker, A.L.; Miyagawa, S. Identification of oligopeptide binding to colon cancer cells separated from patients using laser capture microdissection. J. Drug Target. 2008, 16, 396–404. [Google Scholar] [CrossRef]
- Sánchez-Martín, D.; Sørensen, M.D.; Lykkemark, S.; Sanz, L.; Kristensen, P.; Ruoslahti, E.; Álvarez-Vallina, L. Selection strategies for anticancer antibody discovery: Searching off the beaten path. Trends Biotechnol. 2015, 33, 292–301. [Google Scholar] [CrossRef]
- Zhao, H.; Arnold, F.H. Combinatorial protein design: Strategies for screening protein libraries. Curr. Opin. Struct. Biol. 1997, 7, 480–485. [Google Scholar] [CrossRef]
- Schofield, D.J.; Pope, A.R.; Clementel, V.; Buckell, J.; Chapple, S.D.; Clarke, K.F.; Conquer, J.S.; Crofts, A.M.; Crowther, S.R.E.; Dyson, M.R.; et al. Application of phage display to high throughput antibody generation and characterization. Genome Biol. 2007, 8, R254. [Google Scholar] [CrossRef]
- Gur, D.; Liu, S.; Shukla, A.; Pero, S.C.; Wicha, M.S.; Krag, D.N. Identification of single chain antibodies to breast cancer stem cells using phage display. Biotechnol. Prog. 2009, 25, 1780–1787. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.B.; George, A.J.T.; Ritter, M.A. Selection of antibodies to cell surface determinants on mouse thymic epithelial cells using a phage display library. Immunology 1997, 91, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, C.G.; Rasmussen, N.; Laenkholm, A.-V.; Ditzel, H.J. Phage Display–Derived Human Monoclonal Antibodies Isolated by Binding to the Surface of Live Primary Breast Cancer Cells Recognize GRP78. Cancer Res. 2007, 67, 9507–9517. [Google Scholar] [CrossRef]
- Edwards, B.M.; Main, S.H.; Cantone, K.L.; Smith, S.D.; Warford, A.; Vaughan, T.J. Isolation and tissue profiles of a large panel of phage antibodies binding to the human adipocyte cell surface. J. Immunol. Methods 2000, 245, 67–78. [Google Scholar] [CrossRef]
- Roovers, R.C.; Van der Wall, E.; de Bruïne, A.P.; Arends, J.; Hoogenboom, H.R. Identification of colon tumour-associated antigens by phage antibody selections on primary colorectal carcinoma. Eur. J. Cancer 2001, 37, 542–549. [Google Scholar] [CrossRef]
- Mutuberria, R.; Satijn, S.; Huijbers, A.; van der Linden, E.; Lichtenbeld, H.; Chames, P.; Arends, J.-W.; Hoogenboom, H.R. Isolation of human antibodies to tumor-associated endothelial cell markers by in vitro human endothelial cell selection with phage display libraries. J. Immunol. Methods 2004, 287, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Dorfmueller, S.; Tan, H.C.; Ngoh, Z.X.; Toh, K.Y.; Peh, G.; Ang, H.-P.; Seah, X.-Y.; Chin, A.; Choo, A.; Mehta, J.S.; et al. Isolation of a recombinant antibody specific for a surface marker of the corneal endothelium by phage display. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Van Ewijk, W.; De Kruif, J.; Germeraad, W.T.; Berendes, P.; Röpke, C.; Platenburg, P.P.; Logtenberg, T. Subtractive isolation of phage-displayed single-chain antibodies to thymic stromal cells by using intact thymic fragments. Proc. Natl. Acad. Sci. USA 1997, 94, 3903–3908. [Google Scholar] [CrossRef]
- Radošević, K.; van Ewijk, W. Subtractive Isolation of Single-Chain Antibodies Using Tissue Fragments. In Antibody Phage Display; Humana Press: Totowa, NJ, USA, 2001; Volume 178, pp. 235–243. ISBN 978-1-59259-240-1. [Google Scholar]
- Jarutat, T.; Nickels, C.; Frisch, C.; Stellmacher, F.; Hofig, K.P.; Knappik, A.; Merz, H. Selection of vimentin-specific antibodies from the HuCAL® phage display library by subtractive panning on formalin-fixed, paraffin-embedded tissue. Biol. Chem. 2007, 388. [Google Scholar] [CrossRef]
- Tordsson, J.M.; Brodin, T.N.; Karlström, P.J. Selections on Tissue Sections. In Antibody Engineering; Kontermann, R., Dübel, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 193–205. ISBN 978-3-540-41354-7. [Google Scholar]
- Tordsson, J.; Abrahmsén, L.; Kalland, T.; Ljung, C.; Ingvar, C.; Brodin, T. Efficient selection of scFv antibody phage by adsorption to in situ expressed antigens in tissue sections. J. Immunol. Methods 1997, 210, 11–23. [Google Scholar] [CrossRef]
- Tordsson, J.M.; Ohlsson, L.G.; Abrahmsén, L.B.; Karlström, P.J.; Lando, P.A.; Brodin, T.N. Phage-selected primate antibodies fused to superantigens for immunotherapy of malignant melanoma. Cancer Immunol. Immunother. 2000, 48, 691–702. [Google Scholar] [CrossRef] [PubMed]
- ten Haaf, A.; Gattenlöhner, S.; Tur, M.K. Antibody Selection on FFPE Tissue Slides. In Phage Display: Methods and Protocols; Hust, M., Lim, T.S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 381–391. ISBN 978-1-4939-7447-4. [Google Scholar]
- ten Haaf, A.; Pscherer, S.; Fries, K.; Barth, S.; Gattenlöhner, S.; Tur, M.K. Phage display-based on-slide selection of tumor-specific antibodies on formalin-fixed paraffin-embedded human tissue biopsies. Immunol. Lett. 2015, 166, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Ito, T.; Furuta, M.; Eguchi, C.; Toda, H.; Wakabayashi-Takai, E.; Kaneko, K. In Situ Phage Screening a method for identification of subnanogram tissue componentsin situ. J. Biol. Chem. 2002, 277, 30382–30387. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.; Sassoon, A.; An, F.; Simko, J.P.; Liu, B. Identification of Clinically Significant Tumor Antigens by Selecting Phage Antibody Library on Tumor Cells in Situ Using Laser Capture Microdissection. Mol. Cell. Proteom. 2006, 5, 2364–2373. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Bidlingmaier, S.; Lee, N.-K.; Liu, B. Combine Phage Antibody Display Library Selection on Patient Tissue Specimens with Laser Capture Microdissection to Identify Novel Human Antibodies Targeting Clinically Relevant Tumor Antigens. In Phage Display; Hust, M., Lim, T.S., Eds.; Springer: New York, NY, USA, 2018; Volume 1701, pp. 331–347. ISBN 978-1-4939-7446-7. [Google Scholar]
- Sun, Y.; Shukla, G.S.; Kennedy, G.G.; Warshaw, D.M.; Weaver, D.L.; Pero, S.C.; Floyd, L.; Krag, D.N. Biopanning Phage-Display Libraries on Small Tissue Sections Captured by Laser Capture Microdissection. J. Biotech Res. 2009, 1, 55–63. [Google Scholar] [PubMed]
- Sun, Y.; Shukla, G.S.; Weaver, D.; Pero, S.C.; Krag, D.N. Phage-display selection on tumor histological specimens with laser capture microdissection. J. Immunol. Methods 2009, 347, 46–53. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lykkemark, S.; Mandrup, O.A.; Jensen, M.B.; Just, J.; Kristensen, P. A novel excision selection method for isolation of antibodies binding antigens expressed specifically by rare cells in tissue sections. Nucleic Acids Res. 2017, 45, e107. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.A.; Meldgaard, T.; Lykkemark, S.; Mandrup, O.A.; Kristensen, P. Selection of cell-type specific antibodies on tissue-sections using phage display. J. Cell. Mol. Med. 2015, 19, 1939–1948. [Google Scholar] [CrossRef]
- Sørensen, M.D.; Agerholm, I.E.; Christensen, B.; Kølvraa, S.; Kristensen, P. Microselection–affinity selecting antibodies against a single rare cell in a heterogeneous population. J. Cell. Mol. Med. 2010, 14, 1953–1961. [Google Scholar] [CrossRef]
- Sørensen, M.D.; Kristensen, P. Selection of antibodies against a single rare cell present in a heterogeneous population using phage display. Nat. Protoc. 2011, 6, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.D.; Melchjorsen, C.J.; Mandrup, O.A.; Kristensen, P. Raising antibodies against circulating foetal cells from maternal peripheral blood. Prenat. Diagn. 2013, 33, 284–291. [Google Scholar] [CrossRef]
- Larsen, S.A.; Meldgaard, T.; Fridriksdottir, A.J.; Lykkemark, S.; Poulsen, P.C.; Overgaard, L.F.; Petersen, H.B.; Petersen, O.W.; Kristensen, P. Selection of a breast cancer subpopulation-specific antibody using phage display on tissue sections. Immunol. Res. 2015, 62, 263–272. [Google Scholar] [CrossRef][Green Version]
- Larsen, S.A.; Meldgaard, T.; Fridriksdottir, A.J.R.; Lykkemark, S.; Poulsen, P.C.; Overgaard, L.F.; Petersen, H.B.; Petersen, O.W.; Kristensen, P. Raising an Antibody Specific to Breast Cancer Subpopulations Using Phage Display on Tissue Sections. Cancer Genom. Proteom. 2016, 13, 21–30. [Google Scholar]
- Sørensen, K.M.J.; Meldgaard, T.; Melchjorsen, C.J.; Fridriksdottir, A.J.; Pedersen, H.; Petersen, O.W.; Kristensen, P. Upregulation of Mrps18a in breast cancer identified by selecting phage antibody libraries on breast tissue sections. BMC Cancer 2017, 17. [Google Scholar] [CrossRef]
- Emmert-Buck, M.R.; Bonner, R.F.; Smith, P.D.; Chuaqui, R.F.; Zhuang, Z.; Goldstein, S.R.; Weiss, R.A.; Liotta, L.A. Laser Capture Microdissection. Science 1996, 274, 5. [Google Scholar] [CrossRef]
- Bonner, R.F.; Emmert-Buck, M.; Cole, K.; Pohida, T.; Chuaqui, R.; Goldstein, S.; Liotta, L.A. Laser Capture Microdissection: Molecular Analysis of Tissue. Science 1997, 278, 1481–1483. [Google Scholar] [CrossRef]
- Schütze, K.; Lahr, G. Identification of expressed genes by laser-mediated manipulation of single cells. Nat. Biotechnol. 1998, 16, 6. [Google Scholar] [CrossRef]
- Pinzani, P.; Orlando, C.; Pazzagli, M. Laser-assisted microdissection for real-time PCR sample preparation. Mol. Asp. Med. 2006, 27, 140–159. [Google Scholar] [CrossRef]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the “high-hanging fruit”. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef]
- Jensen, K.B.; Jensen, O.N.; Ravn, P.; Clark, B.F.C.; Kristensen, P. Identification of Keratinocyte-specific Markers Using Phage Display and Mass Spectrometry. Mol. Cell. Proteom. 2003, 2, 61–69. [Google Scholar] [CrossRef]
- Lipman, N.S.; Jackson, L.R.; Trudel, L.J.; Weis-Garcia, F. Monoclonal Versus Polyclonal Antibodies: Distinguishing Characteristics, Applications, and Information Resources. ILAR J. 2005, 46, 258–268. [Google Scholar] [CrossRef]
- Ohtsuki, I.; Maruyama, K.; Ebashi, S. Regulatory and Cytoskeletal Proteins of Vertebrate Skeletal Muscle. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 1986; Volume 38, pp. 1–67. ISBN 978-0-12-034238-9. [Google Scholar]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef]
- Steinwand, M.; Droste, P.; Frenzel, A.; Hust, M.; Dübel, S.; Schirrmann, T. The influence of antibody fragment format on phage display based affinity maturation of IgG. mAbs 2014, 6, 204–218. [Google Scholar] [CrossRef]
- Chan, C.E.Z.; Chan, A.H.Y.; Lim, A.P.C.; Hanson, B.J. Comparison of the efficiency of antibody selection from semi-synthetic scFv and non-immune Fab phage display libraries against protein targets for rapid development of diagnostic immunoassays. J. Immunol. Methods 2011, 373, 79–88. [Google Scholar] [CrossRef]
- Thie, H.; Toleikis, L.; Li, J.; von Wasielewski, R.; Bastert, G.; Schirrmann, T.; Esteves, I.T.; Behrens, C.K.; Fournes, B.; Fournier, N.; et al. Rise and Fall of an Anti-MUC1 Specific Antibody. PLoS ONE 2011, 6, e15921. [Google Scholar] [CrossRef]
- Yang, W.; Yoon, A.; Lee, S.; Kim, S.; Han, J.; Chung, J. Next-generation sequencing enables the discovery of more diverse positive clones from a phage-displayed antibody library. Exp. Mol. Med. 2017, 49, e308. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mairaville, C.; Martineau, P. Antibody Identification for Antigen Detection in Formalin-Fixed Paraffin-Embedded Tissue Using Phage Display and Naïve Libraries. Antibodies 2021, 10, 4. https://doi.org/10.3390/antib10010004
Mairaville C, Martineau P. Antibody Identification for Antigen Detection in Formalin-Fixed Paraffin-Embedded Tissue Using Phage Display and Naïve Libraries. Antibodies. 2021; 10(1):4. https://doi.org/10.3390/antib10010004
Chicago/Turabian StyleMairaville, Célestine, and Pierre Martineau. 2021. "Antibody Identification for Antigen Detection in Formalin-Fixed Paraffin-Embedded Tissue Using Phage Display and Naïve Libraries" Antibodies 10, no. 1: 4. https://doi.org/10.3390/antib10010004
APA StyleMairaville, C., & Martineau, P. (2021). Antibody Identification for Antigen Detection in Formalin-Fixed Paraffin-Embedded Tissue Using Phage Display and Naïve Libraries. Antibodies, 10(1), 4. https://doi.org/10.3390/antib10010004