Refolding Technology for scFv Using a New Detergent, N-Lauroyl-L-glutamate and Arginine

Abstract
:1. Introduction
2. Refolding of Interleukin-6
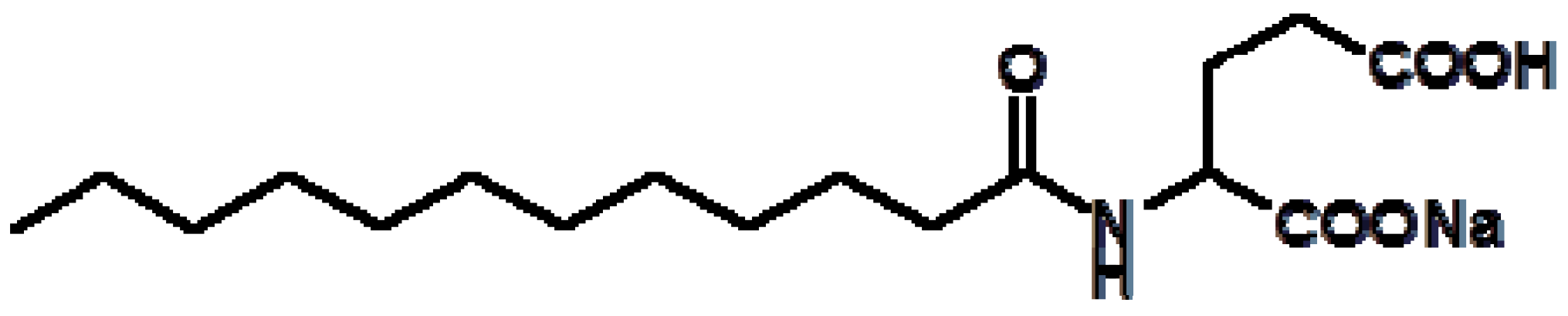
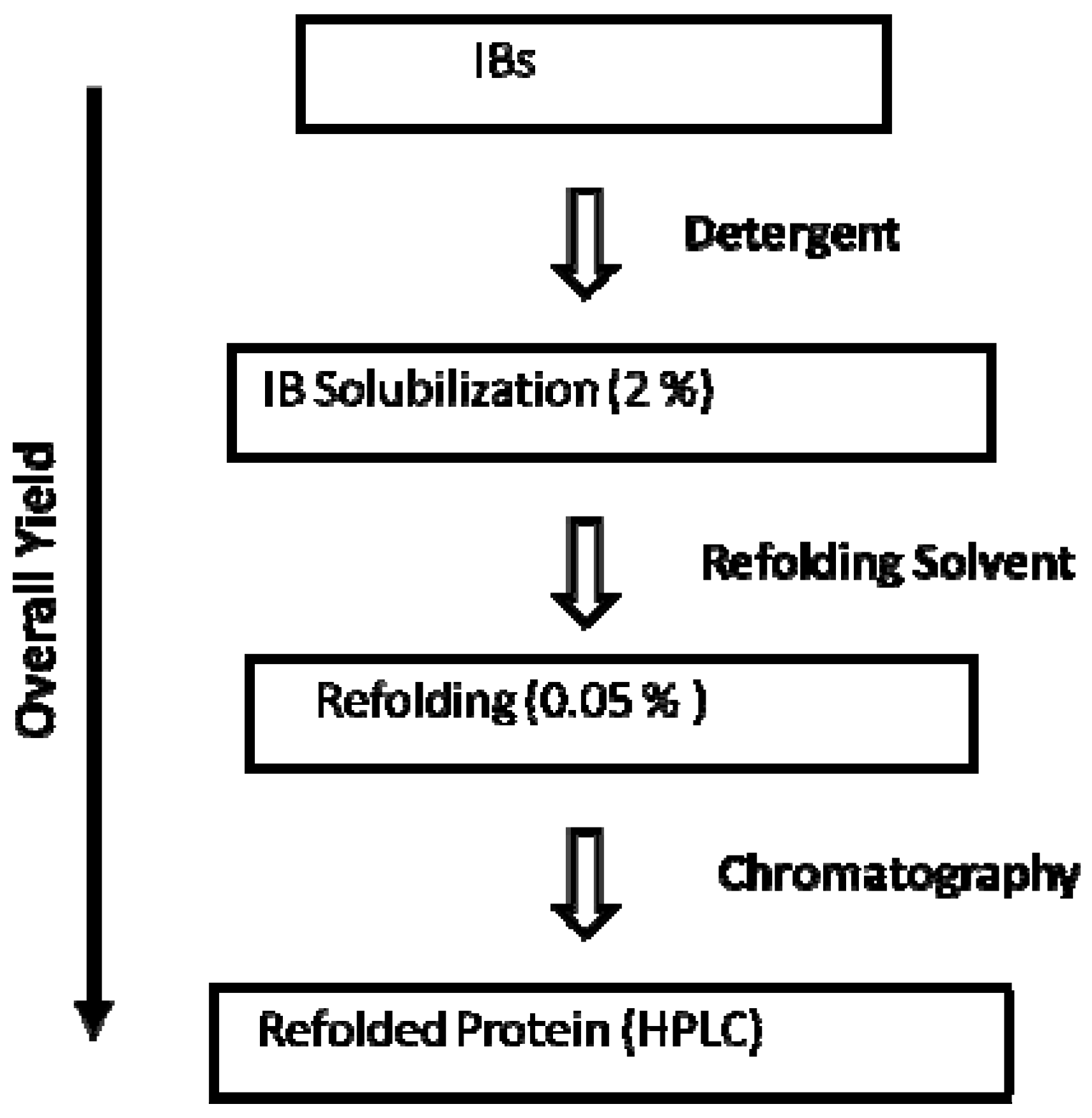
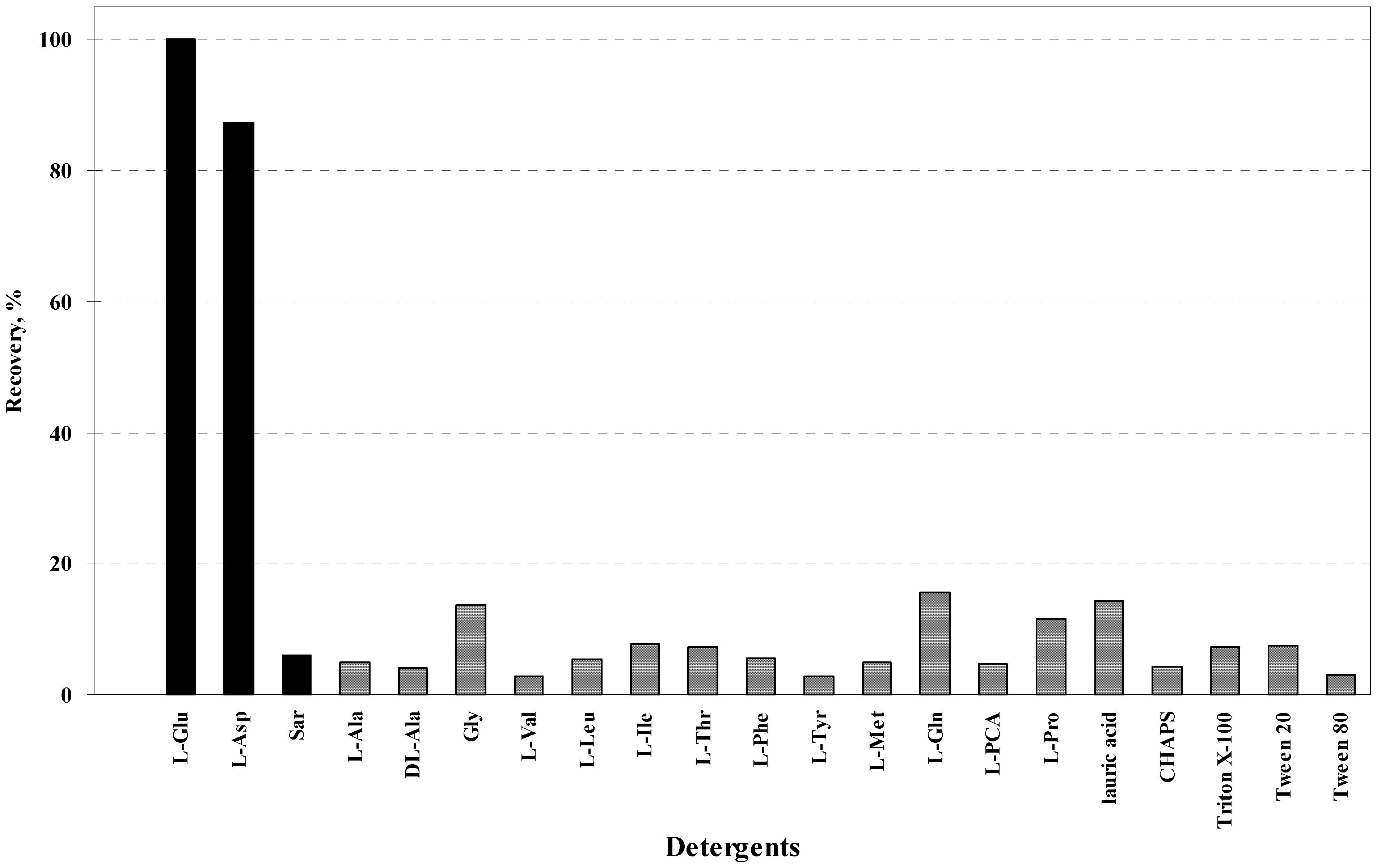
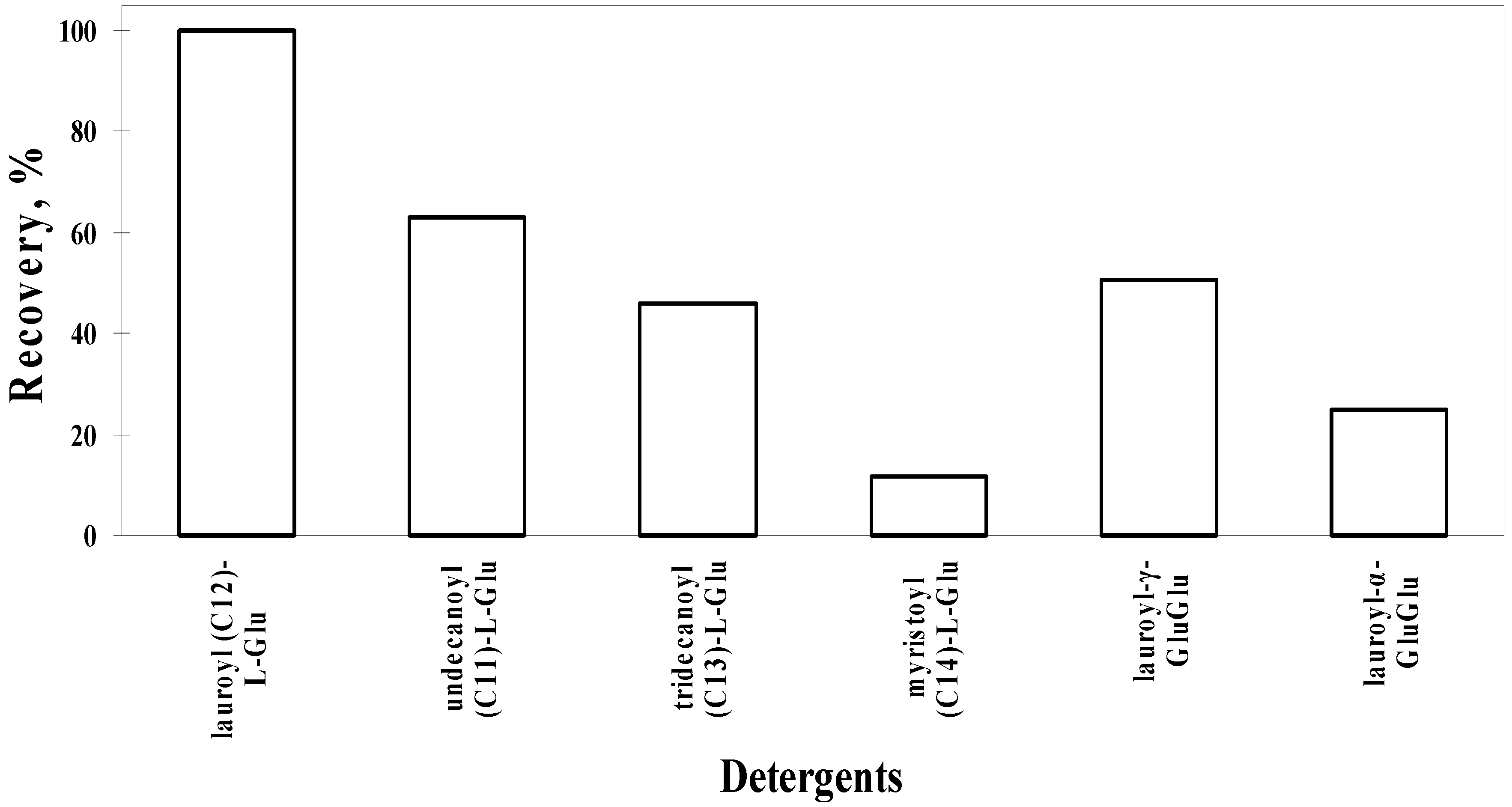
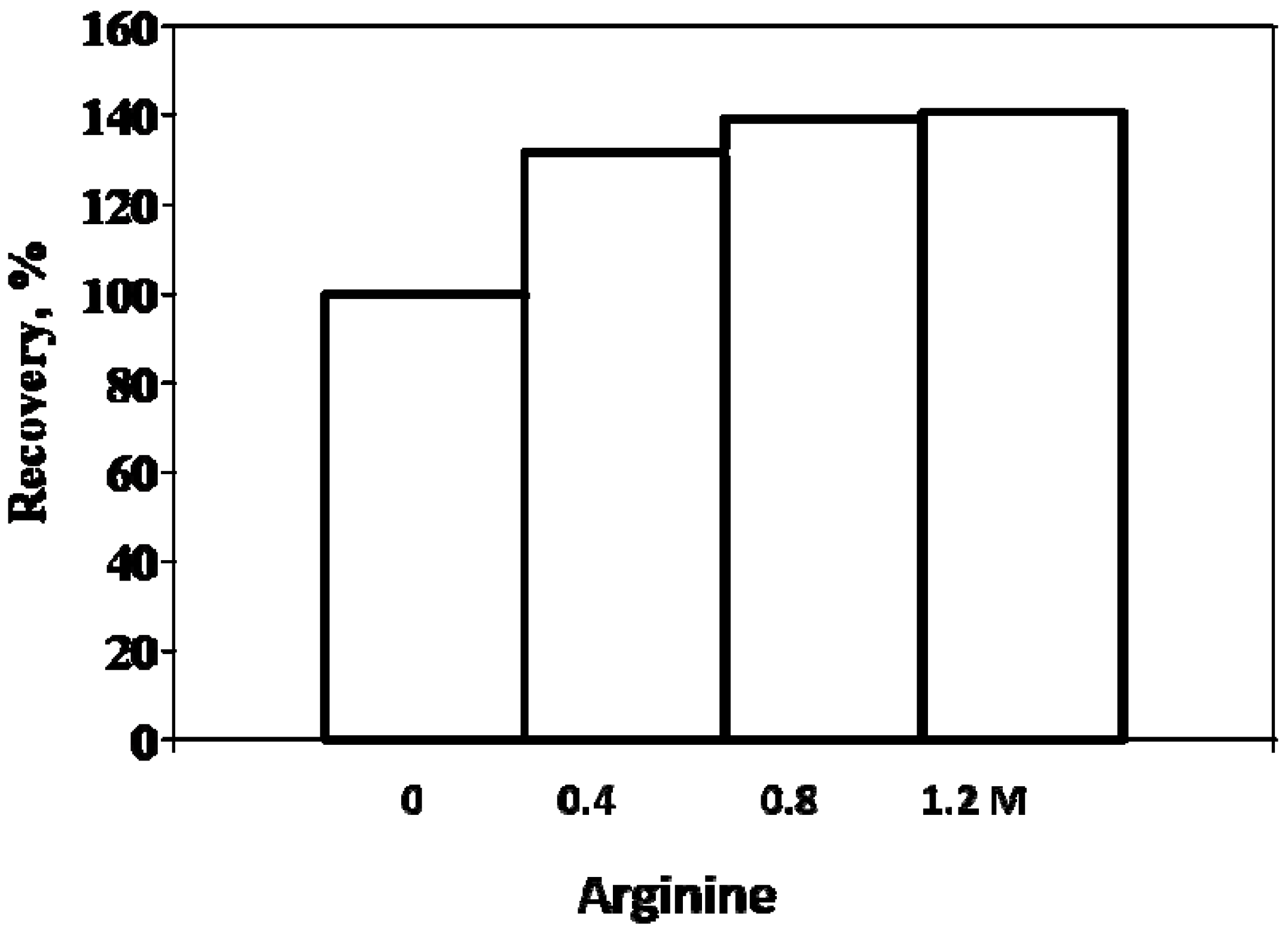
3. Refolding of Transglutaminase
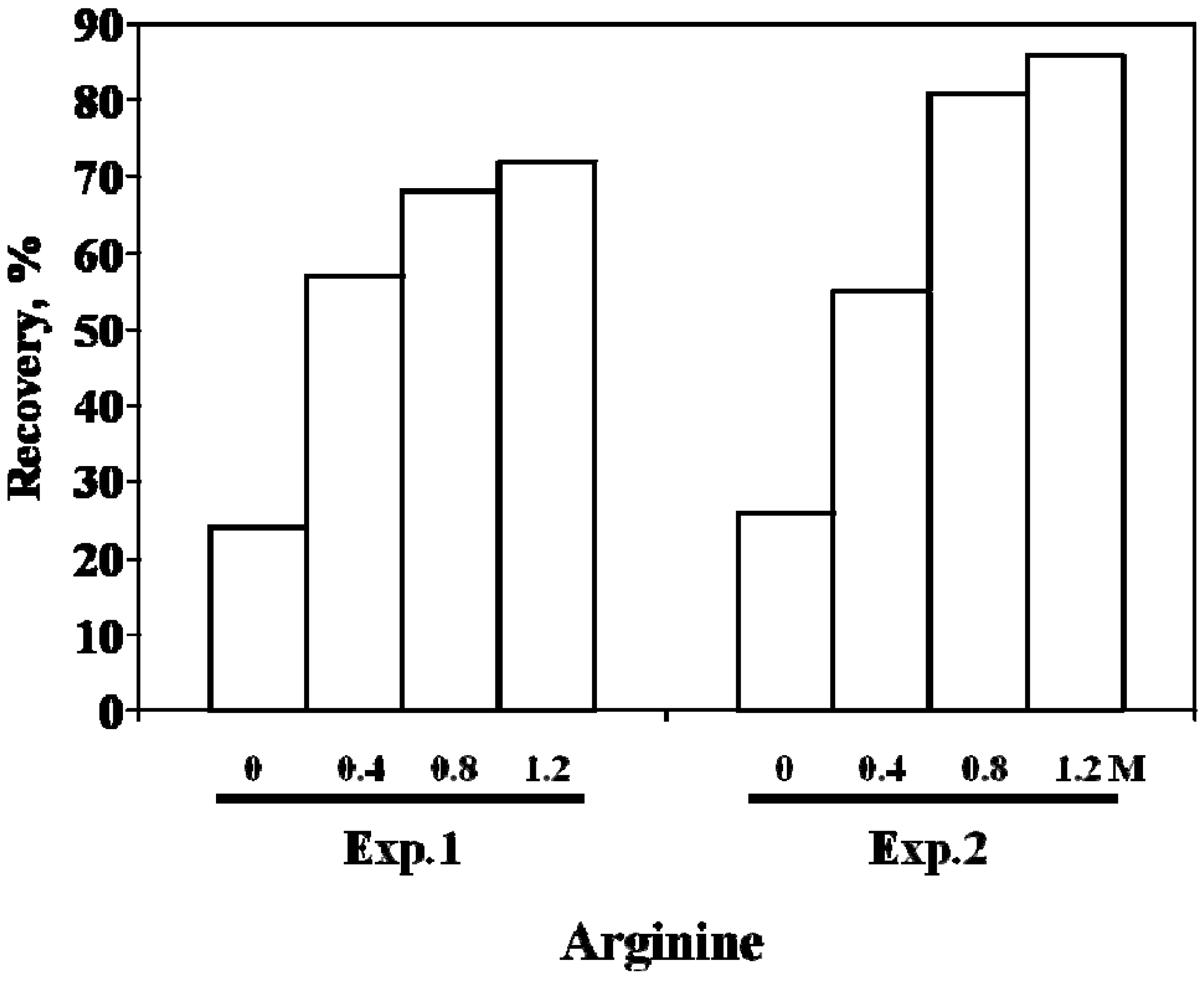
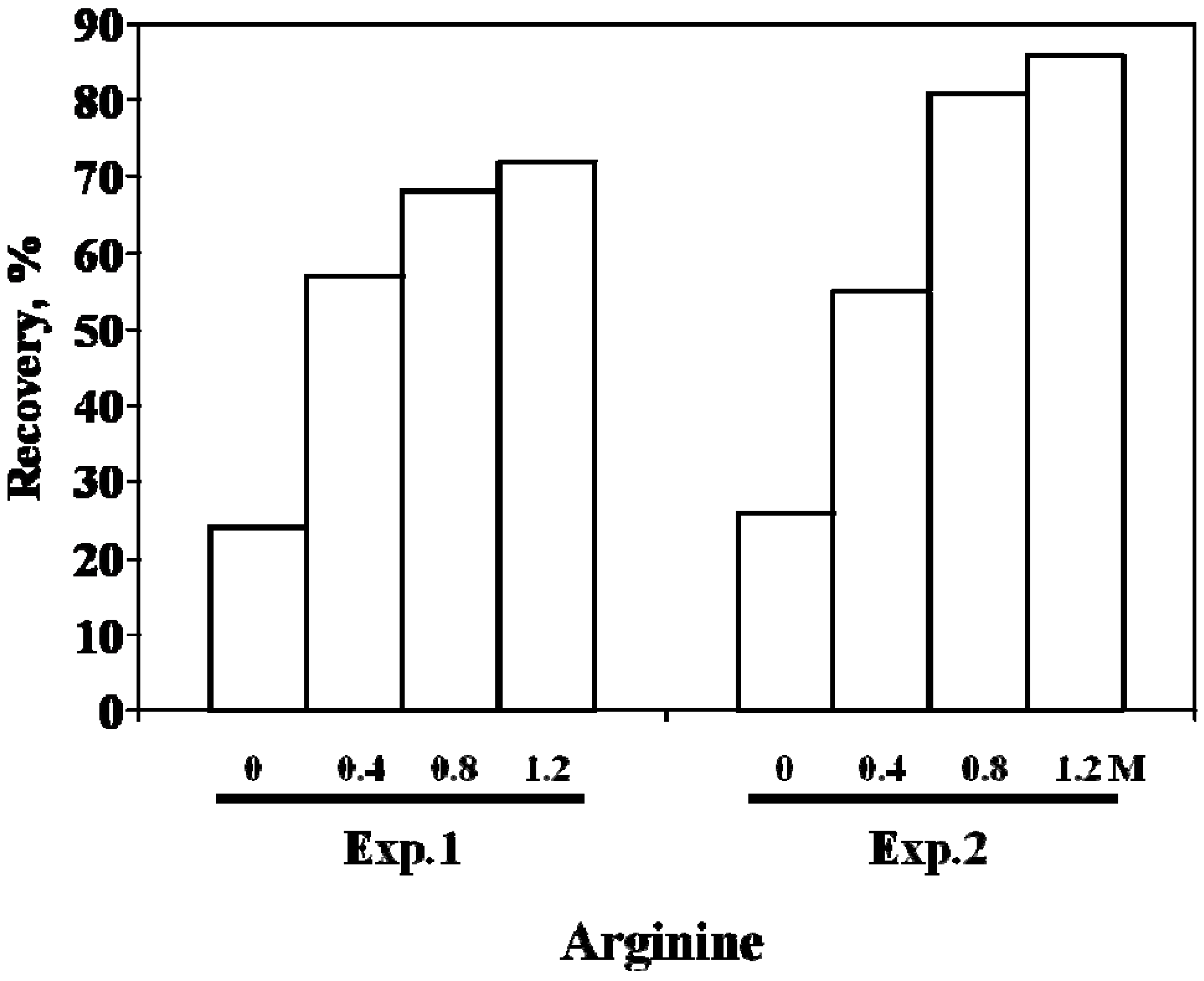
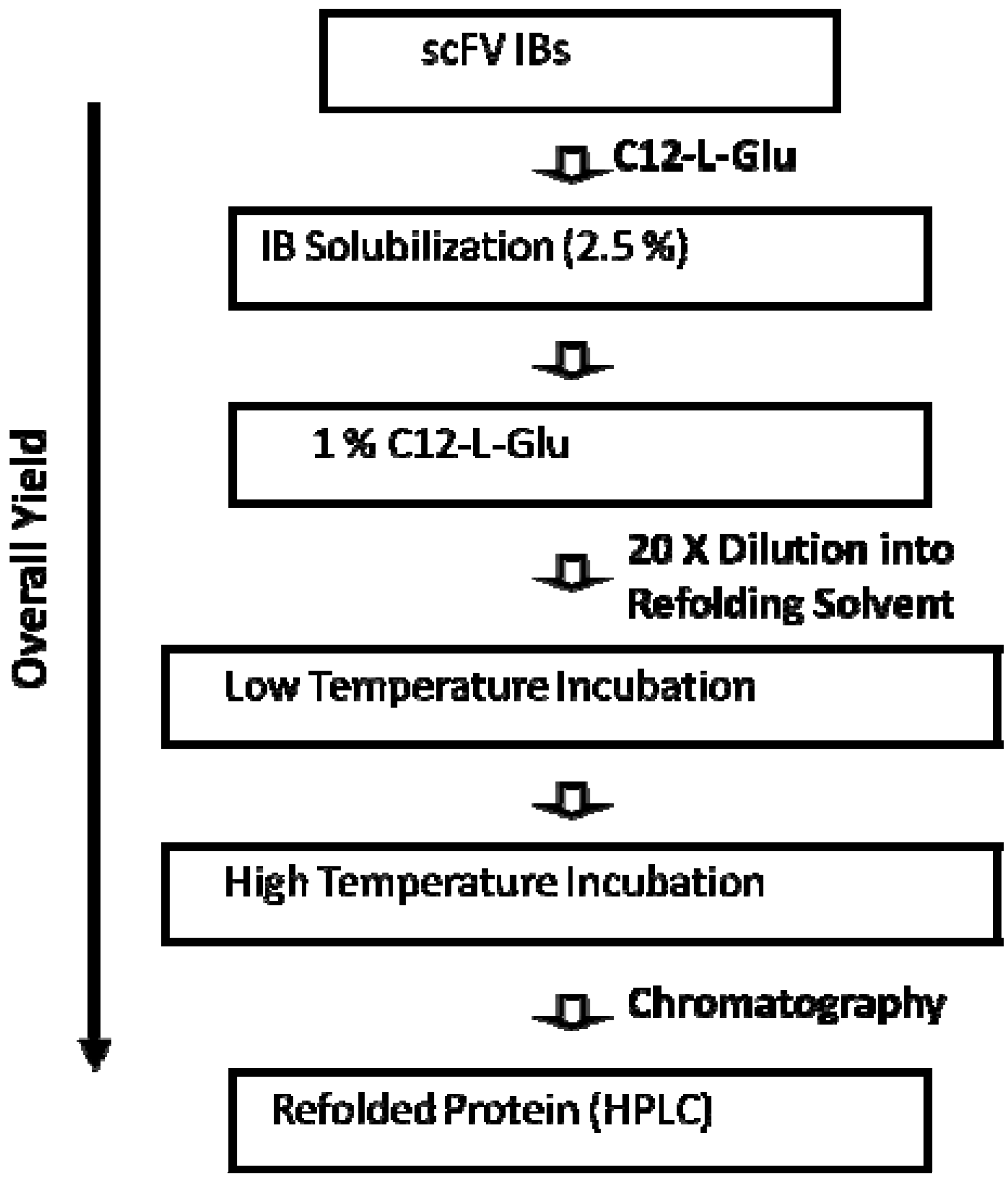
4. scFv
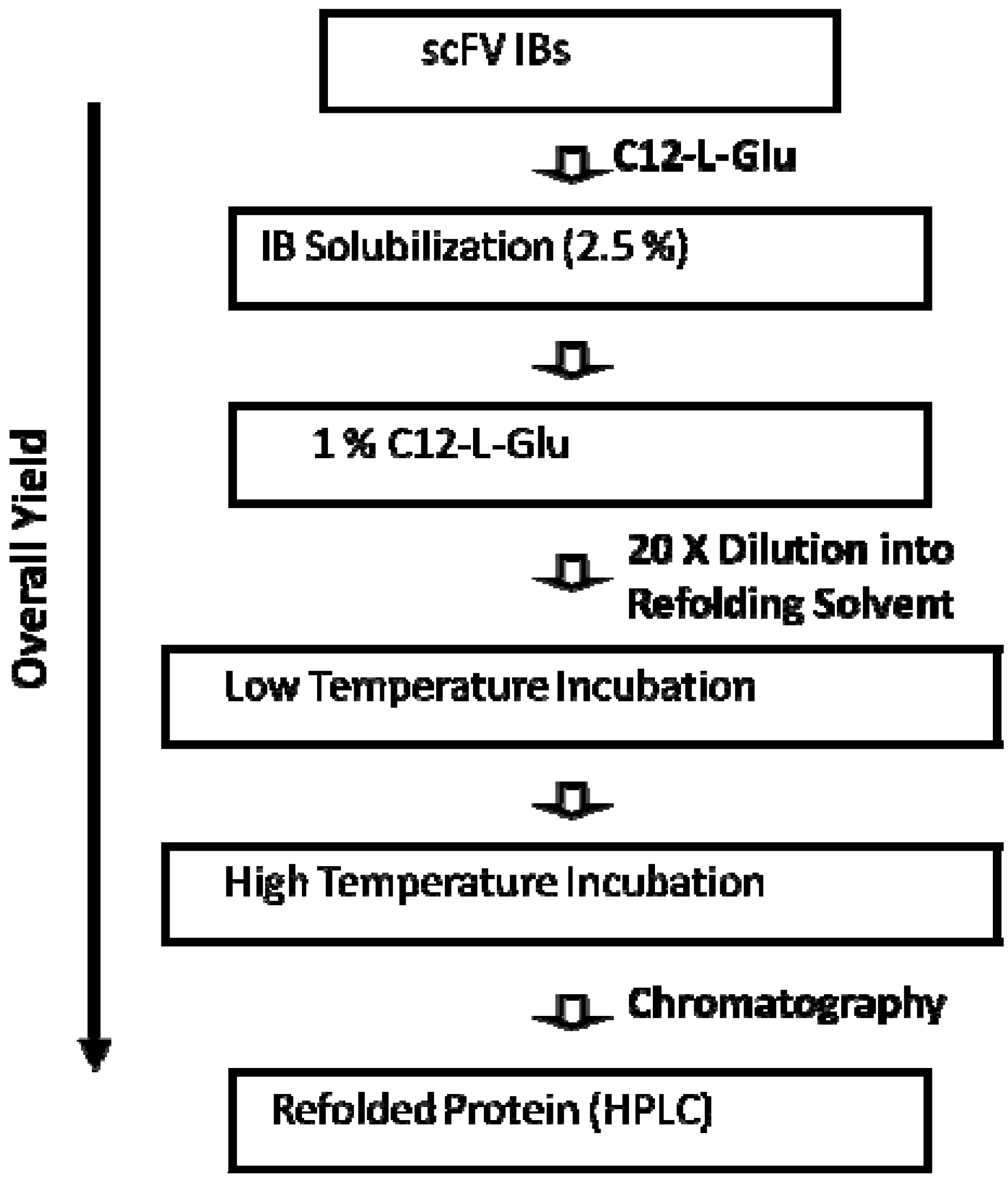
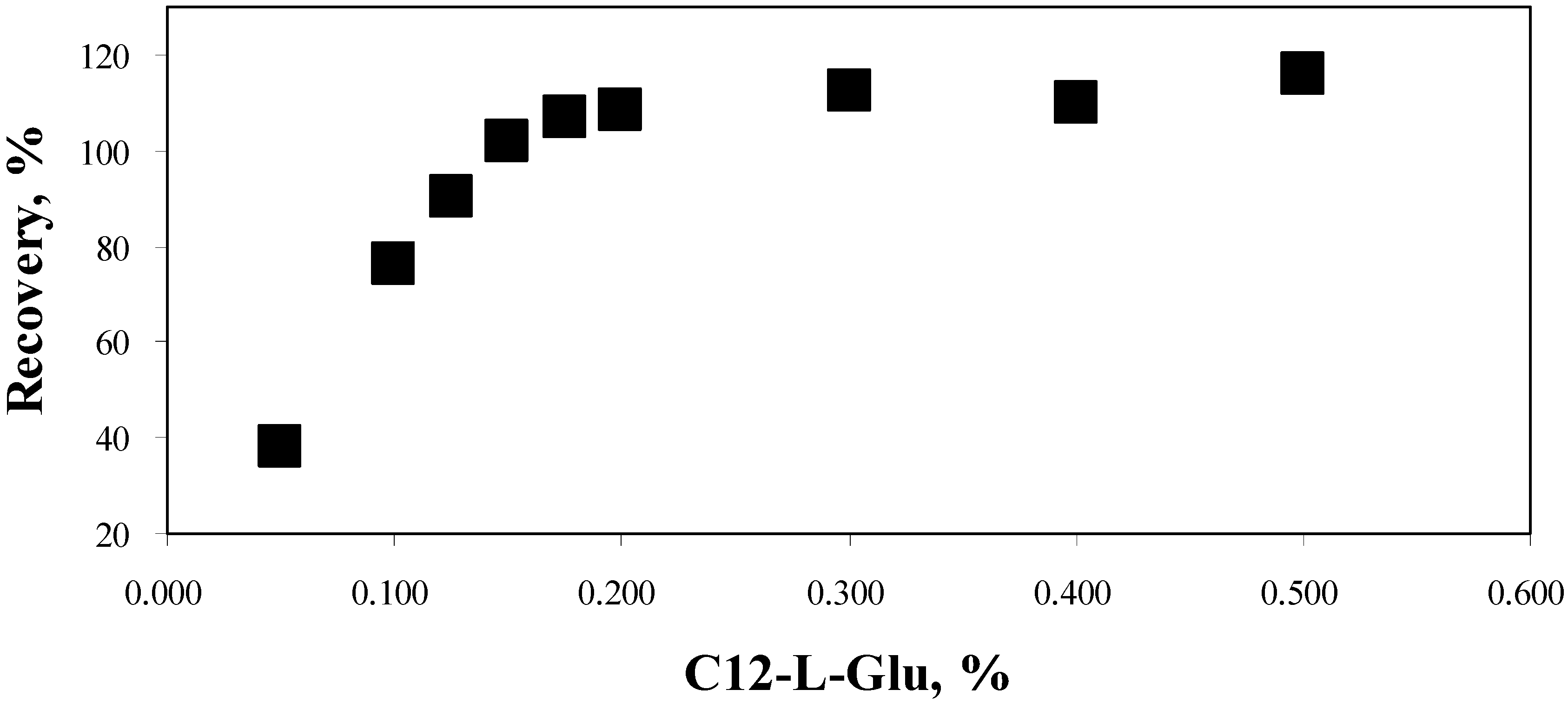
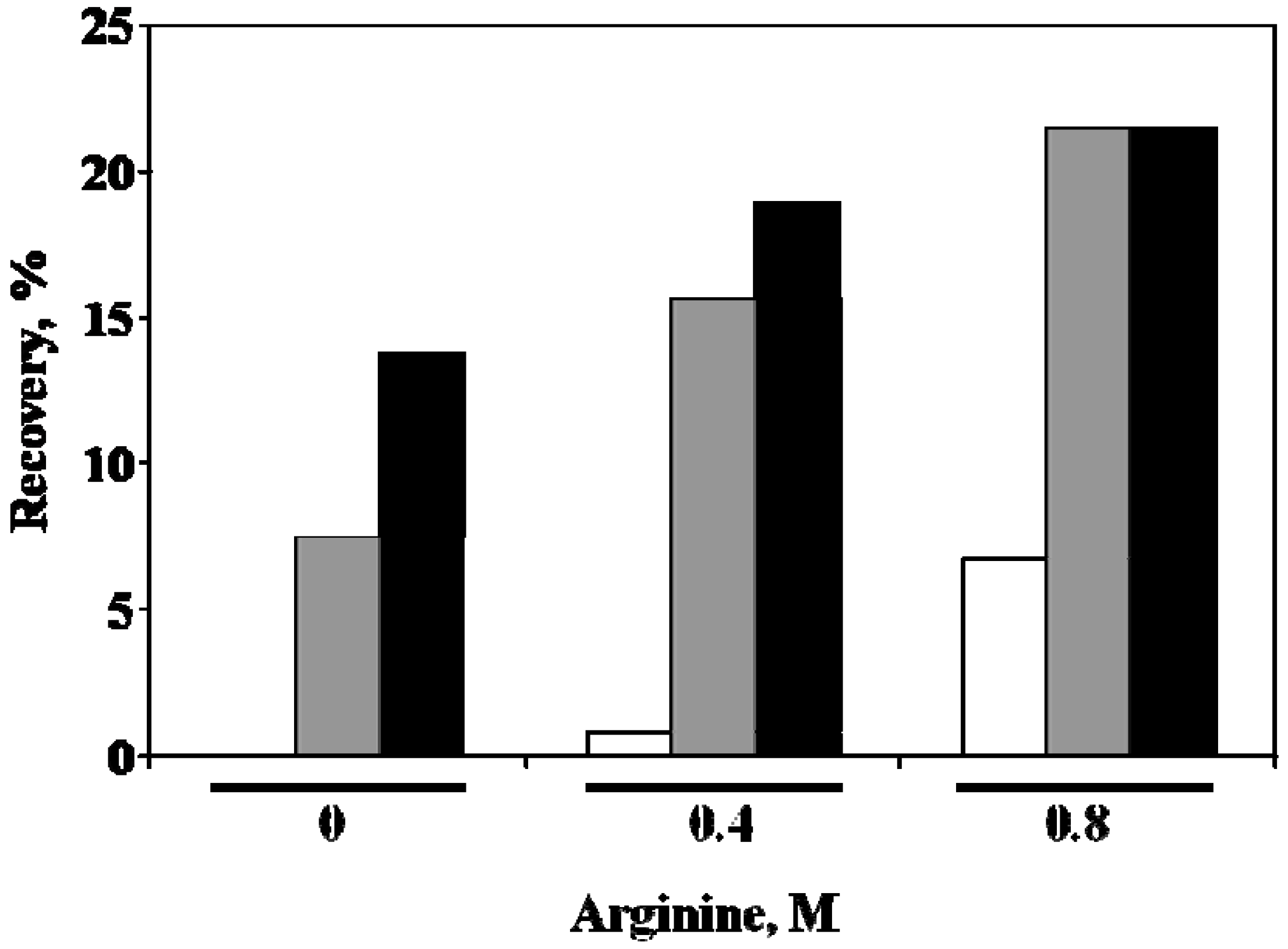
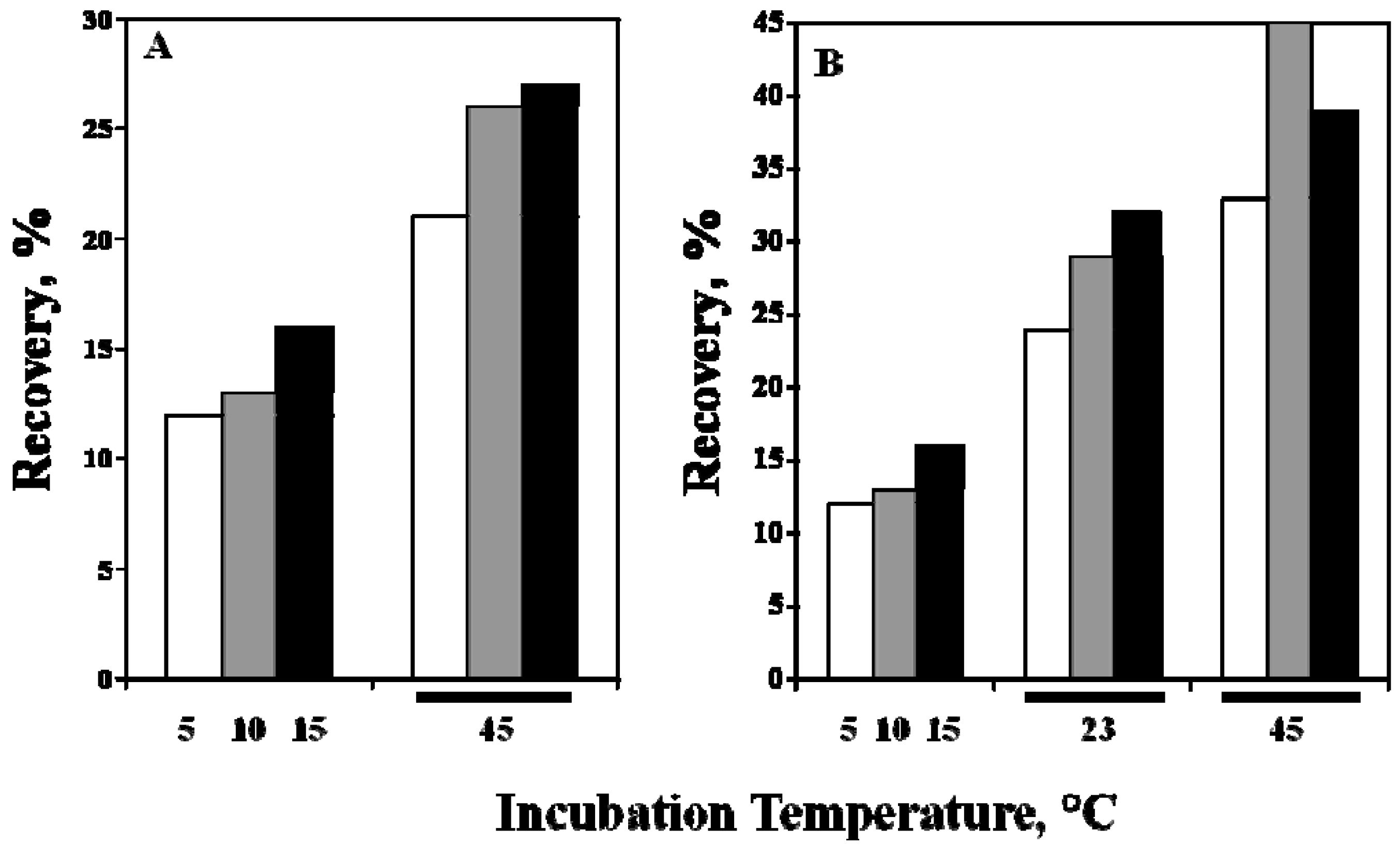
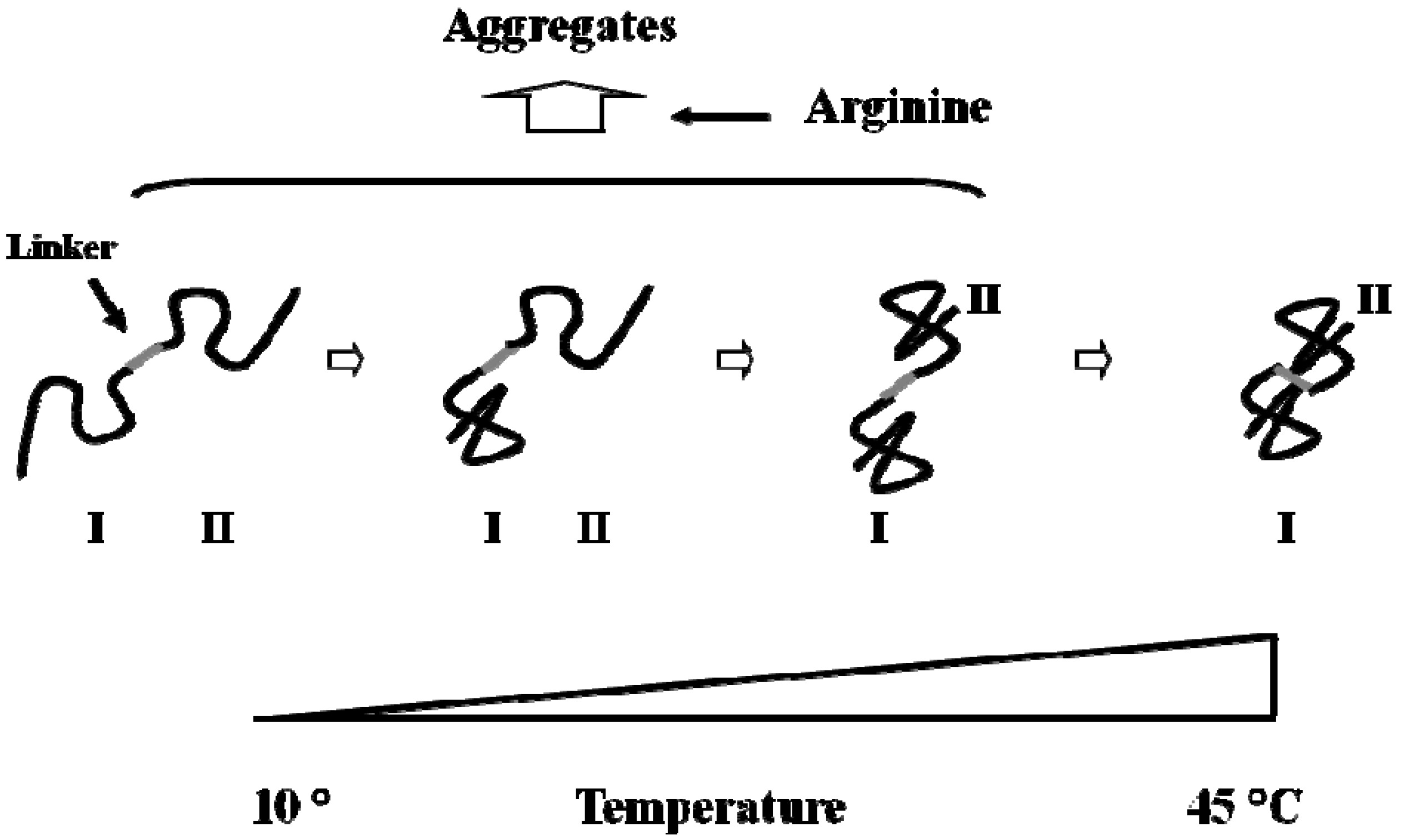
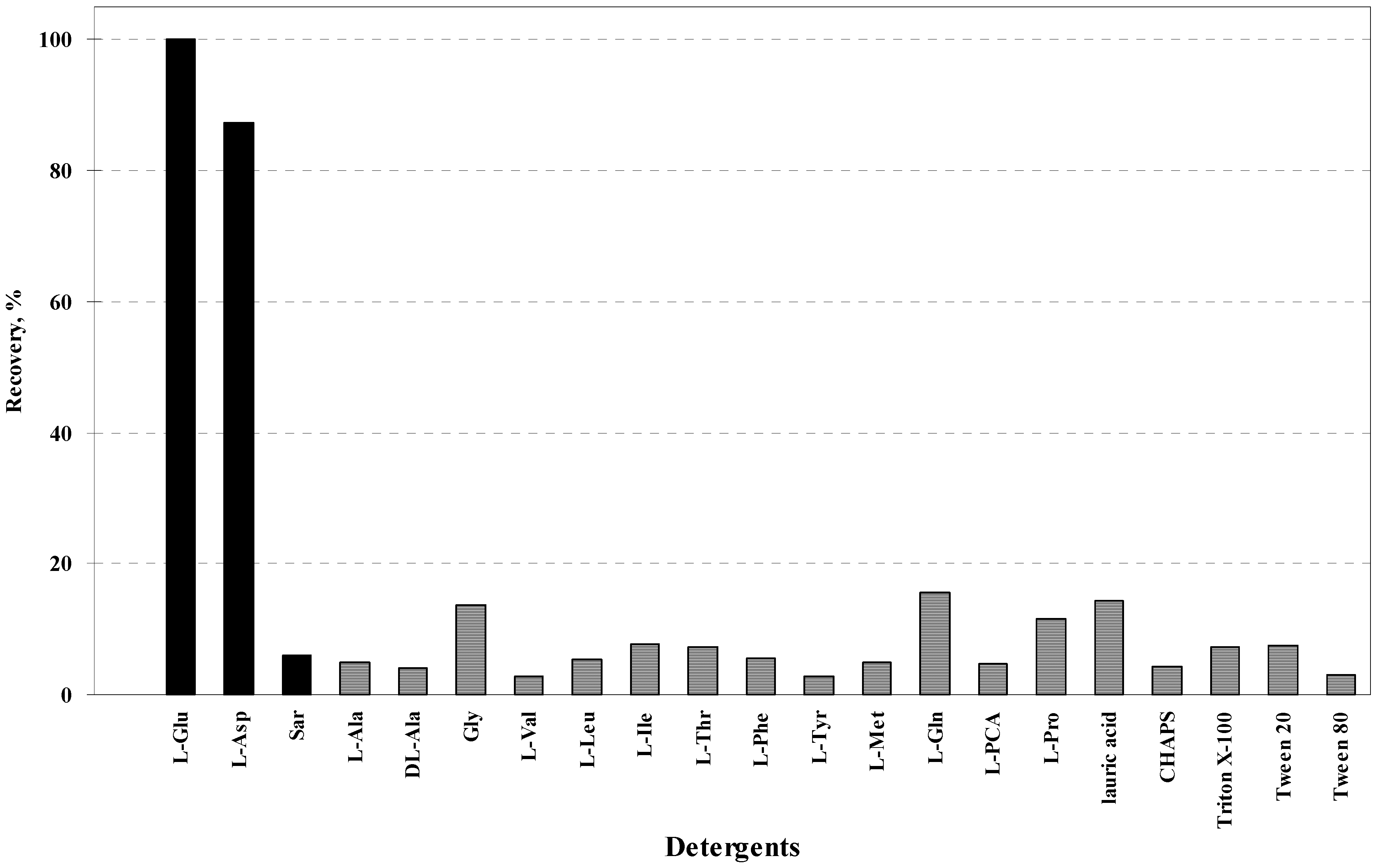
5. Property of C12-L-Glu

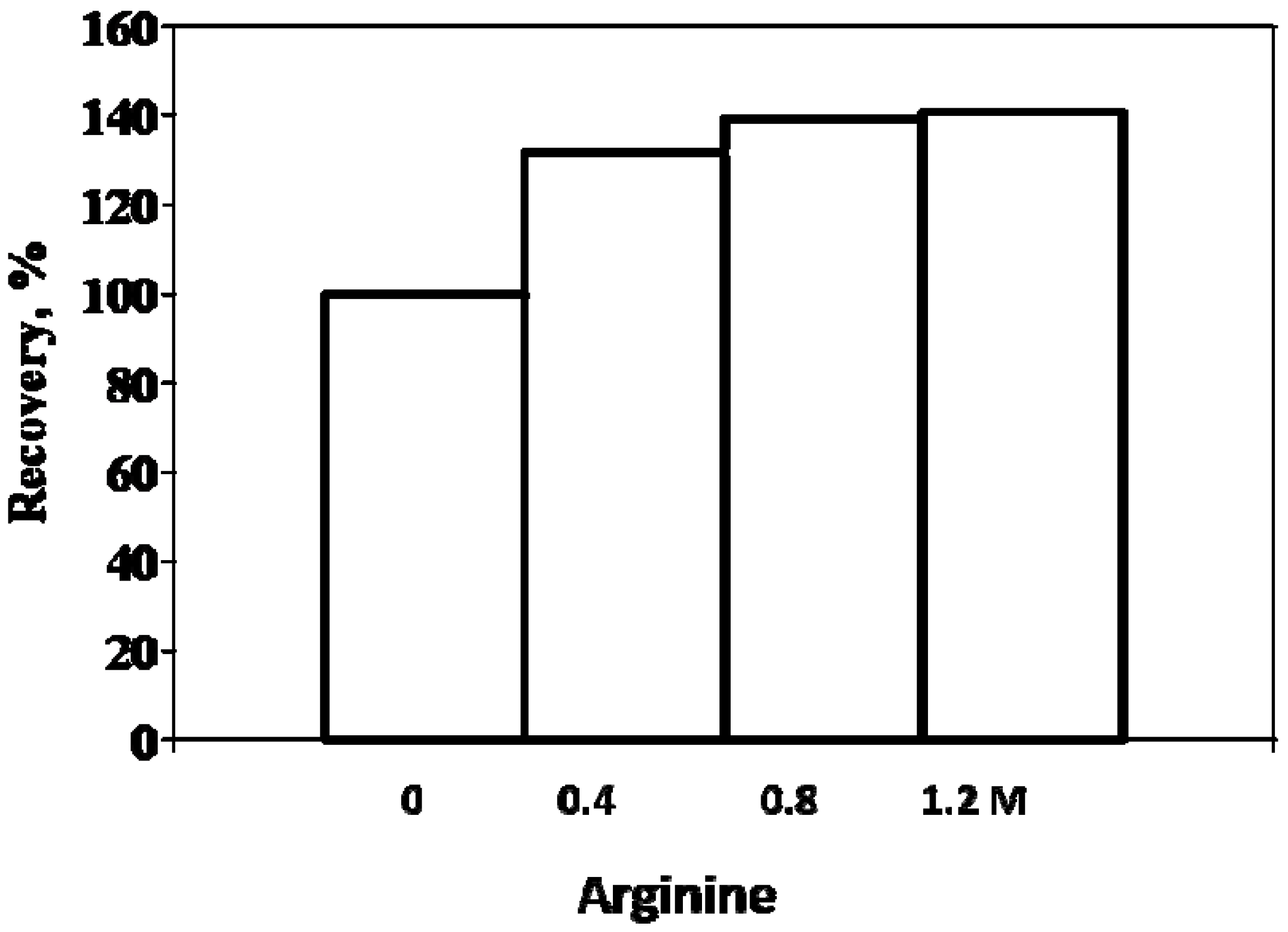
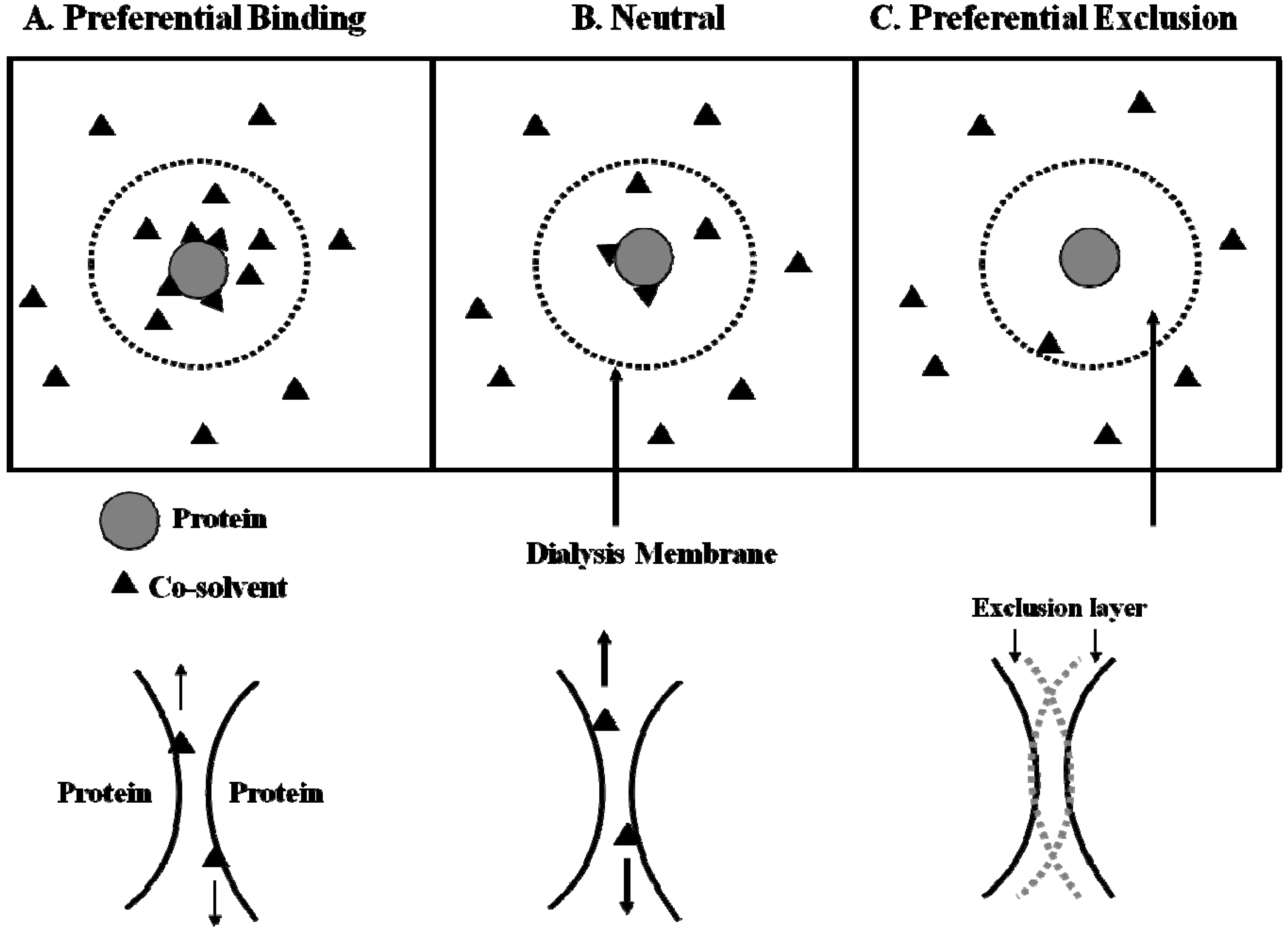
6. Effects of Arginine on Refolding

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Co-solvent | Interaction with Protein | Effects on Protein |
|---|---|---|
| Denaturants (Urea, GdnHCl) | Binding | Denaturation/Salting-in |
| Organic solvents | Exclusion | Precipitation |
| Binding | Denaturation | |
| Sugars/Polyols | Exclusion | Stabilization |
| Salts | Exclusion | Stabilization/Salting-out |
| Binding | Salting-in | |
| Polymers | Exclusion | Precipitation |
| Amino Acids | Exclusion | Stabilization |
| Arginine | ???????? | Aggregation Suppression |

References
- Yamada, T. Therapeutic monoclonal antibodies. Keio J. Med. 2011, 60, 37–46. [Google Scholar] [CrossRef]
- Jenkins, N.; Meleady, P.; Tyther, R.; Murphy, L. Strategies for analysing and improving the expression and quality of recombinant proteins made in mammalian cells. Biotechnol. Appl. Biochem. 2009, 53, 18–25. [Google Scholar]
- Shukla, A.A.; Thommes, J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. 2010, 28, 253–261. [Google Scholar] [CrossRef]
- Shukla, A.A.; Hubbard, B.; Tressel, T.; Guhan, S.; Low, D. Downstream processing of monoclonal antibodies-application of platform approaches. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 848, 28–39. [Google Scholar] [CrossRef]
- Low, D.; O’Leary, R.; Pujar, N.S. Future of antibody purification. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 848, 48–63. [Google Scholar] [CrossRef]
- Chon, J.H.; Zarbis-Papastoitsis, G. Advances in the production and downstream processing of antibodies. N. Biotechnol. 2011, 28, 458–463. [Google Scholar]
- Beck, A.; Wurch, T.; Bailly, C.; Corvaia, N. Challenges for the next generation of therapeutic antibodies. Nat. Rev. Immunol. 2010, 10, 345–352. [Google Scholar] [CrossRef]
- Demarest, S.J.; Glaser, S.M. Antibody therapeutics, antibody engineering, and the merits of protein stability. Curr. Opin. Drug Dev. 2008, 11, 675–687. [Google Scholar]
- Kontermann, R.E. Alternative antibody formats. Curr. Opin. Mol. Ther. 2010, 12, 176–183. [Google Scholar]
- Moffat, F.L.J.; Gulec, S.A.; Serafini, A.N.; Sfakianakis, G.N.; Pop, R.; Robinson, D.S.; Franceschi, D.; Boggs, J.; Livingstone, A.S. A thousand points of light and just dim bulbs? Radiolabeled antibodies and colorectal cancer imaging. Cancer Invest. 1999, 17, 322–334. [Google Scholar] [CrossRef]
- Colcher, D.; Goel, A.; Pavlinkova, G.; Beresford, G.; Booth, B.; Batra, S.K. Effects of genetic engineering on the pharmacokinetics of antibodies. Q. J. Nucl. Med. 1999, 43, 132–139. [Google Scholar]
- Nelson, A.L.; Reichert, J.M. Development trends for therapeutic antibody fragments. N. Biotechnol. 2009, 27, 331–337. [Google Scholar]
- Yazaki, P.J.; Shively, L.; Clark, C.; Cheung, C.-W.; Le, W.; Szpikowska, B.; Shively, J.E.; Raubitschek, A.A.; Wu, A.M. Mammalian expression and hollow fiber biorector production of recombinant anti-CEA diabody and minibody for clinical applications. J. Immunol. Methods 2001, 253, 195–208. [Google Scholar] [CrossRef]
- Accardi, L.; Di Bonito, P. Antibodies in single-chain format against tumour-associated antigens: Present and future applications. Curr. Med. Chem. 2010, 17, 1730–1755. [Google Scholar] [CrossRef]
- Wörn, A.; Plückthun, A. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 2001, 305, 989–1010. [Google Scholar] [CrossRef]
- Stockwin, L.H.; Holmes, S. The role of therapeutic antibodies in drug discovery. Biochem. Soc. Trans. 2003, 31, 433–436. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Mernaugh, R.L.; Zeng, X. Single chain fragment variable recombinant antibody functionalized gold nanoparticles for a highly sensitive colorimetric immunoassay. Biosens. Bioelectron. 2009, 24, 2853–2867. [Google Scholar] [CrossRef]
- Schmidt, S.R. Fusion-proteins as biopharmaceuticals-applications and challenges. Curr. Opin. Drug Discov. Dev. 2009, 12, 284–295. [Google Scholar]
- Makabe, K.; Asano, R.; Ito, T.; Tsumoto, K.; Kudo, T.; Kumagai, I. Tumorpdirected lymphocyte-activating cytokines: Refolding-based preparation of recombinant human interleukin-12 and an antibody variable domain-fused protein by additive-introduced stepwise dialysis. Biochem. Biophys. Res. Commun. 2005, 328, 98–105. [Google Scholar] [CrossRef]
- Sharkey, R.M.; Goldenberg, D.M. Perspective on cancer therapy with radiolabeled monoclonal antibodies. J. Nucl. Med. 2005, 46, 115S–127S. [Google Scholar]
- Kelly, M.P.; Lee, F.T.; Tahtis, K.; Power, B.E.; Smyth, F.E.; Brechbiel, M.W.; Hudson, P.J.; Scott, A.M. Tumor targeting by a multivalent single-chain Fv (scFv) anti-Lewis Y Antibody construct. Cancer Biother. Radiopharm. 2008, 23, 411–423. [Google Scholar] [CrossRef]
- Beckman, R.A.; Weiner, L.M.; Davis, H.M. Antibody constructs in cancer therapy: Protein engineering strategies to improve exposure in solid tumors. Cancer 2007, 109, 170–179. [Google Scholar] [CrossRef]
- Ottiger, M.; Thiel, M.A.; Feige, U.; Lichtlen, P.; Urech, D.M. Efficient intracocular penetration of topical anti-TNF-α single-chain antibody (ESBA105) to anterior and posterior segment without penetration enhancer. Invest. Ophthal. Vis. Sci. 2009, 50, 779–786. [Google Scholar]
- Wittel, U.A.; Jain, M.; Goel, A.; Chauhan, S.C.; Colcher, D.; Batra, S.K. The in vivo characteristics of genetically engineered divalent and tetravalent single-chain antibody constructs. Nucl. Med. Biol. 2005, 32, 157–164. [Google Scholar] [CrossRef]
- Kirschning, C.J.; Dreher, S.; Maass, B.; Fichte, S.; Schade, J.; Köster, M.; Noack, A.; Lindenmaier, W.; Wagner, H.; Böldicke, T. Generation of anti-TLR2 intrabody mediating inhibition of macrophage surface TLR2 expression and TLR2-driven cell activation. BMC Biotechnol. 2010, 10, 31. [Google Scholar] [CrossRef]
- Xiong, H.; Li, S.; Yang, Z.; Burgess, R.R.; Dynan, W.S. E. coli expression of a soluble, active single-chain antibody variable fragment containing a nuclear localization signal. Protein Expr. Purif. 2009, 66, 172–180. [Google Scholar] [CrossRef]
- Poul, M.A. Selection of antibodies able to rapidly enter mammalian cells. Methods Mol. Biol. 2009, 562, 155–163. [Google Scholar] [CrossRef]
- Butler, D.C.; McLear, J.A.; Messer, A. Engineered antibody therapies to counteract mutant huntingtin and related toxic intracellular proteins. Prog. Neurobiol. 2012, 97, 190–204. [Google Scholar] [CrossRef]
- Hasegawa, H.; Wendling, J.; He, F.; Trilisky, E.; Stevenson, R.; Franey, H.; Kinderman, F.; Li, G.; Piedmonte, D.M.; Oslund, T.; Shen, M.; Ketchem, R.R. In vivo crystallization of human IgG in the endoplasmic reticulum of engineered CHO cell. J. Biol. Chem. 2011, 286, 19917–19931. [Google Scholar]
- Wurm, F.M. Production of recombinant protein therapeutics in cultivated mammalian cells. N. Biotechnol. 2004, 22, 1393–1398. [Google Scholar] [CrossRef]
- Humphreys, D.P.; Glover, D.J. Therapeutic antibody production technologies: Molecules, applications, expression and purification. Curr. Opin. Drug Dev. 2001, 4, 172–185. [Google Scholar]
- Andersen, D.C.; Reilly, D.E. Production technologies for monoclonal antibodies and their fragments. Curr. Opin. Biotechnol. 2004, 15, 456–462. [Google Scholar] [CrossRef]
- Arbabi-Ghahroudi, M.; Tanha, J.; MacKenzie, R. Prokaryotic expression of antibodies. Cancer Metastasis Rev. 2005, 24, 501–519. [Google Scholar] [CrossRef]
- Sun, W.; Xie, J.; Lin, H.; Mi, S.; Li, Z.; Hua, F.; Hu, Z. A combined strategy improves the solubility of aggregation-prone single-chain variable fragment antibodies. Protein Expr. Purif. 2012, 83, 21–29. [Google Scholar] [CrossRef]
- Lilie, H.; Schwarz, E.; Rudolph, R. Advances in refolding of proteins produced in E. coli. Curr. Opin. Biotechnol. 1998, 9, 497–501. [Google Scholar] [CrossRef]
- Tsumoto, K.; Ejima, D.; Kumagai, I.; Arakawa, T. Practical consideration in refolding proteins from inclusion bodies. Protein Expr. Purif. 2003, 28, 1–8. [Google Scholar] [CrossRef]
- Zardeneta, G.; Horowits, P.M. Detergent, liposome, and micelle-assisted protein refolding. Anal. Biochem. 1994, 223, 1–6. [Google Scholar] [CrossRef]
- Zardeneta, G.; Horowitz, P.M. Protein refolding at high concentrations using detergent/phospholipid mixtures. Anal. Biochem. 1994, 218, 392–398. [Google Scholar] [CrossRef]
- Michaux, C.; Pomroy, N.C.; Privė, G.G. Refolding SDS-denatured proteins by the addition of amphipathic cosolvents. J. Mol. Biol. 2008, 375, 1477–1488. [Google Scholar] [CrossRef]
- Lu, H.; Clogston, C.L.; Narhi, L.O.; Merewether, L.A.; Pearl, W.R.; Boone, T.C. Folding and oxidation of recombinant human granulocyte colony stimulating factor produced in Escherichia coli. Characterization of the disulfide-reduced intermediates and cysteine-serine analogs. J. Biol. Chem. 1992, 267, 8770–8777. [Google Scholar]
- Kurucz, I.; Titus, J.A.; Jost, C.R.; Segal, D.M. Correct disulfide pairing and efficient refolding of detergent-solubilized single-chain Fv proteins from bacterial inclusion bodies. Mol. Immunol. 1995, 32, 1443–1452. [Google Scholar] [CrossRef]
- Fursova, K.K.; Laman, A.G.; Melnik, B.S.; Semisotnov, G.V.; Kopylov, P.K.; Kiseleva, N.V.; Nesmeyanov, V.A.; Brovko, F.A. Refolding of scFv mini-antibodies using size-exclusion chromatography via arginine solution layer. J. Chromatogr. B 2009, 877, 2045–2051. [Google Scholar] [CrossRef]
- Tsumoto, K.; Shinoki, K.; Kondo, H.; Uchikawa, M.; Juji, T.; Kumagai, I. Highly efficient recovery of functional single-chain Fv fragments from inclusion bodies overexpressed in Escherichia coli by controlled introduction of oxidizing reagents-application to a human single-chain Fv fragment. J. Immunol. Methods 1998, 219, 119–129. [Google Scholar] [CrossRef]
- Fujii, T.; Ohkuri, T.; Onodera, R.; Ueda, T. Stable supply of large amounts of human Fab from the inclusion bodies in E. coli. J. Biochem. 2007, 141, 699–707. [Google Scholar] [CrossRef]
- Kudou, M.; Yumioka, R.; Ejima, D.; Arakawa, T. A novel protein refolding system using lauroyl-L-glutamate as a solubilizing detergent and arginine as a folding assisting agent. Protein Expr. Purif. 2011, 75, 46–54. [Google Scholar] [CrossRef]
- Kudou, M.; Ejima, D.; Sato, H.; Yumioka, R.; Arakawa, T. Refolding single-chain antibody (scFv) using lauroyl-L-glutamate as a solubization detergent and arginine as a refolding additive. Protein Expr. Purif. 2011, 7, 68–74. [Google Scholar]
- Xia, J.; Nnanna, I.A.; Sakamoto, K. Amino acid surfactants: Chemistry, synthesis, and properties. In Protein-Based Surfactants. Synthesis, Physicochemical Properties, and Applications; Nnanna, I.A., Xia, J., Eds.; Marcel Dekker: New York, NY, USA, 2001; pp. 75–122. [Google Scholar]
- Ejima, D.; Watanabe, M.; Sato, Y.; Date, M.; Yamada, N.; Takahara, Y. High yield refolding and purification process for recombinant interleukin-6 expressed in Escherichia coli. Biotechnol. Bioeng. 1999, 62, 301–310. [Google Scholar] [CrossRef]
- Boone, T.; Chazin, V.; Kenney, W.; Swanson, E.; Altrock, B. Construction, purification and biological activities of recombinant human interleukin-2 analogs. Dev. Biol. Stand. 1988, 69, 157–168. [Google Scholar]
- Umetsu, M.; Tsumoto, K.; Hara, M.; Ashish, K.; Goda, S.; Adshiri, T.; Kumagai, I. How additives influence the refolding of immunoglobulin-folded proteins in a stepwise dialysis system. Spectroscopic evidence for highly efficient refolding of a single-chain Fv fragment. J. Biol. Chem. 2003, 278, 8979–8987. [Google Scholar]
- Reddy, K.R.C.; Lilie, H.; Rudolph, R.; Lange, C. L-Arginine increases the solubility of unfolded species of hen egg white lysozyme. Protein Sci. 2005, 14, 929–935. [Google Scholar] [CrossRef]
- Armstrong, N.; de Lencastre, A.; Gouaux, E. A new protein folding screen: Application to the ligand binding domains of a glutamate and kainite receptor and to lysozyme and carbonic anhydrase. Protein Sci. 1999, 8, 1475–1483. [Google Scholar] [CrossRef]
- Oneda, H.; Inouye, K. Refolding and recovery of recombinant human matrix metalloproteinase (matrilysin) from inclusion bodies expressed by Escherichia coli. J. Biochem. 1999, 126, 905–911. [Google Scholar] [CrossRef]
- Arora, D.; Khanna, N. Method for increasing the yield of properly folded recombinant human gamma interferon from inclusion bodies. J. Biotechnol. 1996, 52, 127–133. [Google Scholar]
- Suenaga, M.; Ohmae, H.; Tsuji, S.; Itoh, T.; Nishimura, O. Renaturation of recombinant human neurotrophin-3 from inclusion bodies using a suppressor agent of aggregation. Biotechnol. Appl. Biochem. 1998, 28, 119–124. [Google Scholar]
- Rattenholl, A.; Ruoppolo, M.; Flagiello, A.; Monti, M.; Vinci, F.; Marino, G.; Lilie, H.; Schwarz, E.; Rudolph, R. Pro-sequence assisted folding and disulfide bond formation of human nerve growth factor. J. Mol. Biol. 2001, 305, 523–533. [Google Scholar] [CrossRef]
- Asano, R.; Kudo, T.; Makabe, K.; Tsumoto, K.; Kumagai, I. Antitumor activity of interleukin-21 prepared by novel refolding procedure from inclusion bodies expressed in Escherichia coli. FEBS Lett. 2002, 528, 70–76. [Google Scholar] [CrossRef]
- Bajorunaite, E.; Sereikaite, J.; Bumelis, V. L-arginine suppresses aggregation of recombinant growth hormones in refolding process from E. coli inclusion bodies. Protein J. 2007, 26, 547–555. [Google Scholar] [CrossRef]
- Takahashi, S.; Ogasawara, H.; Watanabe, T.; Kumagai, M.; Inoue, H.; Hori, K. Refolding and activation of human prorenin expressed in Escherichia coli: Application of recombinant human renin for inhibitor screening. Biosci. Biotechnol. Biochem. 2006, 70, 2913–2918. [Google Scholar] [CrossRef]
- Lechtken, A.; Zundorf, I.; Dingermann, T.; Firla, B.; Steinhiber, D. Overexpression, refolding, and purification of polyhistidine-tagged human retinoic acid related orphan receptor RORalpha4. Protein Expr. Purif. 2006, 49, 114–120. [Google Scholar] [CrossRef]
- Hsih, M.H.; Kuo, J.C.; Tsai, H.J. Optimization of the solubilization and renaturation of fish growth hormone produced by Escherichia coli. Appl. Microbiol. Biotechnol. 1997, 48, 66–72. [Google Scholar] [CrossRef]
- Wang, X.T.; Engel, P.C. An optimized system for refolding of human glucose 6-phosphate dehydrogenase. BMC Biotechnol. 2009, 9, 19–29. [Google Scholar] [CrossRef]
- Rudolph, R.; Opitz, U.; Hesse, F.; Riebland, R.; Fischer, S. Reactivation of microbially produced human tissue-type plasminogen activator. In Biotechnology International; North, K., Ed.; Century Press Ltd.: London, UK, 1992; pp. 321–325. [Google Scholar]
- Rinas, U.; Risse, B.; Jaenicke, R.; Abel, K.J.; Zettlmeissl, G. Denaturation-renaturation of the fibrin-stabilizing factor XIII a-chain isolated from human placenta: Properties of the native and reconstituted protein. J. Biol. Chem. 1990, 371, 49–56. [Google Scholar]
- Ahn, J.H.; Lee, Y.P.; Rhee, J.S. Investigation of refolding condition for Pseudomonas fluorescence lipase by response methodology. J. Biotech. 1997, 54, 151–160. [Google Scholar]
- Yancey, P.H.; Clark, M.E.; Hand, S.C.; Bowlus, R.D.; Somero, G.N. Living with water stress: Evolution of osmolyte systems. Science 1982, 217, 1214–1222. [Google Scholar]
- Ishibashi, M.; Tsumoto, K.; Ejima, D.; Arakawa, T.; Tokunaga, M. Charatcerization of arginine as a solvent additive: A halophilic enzyme as a model protein. Protein Pept. Lett. 2005, 12, 649–653. [Google Scholar] [CrossRef]
- Rudolph, R.; Fisher, S. Process for obtaining renatured proteins. US patent 4,933,434, 12 June 1990. [Google Scholar]
- Tischer, A.; Lilie, H.; Rudolph, R.; Lange, C. L-Arginine hydrochloride increases the solubility of folded and unfolded recombinant plasminogen activator rPA. Protein Sci. 2010, 19, 1783–1795. [Google Scholar] [CrossRef]
- Lange, C.; Rudolph, R. Suppression of protein aggregation by L-arginine. Curr. Pharm. Biotechnol. 2009, 10, 408–414. [Google Scholar] [CrossRef]
- Yokoyama, K.; Kunio, O.; Ohtsuka, T.; Nakamura, N.; Seguro, K.; Ejima, D. In vitro refolding process of urea-denatured microbial transgltaminase without pro-peptide sequence. Protein Expr. Purif. 2002, 26, 329–335. [Google Scholar] [CrossRef]
- Hirai, A.; Tanaka, S.; Kawasaki, H.; Nemoto, N.; Suzuki, M.; Maeda, H. Effects of arginine on aggregates of fatty-acid/potassium soap in the aqueous media. Colloid Polym. Sci. 2006, 284, 520–528. [Google Scholar] [CrossRef]
- Nozaki, Y.; Tanford, C. The solubility of amino acids and related compounds in aqueous ethylene glycol solutions. J. Biol. Chem. 1965, 240, 3568–3575. [Google Scholar]
- Nozaki, Y.; Tanford, C. The solubility of amino acids and related compounds in aqueous urea solutions. J. Biol. Chem. 1963, 238, 4074–4081. [Google Scholar]
- Nozaki, Y.; Tanford, C. The solubility of amino acids, diglycine, and triglycine in aqueous guanidine hydrochloride solutions. J. Biol. Chem. 1970, 245, 1648–1652. [Google Scholar]
- Nozaki, Y.; Tanford, C. The solubility of amino acids and two glycine peptides in aqueous ethanol and dioxane solutions. Establishment of a hydrophobicity scale. J. Biol. Chem. 1971, 246, 2211–2217. [Google Scholar]
- Gekko, K. Calorimetric study on thermal denaturation of lysozyme in polyol-water mixtures. J. Biochem. 1982, 91, 1197–1204. [Google Scholar]
- Gekko, K.; Koga, S. Increased thermal stability of collagen in the presence of sugars and polyols. J. Biochem. 1983, 94, 199–205. [Google Scholar]
- Liu, Y.; Bolen, D.W. The peptide backbone plays a dominant role in protein stabilization by naturally occurring osmolytes. Biochemistry 1995, 34, 12884–12891. [Google Scholar] [CrossRef]
- Arakawa, T.; Kita, Y.; Timasheff, S.N. Protein precipitation and denaturation by dimethyl sulfoxide. Biophys. Chem. 2007, 131, 62–70. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S.N. Mechanism of protein salting in and salting out by divalent cation salts: Balance between hydration and salt binding. Biochemistry 1984, 23, 5912–5923. [Google Scholar] [CrossRef]
- Arakawa, T.; Ejima, D.; Tsumoto, K.; Obeyama, N.; Tanaka, Y.; Kita, Y.; Timasheff, S.N. Suppression of protein interactions by arginine: A proposed mechanism of the arginine effects. Biophys. Chem. 2007, 127, 1–8. [Google Scholar] [CrossRef]
- Tanford, C. Contribution of hydrophobic interactions to the stability of the globular conformation of proteins. J. Am. Chem. Soc. 1962, 84, 4240–4247. [Google Scholar] [CrossRef]
- Dioguardi, F.S. To give or not to give. J. Nutrigenet. Nutrigenomics 2011, 4, 90–98. [Google Scholar] [CrossRef]
- Maity, H.; Karkaria, C.; Davagino, J. Mapping of solution components, pH changes, protein stability and the elimination of protein precipitation during freeze-thawing of fibroblast growth factor 20. Int. J. Pharm. 2009, 378, 122–135. [Google Scholar] [CrossRef]
- Maity, H.; Karkaria, C.; Davagnino, J. Effects of pH and arginine on the solubility and stability of a therapeutic protein (Fibroblast Growth Factor 20): Relationship between solubility and stability. Curr. Pharm. Biotechnol. 2009, 10, 609–625. [Google Scholar] [CrossRef]
- Ejima, D.; Yumioka, R.; Arakawa, T.; Tsumoto, K. Arginine as an effective additive in gel permeation chromatography. J. Chromatogr. A 2005, 1094, 49–55. [Google Scholar] [CrossRef]
- Arakawa, T.; Tsumoto, K.; Nagase, K.; Ejima, D. The effects of arginine on protein binding and elution in hydrophobic interaction and ion-exchange chromatography. Protein Expr. Purif. 2007, 54, 110–116. [Google Scholar] [CrossRef]
- Tsumoto, K.; Umetsu, M.; Kumagai, I.; Ejima, D.; Arakawa, T. Solubilization of active green fluorescent protein from inclusion particles by guanidine and arginine. Biochem. Biophys. Res. Commun. 2003, 312, 1383–1386. [Google Scholar] [CrossRef]
- Umetsu, M.; Tsumoto, K.; Nitta, S.; Adschiri, T.; Ejima, D.; Arakawa, T.; Kumagai, I. Nondenaturing solubilization of beta2 microglobulin from inclusion bodies by L-arginine. Biochem. Biophys. Res. Commun. 2005, 328, 189–197. [Google Scholar] [CrossRef]
- Yamasaki, H.; Tsujimoto, K.; Koyama, A.H.; Ejima, D.; Arakawa, T. Arginine facilitates inactivation of enveloped viruses. J. Pharm. Sci. 2008, 97, 3063–3073. [Google Scholar]
- Katsuyama, Y.; Yamasaki, H.; Tsujimoto, K.; Koyama, A.H.; Ejima, D.; Arakawa, T. Butyroyl-arginine as a potent virus inactivation agent. Int. J. Pharm. 2008, 361, 92–98. [Google Scholar] [CrossRef]
- Utsunimoya, H.; Ichinose, M.; Tsujimoto, K.; Katsuyama, Y.; Yamasaki, H.; Koyama, A.H.; Ejima, D.; Arakawa, T. Co-operative thermal inactivation of herpes simplex virus and influenza virus by arginine ans NaCl. Int. J. Pharm. 2009, 366, 99–102. [Google Scholar] [CrossRef]
- Arakawa, T.; Kita, Y.; Koyama, A.H. Synergistic virus inactivation effects of arginine. Biotechnol. J. 2009, 4, 174–178. [Google Scholar]
- Tsujimoto, K.; Uozaki, M.; Ikeda, K.; Yamazaki, H.; Utsunomiya, H.; Ichinose, M.; Koyama, A.H.; Arakawa, T. Solvent-induced virus inactivation by acidic arginine solution. Int. J. Mol. Med. 2010, 25, 433–437. [Google Scholar]
- Naito, T.; Irie, H.; Tsujimoto, K.; Ikeda, K.; Arakawa, T.; Koyama, A.H. Antiviral effect of arginine against herpes simplex virus type 1. Int. J. Mol. Med. 2009, 23, 495–499. [Google Scholar]
- Ohtake, S.; Arakawa, T.; Koyama, A.H. Arginine as a synergistic agent. Molecules 2010, 15, 1408–424. [Google Scholar] [CrossRef]
- Nishide, M.; Tsujimoto, K.; Uozaki, M.; Ikeda, K.; Yamasaki, H.; Koyama, A.H.; Arakawa, T. Effects of electrolytes on virus inactivation by acidic solutions. Int. J. Mol. Med. 2011, 27, 803–809. [Google Scholar]
- Ikeda, K.; Yamasaki, H.; Minami, S.; Naito, T.; Irie, H.; Arakawa, T.; Koyama, A.H. Virucidal ability of arginine and its possible application as an antiherpetic agent. In From the Hallowed Halls of Herpesvirology; Baines, J., Blaho, J., Eds.; Imperial College Press: London, UK, 2012; Chapter 18, pp. 435–450. [Google Scholar]
- Hirano, A.; Kameda, T.; Arakawa, T.; Shiraki, K. Arginine-assisted solubilization system for drug substances: Solubility experiment and simulation. J. Phys. Chem. B 2010, 114, 13455–13462. [Google Scholar] [CrossRef]
- Arakawa, T.; Kita, Y.; Koyama, A.H. Solubility enhancement of gluten and coumarin by arginine. Int. J. Pharm. 2008, 355, 220–223. [Google Scholar] [CrossRef]
- Hirano, A.; Arakawa, T.; Shiraki, K. Arginine increases the solubility of coumarin: Comparison with salting-in and salting-out additives. J. Biochem. 2008, 144, 363–369. [Google Scholar] [CrossRef]
- Arakawa, T.; Uozaki, M.; Koyama, A.H. Modulation of small molecule solubility and protein binding by arginine. Mol. Med. Rep. 2010, 3, 833–836. [Google Scholar]
- Gekko, K.; Morikawa, T. Preferential hydration of bovine serum albumin in polyhydric alcohol-water mixtures. J. Biochem. 1981, 90, 39–50. [Google Scholar]
- Lee, J.C.; Timasheff, S.N. The stabilization of proteins by sucrose. J. Biol. Chem. 1981, 256, 7193–7201. [Google Scholar]
- Gekko, K.; Timasehff, S.N. Mechanism of protein stabilization by glycerol: Preferential hydration in glycerol-water mixtures. Biochemistry 1981, 20, 4667–4676. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S.N. Stabilization of protein structure by sugars. Biochemistry 1982, 21, 6536–6544. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S.N. Preferential interactions of proteins with salts in concentrated solutions. Biochemistry 1982, 21, 6545–6552. [Google Scholar] [CrossRef]
- Kita, Y.; Arakawa, T.; Lin, T.Y.; Timasheff, S.N. Contribution of the surface free energy perturbation to protein-solvent interactions. Biochemistry 1994, 33, 15178–15189. [Google Scholar] [CrossRef]
- Lee, J.C.; Timasheff, S.N. Partial specific volumes and interactions with solvent components in guanidine hydrochloride. Biochemistry 1974, 13, 257–265. [Google Scholar]
- Prakash, V.; Loucheux, C.; Scheufele, S.; Borbunoff, M.J.; Timasheff, S.N. Interaction of proteins with solvent components in 8 M urea. Arch. Biochem. Biophys. 1981, 210, 455–464. [Google Scholar] [CrossRef]
- Hong, J.; Capp, M.W.; Anderson, C.F.; Record, M.T. Preferential interactions in aqueous solution of urea and KCl. Biophys. Chem. 2003, 105, 517–532. [Google Scholar] [CrossRef]
- Timasheff, S.N.; Xie, G. Preferential interactions of urea with lysozyme and their linkage to protein denaturation. Biophys. Chem. 2003, 105, 421–448. [Google Scholar] [CrossRef]
- Bull, H.B.; Breese, K. Interaction of alcohols with proteins. Biopolymers 1978, 17, 2121–2131. [Google Scholar] [CrossRef]
- Inoue, H.; Timasheff, S.N. The interaction of beta-lactoglobulin with solvent components in mixed water-organic solvent systems. J. Am. Chem. Soc. 1968, 90, 1890–1898. [Google Scholar] [CrossRef]
- Timasheff, S.N.; Inoue, H. Preferential binding of solvent components to proteins in mixed water-organic solvent systems. Biochemistry 1968, 7, 2501–2513. [Google Scholar] [CrossRef]
- Inoue, H.; Timasheff, S.N. Preferential and absolute interactions of solvent components with proteins in mixed solvent systems. Biopolymers 1972, 11, 737–743. [Google Scholar] [CrossRef]
- Pittz, E.P.; Timasheff, S.N. Interaction of ribonuclease A with aqueous 2-methyl-2,4-pentanediol at pH 5.8. Biochemistry 1978, 17, 615–623. [Google Scholar] [CrossRef]
- Arakawa, T.; Bhat, R.; Timasheff, S.N. Why preferential hydration does not always stabilize the native structure of globular proteins. Biochemistry 1990, 29, 1924–1931. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S.N. Preferential interactions of proteins with solvent components in aqueous amino acid solutions. Arch. Biochem. Biophys. 1983, 224, 169–177. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S.N. The mechanism of action of Na glutamate, lysine HCl, and piperazine-N,N′-bis(2-ethanesulfonic acid) in the stabilization of rubulin and microtuble formation. J. Biol. Chem. 1984, 259, 4979–4986. [Google Scholar]
- Arakawa, T.; Timasheff, S.N. The stabilization of proteins by osmolytes. Biophys. J. 1985, 47, 411–414. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S.N. Protein stabilization and destabilization by guanidinium salts. Biochemistry 1984, 23, 5923–5929. [Google Scholar]
- Arakawa, T.; Timasheff, S.N. Theory of protein solubility. Methods Enzymol. 1985, 114, 49–77. [Google Scholar]
- Arakawa, T.; Timasheff, S.N. Abnormal solubility behavior of beta-lactoglobulin: Salting-in by glycine and NaCl. Biochemistry 1987, 26, 5147–5153. [Google Scholar] [CrossRef]
- Arakawa, T.; Bhat, R.; Timasheff, S.N. Preferential interactions determine protein solubility in three-component solutions: MgCl2 system. Biochemistry 1990, 29, 1914–1923. [Google Scholar] [CrossRef]
- Schneider, C.P.; Trout, B.L. Investigation of cosolute-protein preferential interaction coefficients: New insight into the mechanism by which arginine inhibits aggregation. J. Phys. Chem. 2009, 113, 2050–2058. [Google Scholar] [CrossRef]
- Ito, L.; Shiraki, K.; Matsuura, T.; Okumura, M.; Hasegawa, K.; Baba, S.; Yamaguchi, H.; Kumasaka, T. High-resolution X-ray analysis reveals binding of arginine to aromatic residues of lysozyme surface: Implication of suppression of protein aggregation by arginine. Protein Eng. Des. Sel. 2011, 24, 269–274. [Google Scholar] [CrossRef]
- Shukla, D.; Trout, B. Interaction of arginine with proteins and the mechanism by which it inhibits aggregation. J. Phys. Chem. B 2010, 114, 13426–13438. [Google Scholar] [CrossRef]
- Woods, A.S. The mighty arginine, the stable quaternary amines, the powerful aromatics, and the aggressive phosphate: Their role in the noncovalent minuet. J. Proteome Res. 2004, 3, 478–484. [Google Scholar] [CrossRef]
- Flocco, M.M.; Mowbray, S.L. Planar stacking interactions of arginine and aromatic side-chains in proteins. J. Mol. Biol. 1994, 235, 709–717. [Google Scholar] [CrossRef]
- Gallivan, J.P.; Dougherty, D.A. Cation-pi interactions in structural biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar] [CrossRef]
- Crowley, P.B.; Golovin, A. Cation-pi interactions in protein-protein interfaces. Proteins 2005, 59, 231–239. [Google Scholar] [CrossRef]
- Dougherty, D.A. Cation-pi interactions involving aromatic amino acids. J. Nutr. 2007, 137, 1504S–1508S. [Google Scholar]
- Nakakido, M.; Tanaka, Y.; Mitsuhori, M.; Kudou, M.; Ejima, D.; Arakawa, T.; Tsumoto, K. Structure-based analysis reveals hydration changes induced by arginine hydrochloride. Biphys. Chem. 2008, 137, 105–109. [Google Scholar] [CrossRef]
- Baynes, B.M.; Wang, D.I.; Trout, B.L. Role of arginine in the stabilization of proteins against aggregation. Biochemistry 2005, 44, 4919–4925. [Google Scholar]
- Arakawa, T.; Tsumoto, K. The effects of arginine on refolding aggregated proteins: Not facilitate refolding, but suppress aggregation. Biochem. Biophys. Res. Commun. 2003, 304, 148–152. [Google Scholar] [CrossRef]
- Shiraki, K.; Kudou, M.; Fujiwara, S.; Imanaka, T.; Takagi, M. Biophysical effect of amino acids on the prevention of protein aggregation. J. Biochem. 2002, 132, 591–595. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arakawa, T.; Kita, Y.; Ejima, D. Refolding Technology for scFv Using a New Detergent, N-Lauroyl-L-glutamate and Arginine. Antibodies 2012, 1, 215-238. https://doi.org/10.3390/antib1020215
Arakawa T, Kita Y, Ejima D. Refolding Technology for scFv Using a New Detergent, N-Lauroyl-L-glutamate and Arginine. Antibodies. 2012; 1(2):215-238. https://doi.org/10.3390/antib1020215
Chicago/Turabian StyleArakawa, Tsutomu, Yoshiko Kita, and Daisuke Ejima. 2012. "Refolding Technology for scFv Using a New Detergent, N-Lauroyl-L-glutamate and Arginine" Antibodies 1, no. 2: 215-238. https://doi.org/10.3390/antib1020215
APA StyleArakawa, T., Kita, Y., & Ejima, D. (2012). Refolding Technology for scFv Using a New Detergent, N-Lauroyl-L-glutamate and Arginine. Antibodies, 1(2), 215-238. https://doi.org/10.3390/antib1020215