Abstract
Peroxynitrite (ONOOH/ONOO−) is increasingly recognized as a key intermediate in advanced oxidation processes (AOPs), yet its role in water treatment remains insufficiently defined. This study provides mechanistic insights into peroxynitrite-mediated AOPs through competition kinetics method, demonstrating that both decomposition and pollutant degradation are strongly pH-dependent, with ONOOH dominating stability and radical pathways across pH 5.0−9.0, while its decay rate decreases from 1.2 s−1 to 0.0022 s−1. The interplay of HO• and diverse reactive nitrogen species (RNS, including reactive nitrogen radicals and peroxynitrite) dictates pollutant-specific degradation efficiencies, with RNS showing a unique reliance in degrading bisphenol A—contributing up to 66.7% at pH 8.0. Buffer chemistry further modulates these pathways: bicarbonate accelerates peroxynitrite decay via CO2 and CO3•−-mediated acceleration (resulting in a 361.9% increase at pH 9.0), while borate promotes reactive nitrogen radical formation but suppresses HO• contributions. Importantly, peroxynitrite was shown to facilitate N-nitrosodimethylamine formation in the presence of dimethylamine, with yields maximized under alkaline conditions and attenuated by bicarbonate. These quantitative findings underscore the critical roles of pH and buffer chemistry in optimizing peroxynitrite-based water treatment while mitigating byproduct risks.
1. Introduction
The widespread presence of endocrine disrupting chemicals (EDCs) in aquatic environments has become a major global concern, particularly due to their persistence, potential for bioaccumulation, and hazards to ecosystems and human health [1]. Among various EDCs, bisphenol A (BPA)—a high-production chemical used in plastics and epoxy resins—exhibits potent estrogenic activity that disrupts hormonal systems. This leads to reproductive disorders, developmental abnormalities, and increased cancer risks in wildlife and humans, even at trace levels [2,3]. Advanced oxidation processes (AOPs) demonstrate superior efficacy in removing BPA compared to conventional water treatment methods, achieving its efficient degradation [4].
In this context, AOPs involving reactive nitrogen species (RNS) have attracted growing attention due to their unique potential to selectively degrade electron-rich phenolic contaminants like BPA [5,6,7,8]. RNS, such as •NO2, •NO, •NH2 and ONOOH/ONOO−, can be generated in situ via ultraviolet (UV) photolysis of nitrites, nitrates and monochloramine, as well as during breakpoint chlorination process and PAA/NO2− reactions. Increasing evidence (e.g., kinetic analysis and nitrogenous oxidized byproducts detection) suggests that these species play important roles in the transformation of electron-rich micropollutants, such as anilines, amines, and phenols [5,7,8,9]. Consequently, RNS-based AOPs are emerging as promising tools for the removal of contaminants of concern in aquatic environments [6,10,11].
Among various RNS, the reactivity and transformation pathways of peroxynitrite (ONOOH/ONOO−) remain relatively understudied in the context of water treatment [9]. Peroxynitrite exists in an acid-base equilibrium (pKa ≈ 6.8) between ONOOH and its conjugate base ONOO−. Since its first identification in biological systems in the early 1990s, peroxynitrite has been recognized as a strong oxidant and a potentially cytotoxic species [12]. ONOOH, the protonated form, is a potent two-electron electrophile with standard redox potentials of E0[(ONOOH, 2H+)/HNO2] = 1.68 V and E0[(ONOOH, 2H+)/•NO2] = 2.14 V, and it also exhibits one-electron oxidizing behavior with significantly higher redox potentials compared to common peroxides [13,14]. Its high reactivity toward electron-rich organic compounds, specifically phenols and aromatic amines which are model contaminants representing large classes of problematic EOCs, has been confirmed via kinetic studies and Hammett analysis [15,16,17]. Critically, peroxynitrite is also recognized as a key intermediate in several AOPs, including breakpoint chlorination, UV/NO2−, UV/NO3−, and CH3C(O)OOH/NO2− systems [5,18,19,20]. In the breakpoint chlorination process, commonly used for complete ammonia removal in drinking water, peroxynitrite has been identified as a primary reactive species [5]. Upon decomposition, peroxynitrite generates a suite of oxidizing radicals, including HO•, •NO2, •NO, and O2•−, which have been detected using CIDNP, electron spin resonance spectrum, and probe-based methods [12,21,22,23]. These radicals are believed to drive the oxidative transformation of contaminants, but their formation pathways and contributions remain inadequately characterized.
The decomposition of peroxynitrite is strongly pH-dependent. ONOOH decomposes rapidly under acidic conditions (k = 0.8–1.4 s−1), whereas ONOO− is more stable at high pH (k ≈ 1.1 × 105 s−1 at pH 14.0) [23,24]. The product distribution also varies with pH: nitrate dominates under acidic and highly alkaline conditions, while nitrite formation peaks near neutral to slightly alkaline pH. Oxygen yields increase markedly with pH, highlighting pathway shifts that are not yet fully understood [23,24,25,26]. Proposed mechanisms include both radical and isomerization pathways, yet their relative importance and environmental relevance remain unclear.
Moreover, in natural and engineered water systems, peroxynitrite can react rapidly with dissolved CO2 to form a labile ONOOCO2− intermediate (k = 3−5.8 × 104 M−1 s−1) [27]. This intermediate decomposes to either nitrate and CO2 or radical species (•NO2 and CO3•−), depending on reaction conditions [23]. Its biological importance has been recognized since 1993 [28,29]. Although bicarbonate is typically abundant in surface waters (61−183 mg/L) and wastewater effluents (38−575 mg/L) are typically high [30], its role in governing peroxynitrite decomposition pathways, radical formation, and pollutant transformation under environmentally relevant conditions remains poorly understood.
In addition to pollutant degradation, recent studies have reported the formation of nitrogenous disinfection byproducts (N-DBPs), particularly N-nitrosodimethylamine (NDMA), in peroxynitrite-involved systems. NDMA formation has been observed during breakpoint chlorination, implicating peroxynitrite as a potential precursor or facilitator. However, the mechanisms underlying NDMA generation in the presence of peroxynitrite are not fully elucidated.
Therefore, the innovation of this study lies in: (1) further elucidating the pH-dependent decomposition kinetics of peroxynitrite under environmentally relevant conditions; (2) evaluating its reactivity toward representative micropollutants and isolating the contribution of ONOOH from RNS; (3) conducting the first comprehensive evaluation of the effects of buffering systems (e.g., phosphate and borate) on radical generation and pollutant degradation; and (4) evaluating the relationship between the potential for NDMA formation and pH in peroxynitrite-related systems. These findings are expected to deepen the understanding of the mechanisms underlying peroxynitrite chemistry and support the safe and effective application of RNS-based AOPs in water treatment.
2. Materials and Methods
2.1. Materials
Reagents used in this study: Sodium nitrite (NaNO2), Hydrogen peroxide (H2O2, 30%), sodium dihydrogen phosphate (NaH2PO4), sodium phosphate dibasic (Na2HPO4), sodium bicarbonate (NaHCO3), sodium hydroxide (NaOH), Sodium thiosulfate (Na2S2O3), perchloric acid (HClO4), hydrochloric acid (HCl, 37%), tert-butyl alcohol (TBA), boric acid (H3BO3), sodium tetraborate decahydrate (Na2B4O7) and acid (BA) were purchased from Sinopharm Chemical Reagent Co., Ltd., (Beijing, China). N,N-diethyl-3-toluamide (DEET, 99%), carbamazepine (CBZ, 99%), Bisphenol A (BPA, 99%), Nitrobenzene (NB, 99%) and p-Chlorophenol (4-CP, 99%) were purchased from Macklin Biochemical Co., Ltd., (Shanghai, China). N-Nitrosodimethylamine (NDMA) were obtained from TCI Chemicals Co., Ltd., (Shanghai, China). High performance liquid chromatography (HPLC)-grade methanol and acetonitrile from TEDIA Co., Ltd., (Anqing, China). High performance liquid chromatography (HPLC)-grade formic acid (HCOOH) and N-Nitrosodimethylamine-d6 (NDMA-d6) were obtained from Aladdin Industrial Co., Ltd., Shanghai, China. The ultrapure water prepared by the UPR-II (18.2 MΩ·cm, ULUPURE, Chengdu, China). All the chemicals are of analytical grade and used as received unless otherwise noted. All these pollutant stock solutions were prepared with ultrapure water and stored at 4 °C in dark place.
The synthesis of peroxynitrite was performed following the procedure reported by Beckman et al. [31], with critical modifications to address ion interference. Specifically, 50 mL of 50 mM H2O2 and 50 mL of 50 mM NaNO2 were thoroughly mixed under an ice-water bath until the system temperature stabilized at 4 °C. Subsequently, 25 mL of prechilled 1 M HClO4 (stored at 4 °C) was rapidly injected, followed by immediate addition of 25 mL of 1.5 M NaOH (stored at 4 °C). Maintaining refrigerated pretreatment reagent solutions is essential to minimize temperature-induced reaction fluctuations. It should be emphasized that, contrary to Beckman’s hydrochloric acid-mediated protocol, HClO4 solution was adopted in this study to circumvent potential Cl− interference while maintaining the required acidic environment. Upon HClO4 addition, an immediate chromogenic transition from colorless to pale yellow indicated substantial peroxynitrite formation. The addition of NaOH aims to promptly establish an alkaline environment (pH = 13.0), thereby suppressing the decomposition of synthesized peroxynitrite. Using the synthesis method described above typically yields peroxynitrite at a concentration of 9–11 mM. Quantification of peroxynitrite was achieved through UV-Vis measurements at 302 nm (ε302 = 1670 M−1 cm−1) using 1.5 M NaOH as diluent. Due to the inherent instability of peroxynitrite, the working solution was always freshly prepared for each experiment, stored at 4 °C, and used within 3 h to prevent experimental interference caused by its decomposition.
2.2. Experimental Procedures
The pH-dependent decay kinetics of peroxynitrite were investigated using a stopped-flow spectrometer (SFS, Model SX20, Applied Photophysics Ltd., Leatherhead, UK). Prior to each experiment, the peroxynitrite stock solution was diluted with 0.2 M NaOH to the desired concentration. An acidic working solution was prepared separately by combining perchloric acid with phosphate buffer to achieve the desired final pH upon mixing. The reaction was initiated by simultaneously injecting equal volumes of the alkaline and acidic solutions into the optical cell of the stopped-flow spectrometer using two auto-injectors powered by compressed nitrogen. The concentration of peroxynitrite was continuously monitored at 302 nm (ε302 = 1670 M−1 cm−1).
Contaminant degradation experiments were conducted in 250 mL conical flasks with continuous stirring using a magnetic stirrer at 300 rpm. Each reaction was initiated by adding 2 mg/L as N of peroxynitrite stock solution to 100 mL of working solution containing the target organic contaminant, buffer, and other coexisting substances as required. At designated time points, 1.0 mL of the reaction mixture was withdrawn and immediately quenched in a sample vial containing 20 μL of 0.5 M sodium thiosulfate (Na2S2O3) solution. Residual contaminant concentrations were subsequently analyzed by high-performance liquid chromatography (HPLC). For radical quenching experiments, 100 mM tert-butyl alcohol (TBA) was added to the working solution prior to initiating the reaction. NDMA formation experiments were carried out following the same protocol as contaminant degradation tests, with dimethylamine (DMA) spiked into the working solution in advance. At the end of the reaction, 1.0 mL of 1 M ascorbic acid was added directly to the conical flask to terminate residual oxidants. A 100 mL aliquot of the quenched solution was then transferred to a 150 mL polytetrafluoroethylene-lined screw-cap bottle for further extraction and analysis. Unless otherwise specified, all experiments were conducted in ultrapure water at ambient temperature (20.0 ± 2 °C) and repeated at least twice. The results are reported as the average values with corresponding standard deviations.
2.3. Analytical Methods
2.3.1. HPLC Analysis of Organic Pollutants
The concentrations of organic compounds were determined using an HPLC system (HPLC, Thermo Ultimate 3000) with UV detection. Chromatographic separations were performed using a C18 column (4.6 × 250 mm, 5 μm; Thermo, Waltham, MA, USA) in an isocratic mode of elution at 35 ± 0.5 °C. The injection volume of a sample was 10 μL. NB, CBZ, DMOB and DEET were detected by using methanol and water formate as mobile phases in the ratio of 70:30 at a flow rate of 1.0 mL/min at a detection wavelength of 265 nm. 4-CP was detected by using acetonitrile and water formate as mobile phases in the ratio of 60:40 at a flow rate of 1.0 mL/min at a detection wavelength of 265 nm. The detection wavelength was 265 nm. The pH of the reaction solution was monitored with Shanghai Leici pH meter (Leici Co., Ltd., Shanghai, China).
2.3.2. GC-MS Analysis of NDMA
A volume of 100 mL of the solution was transferred into a 150 mL sample vial, followed by the addition of deuterated NDMA (NDMA-d6) as an internal standard and thorough mixing. Subsequently, 20 mL of dichloromethane (DCM) was added to the vial, immediately followed by the addition of 10 g anhydrous sodium sulfate. The mixture was vigorously shaken until complete dissolution of sodium sulfate. The sample was then vortex-mixed for 5 min and allowed to phase-separate for 30 min. After separation, 15 mL of the organic layer was collected and concentrated to 1 mL using a nitrogen evaporator under a gentle nitrogen stream. Prior to analysis, samples were transferred to vials and analyzed using gas chromatography-mass spectrometry (GC-MS, GCMS-QP2010, Shimadzu, Kyoto, Japan) in single-injection mode for the identification and quantification of NDMA and NDMA-d6 in blanks, standards, and samples. Quantification was achieved through six-point calibration curves (5–200 μg L−1 for both NDMA and NDMA-d6), which exhibited correlation coefficients (R2) exceeding 0.999. The GC-MS system was operated with an inlet temperature of 250 °C and a DB-Wax capillary column (30 m × 0.25 mm × 0.25 μm). The temperature program Initiated at 50 °C (8 min hold), followed by ramping at 8 °C min−1 to 170 °C, then at 15 °C min−1 to 250 °C (1 min hold). MS detection employed selected ion monitoring (SIM) with characteristic m/z ratios: 74 (quantitative) and 42 (qualitative) for NDMA, and 80 (quantitative) with 46 (qualitative) for NDMA-d6.
2.3.3. UPLC-QTOF-MS Analysis of Products
Samples were analyzed by ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry (UPLC-QTOF-MS, 1290 Infinity II, Agilent, Santa Clara, CA, USA). The column was a Waters BEH C18 (150 mm × 3.0 mm, 1.7 μm) maintained at 30 °C, with an injection volume of 20 μL. Mobile phase A was 0.1% formic acid in water, and B was acetonitrile, with a flow rate of 0.3 mL min−1. The gradient started at 95% A for 2 min, linearly decreased to 0% A over 3 min and held for 3 min, then rapidly recovered to 95% A within 0.05 min, followed by a 2.95 min re-equilibration period. Mass spectrometry scanning range: m/z 50–1100.
2.3.4. Calculation of the Exposure of HO•, CO3•− and ONOO−
NB, 4-CP were chosen as probe compounds, while their rate constants with HO• and CO3•− were shown in Table S1.
- (1)
- Calculation of the exposure of HO•.
Equation (1) are rearranged and integrated to yield the following expression:
Rearranging Equation (2) gives the exposure of HO•:
- (2)
- Calculation of the exposure of CO3•−:
Equation (4) are rearranged and integrated to yield the following expression:
Rearranging Equation (5) gives the CO3•− exposure:
- (3)
- Calculation of the exposure of ONOO−:
Consequently,
2.3.5. Relative Contributions of HO•, CO3•− and Reactive Nitrogen Radicals to Micropollutant Degradation
RNS include ONOOH and reactive nitrogen radicals.
- (1)
- The removal of HO• to micropollutant degradation can be estimated by:
- (2)
- The removal of CO3•− to micropollutant degradation can be estimated by:
- (3)
- Calculation of the removal of reactive nitrogen radicals to micropollutant degradation:
2.3.6. Influence of BPA on Peroxynitrite Decay
Peroxynitrite decay was monitored by measuring absorbance at 302 nm at pH 9.0. Since the absorption spectra of BPA and peroxynitrite overlap at this wavelength, samples were withdrawn at fixed time intervals and immediately quenched with sodium thiosulfate. The absorbance attributable to peroxynitrite was determined from the difference between the values before and after quenching.
3. Results and Discussion
3.1. Decomposition of Peroxynitrite at Different pH
Figure S1 presents the decay kinetics of peroxynitrite across different pH values, with phosphate buffer used in all reactions. The decomposition rate decreased monotonically as pH increased from 5.0 to 9.0. Due to the relatively slow reaction rate at pH 9.0, the potential influence of phosphate on peroxynitrite decomposition was specifically examined at this pH. In control experiments without phosphate, pH was maintained by adding HClO4 or NaOH as needed. As shown in Figure S2a, phosphate had a negligible effect on peroxynitrite decay at pH 9.0, and was therefore considered to have minimal influence across the pH range 5.0–9.0.
Peroxynitrite decomposition proceeds via two primary pathways, depending on its protonation state [23]: (1) ONOOH undergoes O–O bond homolysis, producing hydroxyl (HO•) and nitrogen dioxide radicals (•NO2) (Equation (14)). (2) ONOOH reacts bimolecularly with ONOO− to yield nitrite and dioxygen in a 2:1 stoichiometric ratio (Equation (15)).
ONOOH ⇌ [HO• + •NO2]cage → HO• + •NO2
ONOO− + ONOOH → H+ + 2NO2− + O2
The pKa of ONOOH is ~6.8, and ONOO− is thermally more stable than ONOOH. As shown in Figure 1a–c, the decay rate of peroxynitrite at low pH is substantially higher than at pH 8.0−9.0, confirming the relative inertness of ONOO−. In Figure 1a,b, the decay follows pseudo-first-order kinetics at pH 5.0–8.0, indicating that Equation (14) dominates under these conditions. Thus, the decay kinetics of peroxynitrite can be expressed as:
where [peroxynitrite] = [ONOOH] + [ONOO−].
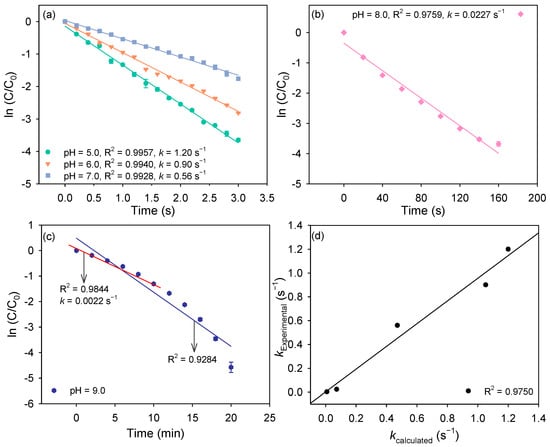
Figure 1.
(a–c) Decomposition kinetics of peroxynitrite at different pH. (d) The relationship between kcalculated and kExperimental. Experimental conditions: [peroxynitrite]0 = 2.0 mg/L as N, [phosphate buffer] = 20 mM.
Rearranging and integrating Equation (16) yields:
At pH 5.0, ONOOH is the predominant species, and approximates the observed rate constant (k) which was determined to be 1.20 s−1. The pseudo-first-order rate constants (k) of peroxynitrite decomposition at pH 6.0−8.0 were then calculated. As shown in Figure 1d, there is excellent agreement between calculated and experimentally observed rate constants, with a slope of 0.95, confirming that ONOOH decomposition is the main decay pathway in the pH range of 5.0−8.0.
At pH 9.0, peroxynitrite decomposes much more slowly, but an auto-accelerating trend was observed. This behavior suggests involvement of reactive intermediates, including HO• and reactive nitrogen radicals, which may promote further peroxynitrite decomposition (Table S2). To probe this, excess TBA, a HO• scavenger, was added [32]. As shown in Figure S2b, TBA had negligible effect, ruling out HO• as the cause of accelerated decay. Phenol, a scavenger of reactive nitrogen radicals [7,33], was then employed. As shown in Figure S2c, the auto-acceleration disappeared, peroxynitrite decomposition reverted to pseudo-first-order kinetics, confirming that reactive nitrogen radicals drive the autocatalytic process. Kinetic fitting of the first 10 min in the absence of phenol yielded a pseudo-first-order rate constant of 0.0022 s−1, consistent with the calculated value (Figure 1d). These results demonstrate that ONOOH decomposition remains the dominant pathway at pH 9.0, with deviations attributable to the accumulation of reactive nitrogen radicals and their subsequent reactions with peroxynitrite.
3.2. Roles of HO• and RNS in Micropollutant Degradation
To comparatively evaluate the reactivity of peroxynitrite toward different pollutant categories, bisphenol A (BPA, a phenolic pollutant), carbamazepine (CBZ, a nitrogen-containing heterocyclic pollutant), and N,N-diethyl-m-toluamide (DEET, a polar repellent containing an aliphatic amine group) were selected as target pollutants. Figure 2 shows that micropollutant removal occurred concurrently with peroxynitrite decomposition, implying the participation of reactive species in this process. Table S2 summarizes the reactions involved in peroxynitrite decomposition, indicating that HO• and reactive nitrogen species (RNS, including both nitrogen radicals and peroxynitrite itself) are the primary contributors to micropollutant abatement.
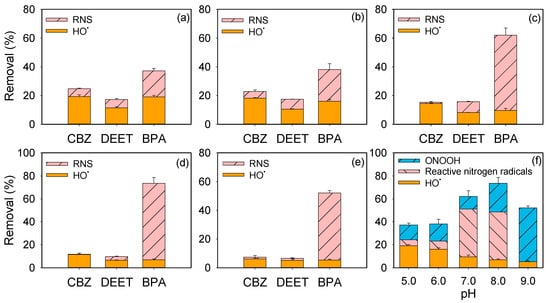
Figure 2.
Contributions of different reactive species to micropollutant removal: (a–d) within 5 min at pH 5.0–8.0; (e) within 10 min at pH 9.0; (f) contributions of reactive species to BPA degradation at various pH. Experimental conditions: [phosphate buffer] = 20 mM, [micropollutant]0 = 5 μM, [peroxynitrite]0 = 2 mg/L as N.
Role of HO•. NB was used as a probe compound since its degradation can only be attributed to HO• attack in peroxynitrite solution (Equation (18)) [34,35]:
From the experimentally measured removal efficiency and the literature-reported second-order rate constant of HO• toward NB, the time-integrated HO• exposure () was calculated at different pH values (Figure 3). Peroxynitrite decomposition was relatively slow at pH 9.0, so HO• exposure was tracked for 10 min, whereas 5 min was sufficient at pH 5.0–8.0. Figure 3 shows that HO• exposure decreased with increasing pH. Since ONOOH decomposition (Equation (14)) is the primary HO• source, and the initial peroxynitrite concentration was constant across all tests, the decline in HO• exposure at higher pH is likely due to the increased scavenging effect. This is consistent with the shift from ONOOH to ONOO− as pH rises, which accelerates HO• consumption through the following reactions [36,37,38]:
ONOO− + HO• → O2 + •NO + HO− k = 4.80 × 109 M−1 s−1
ONOOH + HO• → O2 + •NO + H2O k = 2.00 × 107 M−1 s−1
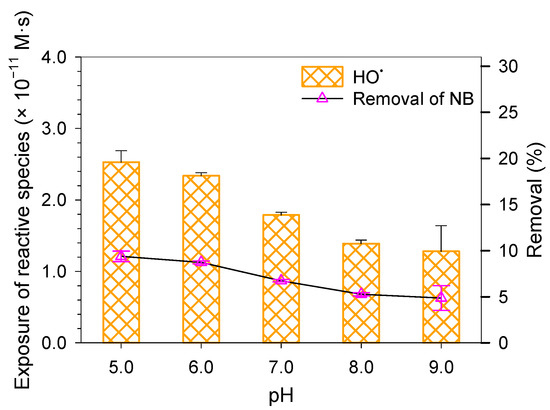
Figure 3.
The exposure of HO• (left Y-axis) and removal of NB (right Y-axis) at pH 5.0–8.0 (5 min) and pH 9.0 (10 min). Experimental conditions: [phosphate buffer] = 20 mM, [micropollutant]0 = 5 μM, [peroxynitrite]0 = 2 mg/L as N.
Using the determined HO• concentrations, its contribution to pollutant degradation was calculated (Figure 2). HO• was found to be the dominant oxidant for CBZ and DEET.
Role of RNS. The contribution of RNS, encompassing the combined effects of reactive nitrogen radicals and peroxynitrite, is calculated by subtracting the contribution of HO•. Figure 2 shows that RNS made only minor contributions to CBZ and DEET degradation at pH 5.0–7.0, and this contribution decreased further at pH 8.0–9.0. This trend is attributed to the reaction of reactive nitrogen radicals with ONOO−, which reduces concentrations as peroxynitrite shifts from ONOOH to ONOO−. Interestingly, the RNS contribution differed between the two pollutants: for CBZ it decreased from pH 5.0 to 7.0, whereas for DEET it increased, suggesting that different reactive nitrogen radicals with distinct reactivities toward CBZ and DEET were involved.
In contrast, RNS played a dominant role in BPA degradation. Their contribution increased from pH 5.0 to 8.0 and then declined at pH 9.0, with a maximum (~66.7%) at pH 8.0. Figure S3 shows that BPA strongly accelerated peroxynitrite decay, indicating the high reactivity of peroxynitrite towards BPA. In the presence of BPA, peroxynitrite kinetics can be described by:
At pH 11.0, BPA had negligible impact (Figure S4), confirming that ONOO− is unreactive toward BPA and that ONOOH is the reactive species. Accordingly, Equation (21) was revised to:
where is the fraction of [ONOOH] in total peroxynitrite. Rearrangement of Equation (22) gives:
where k is the pseudo-first-order rate constant of peroxynitrite self-decomposition. represents the apparent rate constant in the presence of excess BPA, which was experimentally measured to be 9.6 × 10−3 s−1. The second-order rate constant for the reaction between BPA and ONOOH was thus calculated as 1.18 × 103 M−1 s−1. Using this value and peroxynitrite decay kinetics at different pH, the contribution of peroxynitrite to BPA degradation was estimated with:
Rearranging and integrating Equation (24) yield:
Given the second-order rate constant and dosed BPA concentration (5 μM), peroxynitrite consumption by BPA was negligible at pH 5.0–9.0. At pH 9.0, peroxynitrite decay appeared nearly linear with time (Figure S5) and was fitted as:
With k = 0.00122 s−1. Substituting Equation (26) into Equation (25) gives:
Consequently,
With the reaction of 10 min, ~46.8% of BPA was removed by ONOOH at pH 9.0, with negligible contributions from other RNS. This suppression likely resulted from substantial consumption of reactive nitrogen radicals by ONOO− at high pH.
As discussed in part 3.1, peroxynitrite decay at pH 5.0−8.0 followed pseudo-first-order kinetics, described by Equation (29):
Substituting Equation (29) into Equation (25) gives:
Consequently,
Based on this model, the contributions of peroxynitrite to BPA degradation were estimated as 12.9%, 14.9%, 11.0%, and 25.0% at pH 5.0, 6.0, 7.0, and 8.0, respectively (Figure 2f). The contribution at pH 8.0–9.0 was markedly higher than at pH 5.0–7.0. This difference arises because, although [ONOOH] is greater at lower pH, its rapid decomposition limits its effective availability. In contrast, the slower decomposition of ONOOH at pH 8.0–9.0 prolongs its contact with BPA, resulting in a substantially larger direct contribution to degradation.
After subtracting the contributions of peroxynitrite and HO•, the remaining portion was attributed to other reactive nitrogen radicals. As shown in Figure 2f, the contribution of reactive nitrogen radicals to BPA degradation was minor at pH 5.0−6.0 but increased sharply to 41.5% and 41.7% at pH 7.0 and 8.0, respectively. These reactive nitrogen radicals are likely produced from the reaction of primary radicals (e.g., HO• and •NO2) with ONOO− (Equations (19) and (32)). At 5.0−6.0, peroxynitrite predominantly exists as ONOOH, limiting the formation of reactive nitrogen radicals. As pH increased to 7.0−8.0, the shift toward ONOO− enhanced the generation of reactive nitrogen radicals. However, at pH 9.0, the strong quenching effect of ONOO− on the reactive nitrogen radicals outweighed their formation, thereby suppressing their overall contribution to BPA degradation (Equation (32) source: ref [36]).
ONOO− + •NO2 → NO2− + •NO + O2 k = 2.5 × 104 M−1 s−1
Overall, in advanced oxidation processes utilizing peroxynitrite as a reaction intermediate, water bodies with a pH of 5.0–6.0 rely on hydroxyl radical-dominated hydroxylation reactions, while those with a pH of 7.0–9.0 are more conducive to pollutant degradation through electrophilic reactions dominated by ONOOH and reactive nitrogen species.
3.3. Impact of HCO3− on Peroxynitrite-Mediated Pollutant Degradation
Figure 4 demonstrates that 1 mM HCO3− significantly enhanced BPA degradation by peroxynitrite across pH 5.0−9.0, with extent of enhancement showing strong pH dependence. This differs from the common understanding that HCO3− typically reduces the degradation rate of pollutants. To elucidate the mechanism of this effect, the contributions of different reactive species were systematically evaluated.
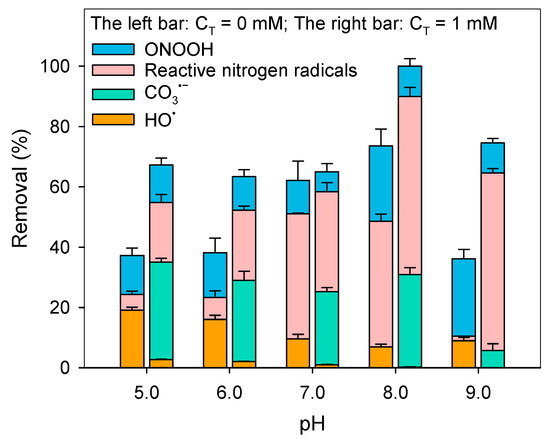
Figure 4.
Influence of HCO3− on BPA removal in peroxynitrite solution at different pH and the corresponding contributions of reactive species. Experimental conditions: [phosphate buffer] = 20 mM, [micropollutant]0 = 5 μM, [peroxynitrite]0 = 2 mg/L as N, reaction time = 5 min. (CT = [H2CO3*] + [HCO3−] + [CO32−], with [H2CO3*] = [CO2(aq)] + [H2CO3]).
Influence on peroxynitrite decay. In the presence of HCO3−, the reactive species involved in BPA degradation included ONOOH, HO•, CO3•− and reactive nitrogen radicals. As shown in Figure S6, 1 mM HCO3− substantially accelerated peroxynitrite decay, which can be attributed to the reaction between ONOO− and CO2—a pathway known to dominate in vivo [12,39]. The kinetic of peroxynitrite decay under these conditions can be described as:
where is the second-order rate constant for ONOO− + CO2. Since the dosed HCO3− distributes among CO2, H2CO3, HCO3− and CO32−, CT was used to represent total carbonate species. Equation (33) can be rewritten as,
where is the fraction of CO2 in CT, and is the fraction of [ONOO−] in total peroxynitrite. Rearrangement gives:
As shown in Figure S6, 1 mM HCO3− increased the peroxynitrite decay rate from 2.15 × 10−3 s−1 to 9.93 × 10−3 s−1, confirming that decomposition was shifted toward the CO2 pathway (Scheme 1). The calculated was 3.67 × 103 M−1 s−1, about one order of magnitude lower than the literature values [40]. This discrepancy likely reflects differences in experimental conditions: previous studies were typically performed at 37 °C and 25 mM bicarbonate (equilibrated with ~1.3 mM CO2) to mimic plasma environments. Nevertheless, it is generally accepted that the ONOO− + CO2− reaction rate is pH-independent [40]. Accordingly, the pseudo-first-order decay constants at different pH under CO2 presence were calculated and summarized in Table S3. Overall, dosed HCO3− markedly enhanced peroxynitrite decay, with the effect intensifying at higher pH (Figure S7). Consequently, peroxynitrite’s direct contribution to BPA degradation diminished (Figure 4). At pH 8.0 and 9.0, decay rates increased by 331.4% and 361.9%, respectively, leading to sharp reductions in peroxynitrite’s role in BPA degradation.

Scheme 1.
The reaction pathway of ONOO− with CO2.
Formation of secondary oxidants. The contributions of HO•, CO3•− and reactive nitrogen radicals were calculated and the details were presented in Texts S4 and S5.
CO3•−:Figure 4 shows that CO3•− considerably contributed to BPA degradation at pH 5.0−8.0. CO3•− may form via (1) HO• + HCO3−, or (2) ONOO− + CO2− ONOOC(O)O− •NO2 + CO3•−. To distinguish between these, TBA (a HO• scavenger) was applied at 100 mM. As shown in Figure S8, TBA had negligible influence on CO3•− contribution, indicating that HO• played little role in CO3•− formation. Instead, the ONOO− + CO2 pathway (pathway 2) was the dominant source of CO3•−.
Reactive nitrogen radicals: At pH 5.0−6.0, HCO3− enhanced the role of reactive nitrogen radicals in BPA degradation, consistent with •NO2 generation during ONOOC(O)O− decomposition (Scheme 1). However, at pH 7.0, reactive nitrogen radicals contribution decreased. This reduction might be due to scavenging of reactive nitrogen radicals by HCO3− as the equilibrium shifted from H2CO3 to HCO3− (pKa1 = 6.38).
At pH 8.0−9.0, the main oxidant responsible for BPA removal is reactive nitrogen radicals. As shown in Figure S7, majority of peroxynitrite decomposed through the reaction with CO2 and form ONOOCOO−, which then decomposed to •NO2 and CO3•−. Under this pH range, peroxynitrite mainly existed in the form of ONOO−. The high exposure of ONOO− resulted in the significantly transformation of CO3•−, along with the increased formation of RNS (Equation (36)) [41]. Consequently, the boosted RNS served as the main oxidant responsible for BPA degradation.
ONOO− + CO3•− → •NO + O2 + CO32− k = 7.7 × 106 M−1 s−1
In water bodies containing bicarbonate ions, while HO• are quenched, the introduction of CO3•− enhances pollutant removal efficiency. Under appropriate pH conditions, this process also promotes the conversion of peroxynitrite into nitrogen-containing radicals, further boosting the pollutant removal efficacy of reactive nitrogen species.
3.4. Impact of Borate Buffer on Peroxynitrite-Mediated Pollutant Degradation
Borate buffer is frequently used in laboratory experiments in the pH range of 7.0–9.0. During buffer selection, borate was tested, and the results revealed that it unexpectedly promoted BPA degradation (Figure 5). To clarify the underlying mechanism, the contributions of different reactive species were evaluated. NB was employed to calculate HO• contribution and the results were shown in Figure 5. Compared to the system with phosphate buffer, the HO• contribution decreased in borate-buffered systems. This inhibition might be ascribed to the consumption of HO• by borate ( = 3.6 × 104 M−1 s−1, = 6.8 × 106 M−1 s−1) [42,43]. However, Figure S9 shows that borate had little effect on BPA removal in the UV/H2O2 process, where HO• is the primary oxidant, indicating that HO• scavenging by borate only minimally influenced overall HO• exposure.
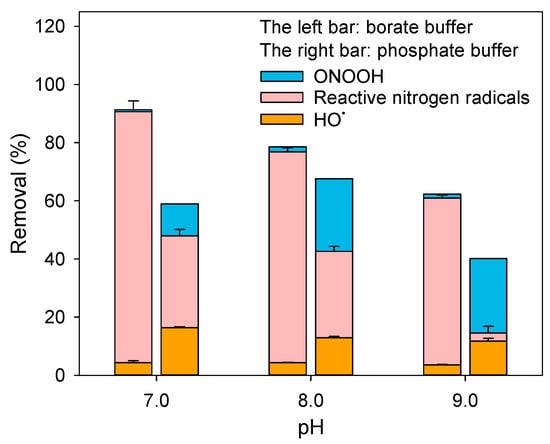
Figure 5.
Contributions of HO•, ONOOH and reactive nitrogen radicals to BPA removal during peroxynitrite treatment in the presence of borate buffer (left bar) or phosphate buffer (right bar). Experimental conditions: [phosphate buffer] = 20 mM, [borate buffer] = 20 mM, [BPA]0 = 5 μM, [peroxynitrite]0 = 2 mg/L as N.
Figure S10 shows that borate significantly accelerated peroxynitrite decomposition at pH 7.0−9.0. The exposure of ONOOH was quantified, and its contribution to BPA removal was calculated. As shown in Figure 5, ONOOH’s contribution to BPA degradation markedly decreased in the presence of borate. Thus, borate-induced acceleration of peroxynitrite decay reduced HO• formation, thereby accounting for the lower HO• contribution.
Sikora et al. reported that peroxynitrite reacts rapidly with aromatic and aliphatic boronic acids (k ≈ 106 M−1 s−1), involving a radical pathway in which ONOO− reacts to generate •NO2 [44]. Consistent with this, our results showed an increased contribution of reactive nitrogen radicals accompanied by a decreased contribution from ONOOH, suggesting that peroxynitrite reacts with the borate buffer. To identify the specific reactive species involved in ONOOH/ONOO− interactions with H3BO3/B(OH)4−, we performed systematic calculations. Given that the borate buffer system maintains an equilibrium between H3BO3 and B(OH)4−, BT denotes the total concentration of borate species. As the peroxynitrite decay rate decreased markedly with increasing pH from 7.0 to 9.0, we assumed that the reaction of ONOOH with H3BO3 dominates peroxynitrite loss, expressed as:
where k is the pseudo-first-order rate constant for peroxynitrite self-decomposition and is the second-order rate constant for H3BO3 + ONOOH. Equation (37) can be rewritten as,
where is the fraction of H3BO3 in BT, and is the fraction of [ONOOH] in total peroxynitrite. Rearranging and integrating Equation (37) yields:
Consequently,
Kinetic analysis revealed an excellent linear correlation between and , as shown in Figure S11a. In contrast, no clear linearity was observed for B(OH)4−/ONOOH or H3BO3/ONOO− (Figure S11b,c), confirming that H3BO3 + ONOOH is the primary pathway promoting BPA degradation.
Overall, the efficiency of BPA removal by peroxynitrite in the presence of borate decreased with increasing pH (Figure 5). The dominant peroxynitrite species shifted from ONOOH to ONOO− with increasing pH from 7.0 to 9.0, thereby enhancing the scavenging of reactive nitrogen radicals and reducing their role in pollutant degradation.
3.5. Peroxynitrite-Driven Degradation Pathways of Bisphenol A
Degradation by-products of bisphenol A (BPA) formed under peroxynitrite treatment were identified by UPLC-QTOF-MS (Scheme 2 and Figure S14). A variety of products were detected (Table S5), consistent with reported RNS-driven BPA degradation [45]. Based on the identified intermediates and literature mechanisms, the following pathway is proposed:
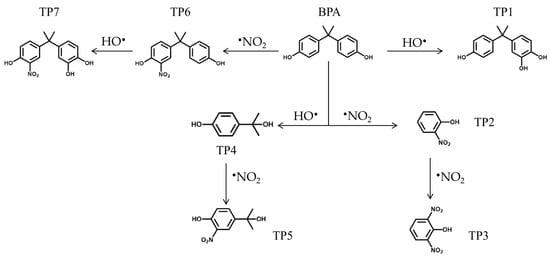
Scheme 2.
The proposed degradation pathways of BPA.
- (i)
- Hydroxylation and Nitration Pathways: BPA can undergo hydroxylation by HO• to form TP1 (m/z 243.1024) or be attacked by •NO2 to form TP6 (m/z 272.0953). TP6 (m/z 272.0953) subsequently undergoes further hydroxylation to yield TP7 (m/z 288.0879). Studies indicate that carbon atoms in BPA connected to the hydroxyl group and its adjacent carbon exhibit relatively high FED2 values, suggesting that the aromatic ring is more susceptible to substitution by both HO• and •NO2 [46].
- (ii)
- β-Scission pathway: Following the cleavage of the β C–C bond in BPA, a phenoxy radical and a phenol cation are formed [47]. The phenoxy radical undergoes hydroxylation to produce TP4 (m/z 151.076). TP4 (m/z 151.076) can react with •NO2 to yield TP5 (m/z 196.0612). Previous density functional theory calculations on BPA molecules have revealed that the carbon atom adjacent to the hydroxyl group is the most vulnerable site for radical attack [48]. The product structures align with existing research findings that the phenolic hydroxyl group can direct HO•/•NO2 addition to the ortho and/or para positions. Direct attack by NO2 on the aromatic ring of the phenol cation yields two nitrophenol derivatives: TP2 (m/z 138.0207) and TP3 (m/z 183.0048). Detection of these nitration products confirms the pivotal role of NO2 in redirecting the BPA degradation pathway [45].
Meanwhile, studies indicate that ONOOH decomposes via a protonation process to form NO+, which can oxidize phenolic compounds into corresponding radical anions before deprotonating to form phenoxy radicals [49]. These phenoxy radicals are readily susceptible to further oxidation and nitration. Since the products obtained through this pathway resemble those from the •NO2 nitrification pathway, subsequent isotope labeling experiments and quenching assays can be employed to distinguish between different pathways. This approach facilitates a more precise determination of the formation pathways for various nitrogen-containing products.
Additionally, regarding the toxicity of intermediate products, studies similar to this paper have evaluated the toxicity of BPA degradation products [47]. The results indicate that the toxicity of intermediate products is lower than that of BPA.
3.6. Formation of Nitrosamine Disinfection Byproducts
A sharp increase in NDMA during the breakpoint chlorination process was observed [50,51,52]. Given the critical role of peroxynitrite in this process, it is likely that peroxynitrite contributes to NDMA formation. To explore this, we examined the reaction of peroxynitrite with dimethylamine (DMA, the common precursor of NDMA) under varying pH conditions.
As shown in Figure 6, NDMA was consistently detected after the reaction of peroxynitrite with DMA. The NDMA yield decreased as pH increased from 5.0 to 7.0, then rose again with further increases in pH up to 9.0. This trend differed markedly from that observed for BPA removal, suggesting that different reactive species govern NDMA formation and pollutant degradation. When 1 mM NB was introduced as HO• scavenger, NDMA production was slightly suppressed at pH 5.0–6.0, had a negligible impact at pH 7.0, but unexpectedly enhanced at pH 8.0–9.0. These results differ from Masuda et al. [53], who proposed a HO•-driven nitrosation mechanism for secondary amines under acidic to neutral conditions. Instead, our data show that HO• contributes only marginally at pH 5.0–6.0 and becomes negligible from neutral to alkaline pH. Therefore, peroxynitrite itself and the reactive nitrogen radicals generated during its decomposition are more likely responsible for NDMA formation. Figure S12 shows that NDMA production at pH 11.0 within 30 min exceeded that at pH 9.0 by two orders of magnitude, suggesting that ONOO− might dominate NDMA formation, especially at high pH.
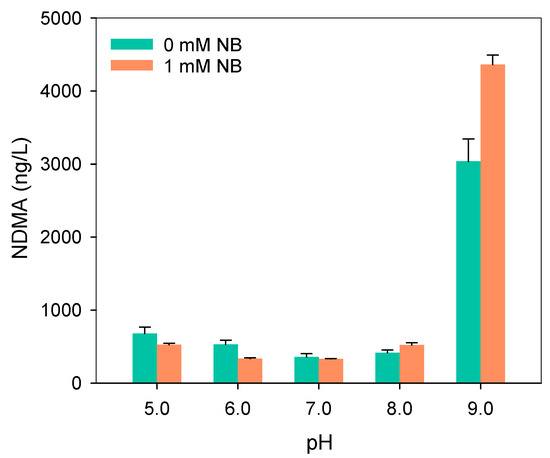
Figure 6.
NDMA formation under different conditions on. Experimental conditions: [DMA]0 = 10 μM, [phosphate buffer] = 20 mM, [peroxynitrite]0 = 2.0 mg/L as N.
Given the strong effect of HCO3− on peroxynitrite decay, its addition was expected to suppress NDMA formation during breakpoint chlorination. As shown in Figure S13, substantial NDMA was produced at pH 7.0, but the presence of 50 mM HCO3− markedly reduced the yield. This suppression is attributable to reduced exposure to peroxynitrite, highlighting the central role of peroxynitrite-driven pathways in NDMA formation under breakpoint chlorination conditions. This study primarily applies to water bodies with a pH not exceeding 9.0. For industrial wastewater with higher pH levels, the process may undergo changes, which warrants further investigation in future research. As a known persistent, recalcitrant, and carcinogenic substance [54], the formation of NDMA indicates that when applying processes that generate peroxynitrite to treat amine-containing wastewater, rigorous NDMA monitoring and targeted control of the effluent are required.
4. Conclusions and Engineering Implications
This study systematically elucidates the intrinsic mechanism of peroxynitrite-mediated advanced oxidation processes under environmentally relevant conditions. Based on the pH dependence of peroxynitrite decomposition, this study first established that ONOOH dominates the decomposition process across the pH range of 5.0–9.0. The decay rate constant was determined to be approximately 1.20 s−1 at pH 5.0, decreasing to 0.0022 s−1 at pH 9.0—spanning three orders of magnitude. Addressing pH-dependent degradation variations, we distinguished the specific contributions of HO•, ONOOH, and reactive nitrogen radicals. We conclusively demonstrate that BPA degradation at pH 7.0–8.0 relies heavily on reactive nitrogen radicals, whereas ONOOH dominates at pH 9.0. The secondary rate constant for the BPA–ONOOH reaction was measured as k = 1.18 × 103 M−1 s−1. Furthermore, the study validated the regulatory role of buffering chemistry on reaction pathways: bicarbonate accelerated peroxynitrite decomposition by over 300% at pH 8.0–9.0, suppressing the direct contribution of ONOOH. At pH 9.0, abundant CO3•− acted on unreacted ONOO−, promoting reactive nitrogen radicals as the primary degradation pathway for the pollutant. Notably, despite literature suggesting borate accelerates peroxynitrite decay, this study found it significantly enhances pollutant degradation by shifting the reaction pathway toward reactive nitrogen species. In the presence of dimethylamine, peroxynitrite was confirmed as the key driver for NDMA formation, with its yield peaking under alkaline conditions but being inhibited by bicarbonate.
Collectively, these findings fill gaps in understanding the detailed degradation mechanism of peroxynitrite while revealing its dual role: as both a potentially efficient oxidant for removing trace organic pollutants and a potential precursor for harmful nitrosamine byproducts. Compared to HO•, ONOOH exhibits a higher oxidation potential as a two-electron oxidant, offering greater advantages for the direct oxidation of electron-rich pollutants. Moreover, the derived reactive nitrogen species possess distinct selectivity characteristics. This contributes to heightened academic recognition of peroxynitrite as a significant reactive nitrogen species.
The findings of this study provide critical scientific basis for evaluating the economic and engineering feasibility of peroxynitrite-related processes. From an engineering perspective, controlling solution chemistry—particularly pH and buffering systems—is central to maximizing oxidation efficiency while mitigating byproduct risks. This work offers guidance for selecting and designing processes to treat water bodies containing characteristic pollutants like phenols. By precisely regulating reaction conditions, it holds promise for enhancing oxidation efficiency and optimizing operational costs.
However, implementing these technologies in actual water bodies requires careful consideration of their limitations: First, complex matrix components in water (such as natural organic matter and various ions) may quench active species or compete for reaction pathways, leading to treatment outcomes that deviate from expectations. Second, maintaining the pH range required for optimal performance poses precise control challenges in continuous-flow operations, increasing process complexity. Third, research confirms that the risk of harmful byproducts like NDMA forming in actual wastewater systems may be further influenced by water quality. Systematic management through integrated strategies—combining source control, process optimization, and end-of-pipe treatment—is necessary, though this may increase treatment processes and costs.
Despite the aforementioned progress, the specific mechanisms of action for the relevant reactive nitrogen species remain poorly understood. Further research should elucidate the formation pathways, steady-state concentrations, and reaction selectivity of specific nitrogen radicals.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/w18010097/s1, Figure S1: Time course of peroxynitrite decay at different pH; Figure S2: Influence of (a) buffer solution (b) TBA and (c) phenol on the decay of peroxynitrite at pH 9.0; Figure S3: Effect of BPA on the decomposition kinetics of peroxynitrite at pH 9.0; Figure S4: Effect of BPA on the decomposition kinetics of peroxynitrite at pH 11.0; Figure S5: Decay of peroxynitrite at pH = 9.0; Figure S6: Influence of HCO3− on the decay of peroxynitrite at pH 9.0; Figure S7: Influence of HCO3− on the decomposition pathway of peroxynitrite at pH 9.0; Figure S8: Influence of TBA on BPA removal by peroxynitrite in the presence of HCO3−; Figure S9: The degradation of BPA by UV/H2O2 at different pH conditions; Figure S10: Decay of peroxynitrite in different buffer systems over pH 7.0–9.0; Figure S11: The relationship between between and (a) , (b) . (c) ; Figure S12: NDMA formation in the reaction of DMA with peroxynitrite at different pH; Figure S13: Effects of HCO3− on NDMA formation in breakpoint chlorination process; Figure S14: Secondary mass spectrometry analysis of the intermediate compounds generated during BPA degradation (TP1-TP7); Table S1: Second-order rate constants of several reactive species and target organic matters; Table S2: The reactions relevant to RNS; Table S3: The pseudo-first-order rate constants of peroxynitrite decomposition through different routes; Table S4: Exposure of ONOO− in the presence of 1 mM of HCO3− over pH 5.0−9.0; Table S5: Text for BPA degradation intermediates.
Author Contributions
Z.C.: Investigation, Data curation, Writing—Original Draft Preparation. D.R.: Conceptualization, Writing Editing. J.Z.: Supervision. B.S.: Supervision, Funding Acquisition, Writing—Reviewing and Editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Taishan Scholars Program of Shandong Province (No. tsqn201909019).
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Acknowledgments
We thank Zhifeng Li and Xiangmei Ren of the Core Facilities for Life and Environmental Sciences, State Key laboratory of Microbial Technology of Shandong University for GC-MS and stopped-flow spectrometer analysis. We appreciate the GC-MS measurements assisted by Yiqiao Zhang from Analytical Testing Center, School of Environmental Science and Engineering, Shandong University.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zou, R.; Yang, W.; Rezaei, B.; Bendtsen, E.B.; Guo, K.; Tang, K.; Andersen, H.R.; Keller, S.S.; Zhang, Y. On the mechanism and selectivity of a novel iodine/peracetic acid process for the efficient and rapid elimination of micropollutants. Chem. Eng. J. 2024, 479, 147815. [Google Scholar] [CrossRef]
- Peng, J.; Zhao, H.; Wang, H.; Feng, Y.; Yang, Y.; Zhou, Y.; An, N.; Zhao, X. Biochar matrix anchoring pure phase Fe3C to promote advanced oxidation: A reliable pathway for organic wastewater purification. Sep. Purif. Technol. 2025, 362, 131845. [Google Scholar] [CrossRef]
- Li, Z.; Wu, X.; Sun, J.; Wei, B.; Ma, Y.; Liu, H. Computational study on degradation mechanisms, efficiencies and toxicity of BPA and its analogs in UV/H2O2 and fenton/ H2O2 systems. Chem. Eng. J. 2025, 526, 171542. [Google Scholar] [CrossRef]
- Yang, S.; Duan, X.; Liu, J.; Wu, P.; Li, C.; Dong, X.; Zhu, N.; Dionysiou, D.D. Efficient peroxymonosulfate activation and bisphenol a degradation derived from mineral-carbon materials: Key role of double mineral-templates. Appl. Catal. B Environ. 2020, 267, 118701. [Google Scholar] [CrossRef]
- Lu, S.; Shang, C.; Sun, B.; Xiang, Y. Dominant Dissolved Oxygen-Independent Pathway to Form Hydroxyl Radicals and the Generation of Reactive Chlorine and Nitrogen Species in Breakpoint Chlorination. Environ. Sci. Technol. 2023, 57, 150–159. [Google Scholar] [CrossRef]
- Wu, L.; Patton, S.D.; Liu, H. Mechanisms of oxidative removal of 1,4-dioxane via free chlorine rapidly mixing into monochloramine: Implications on water treatment and reuse. J. Hazard. Mater. 2022, 440, 129760. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, C.; Zhu, B.-Z.; Huang, C.-H.; An, T.; Meng, F.; Fang, J. Reactive nitrogen species are also involved in the transformation of micropollutants by the UV/monochloramine process. Environ. Sci. Technol. 2019, 53, 11142–11152. [Google Scholar] [CrossRef]
- Sun, P.; Meng, T.; Wang, Z.; Zhang, R.; Yao, H.; Yang, Y.; Zhao, L. Degradation of organic micropollutants in UV/NH2Cl advanced oxidation process. Environ. Sci. Technol. 2019, 53, 9024–9033. [Google Scholar] [CrossRef]
- Sharma, V.K.; Manoli, K.; Ma, X. Reactivity of nitrogen species with inorganic and organic compounds in water. Chemosphere 2022, 302, 134911. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Qiu, W.; Li, J.; Pang, S.; Zhang, Z.; Jiang, J. Mechanism, kinetics and DBP formation of UV/NH2Cl process on contaminant removal in aqueous solution: A review. Chem. Eng. J. 2021, 420, 130405. [Google Scholar] [CrossRef]
- Cao, Z.; Yu, X.; Zheng, Y.; Aghdam, E.; Sun, B.; Song, M.; Wang, A.; Han, J.; Zhang, J. Micropollutant abatement by the UV/chloramine process in potable water reuse: A review. J. Hazard. Mater. 2022, 424, 127341. [Google Scholar] [CrossRef]
- Lobachev, V.L.; Rudakov, E.S. The chemistry of peroxynitrite. Reaction mechanisms and kinetics. Russ. Chem. Rev. 2006, 75, 375–396. [Google Scholar] [CrossRef]
- Merényi, G.; Lind, J. Thermodynamics of peroxynitrite and its CO2 adduct. Chem. Res. Toxicol. 1997, 10, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Alcolado, C.I.; Jiménez, E.; García-Río, L.; Poblete, F.J. Aqueous-phase degradation mechanism of parabens, emerging contaminants, by peroxynitrite. ACS EST Water 2025, 5, 4147–4155. [Google Scholar] [CrossRef]
- Alcolado, C.I.; Garcia-Rio, L.; Mejuto, J.C.; Moreno, I.; Poblete, F.J.; Tejeda, J. Oxidation of aldehydes used as food additives by peroxynitrite. Antioxidants 2023, 12, 743. [Google Scholar] [CrossRef]
- Vione, D.; Maurino, V.; Minero, C.; Lucchiari, M.; Pelizzetti, E. Nitration and hydroxylation of benzene in the presence of nitrite/nitrous acid in aqueous solution. Chemosphere 2004, 56, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, N.; Long, Y.-T.; Qian, X.; Yang, Y. Understanding the selectivity of a multichannel fluorescent probe for peroxynitrite over hypochlorite. Bioconjug. Chem. 2016, 27, 341–353. [Google Scholar] [CrossRef]
- Duan, J.; Cao, Y.; Yang, Q.; Li, W.; Huang, Q.; Guo, Q.; Jiang, J. Involvement of inorganic nitrogen species (NOX−(x = 2, 3)) in the degradation of organic contaminants in environmental waters via UV irradiation or chemical oxidation: A dual-edged approach. Sci. Total Environ. 2025, 963, 178500. [Google Scholar] [CrossRef]
- Kodamatani, H.; Kubo, S.; Takeuchi, A.; Kanzaki, R.; Tomiyasu, T. Sensitive detection of nitrite and nitrate in seawater by 222 nm UV-irradiated photochemical conversion to peroxynitrite and ion chromatography-luminol chemiluminescence system. Environ. Sci. Technol. 2023, 57, 5924–5933. [Google Scholar] [CrossRef]
- Liu, T.; Chen, J.; Li, N.; Xiao, S.; Huang, C.-H.; Zhang, L.; Xu, Y.; Zhang, Y.; Zhou, X. Unexpected role of nitrite in promoting transformation of sulfonamide antibiotics by peracetic acid: Reactive nitrogen species contribution and harmful disinfection byproduct formation potential. Environ. Sci. Technol. 2022, 56, 1300–1309. [Google Scholar] [CrossRef]
- Lehnig, M. Radical mechanisms of the decomposition of peroxynitrite and the peroxynitrite–CO2 adduct and of reactions with l-tyrosine and related compounds as studied by 15N chemically induced dynamic nuclear polarization. Arch. Biochem. Biophys. 1999, 368, 303–318. [Google Scholar] [CrossRef]
- Lehnig, M.; Jakobi, K. 15N CIDNP during reaction of the oxoperoxonitrate–CO2 adduct with bovine albumin and L-tyrosine derivatives. J. Chem. Soc. Perkin Trans. 2000, 2, 2016–2021. [Google Scholar] [CrossRef]
- Kirsch, M.; Korth, H.-G.; Wensing, A.; Sustmann, R.; De Groot, H. Product formation and kinetic simulations in the pH range 1–14 account for a free-radical mechanism of peroxynitrite decomposition. Arch. Biochem. Biophys. 2003, 418, 133–150. [Google Scholar] [CrossRef]
- Merényi, G.; Lind, J.; Goldstein, S.; Czapski, G. Mechanism and thermochemistry of peroxynitrite decomposition in water. J. Phys. Chem. A 1999, 103, 5685–5691. [Google Scholar] [CrossRef]
- Bonini, M.G.; Mason, R.P.; Augusto, O. The mechanism by which 4-hydroxy-2,2,6,6-tetramethylpiperidene-1-oxyl (tempol) diverts peroxynitrite decomposition from nitrating to nitrosating species. Chem. Res. Toxicol. 2002, 15, 506–511. [Google Scholar] [CrossRef]
- Ferkous, H.; Hamdaoui, O.; Pétrier, C. Sonochemical formation of peroxynitrite in water: Impact of ultrasonic frequency and power. Ultrason. Sonochem. 2023, 98, 106488. [Google Scholar] [CrossRef]
- Robert Carola, J.P.H.; Noback, C.R. Human Anatomy & Physiology, 2nd ed.; McGraw-Hill College: New York, NY, USA, 1992. [Google Scholar]
- Radi, R.; Cosgrove, T.P.; Beckman, J.S.; Freeman, B.A. Peroxynitrite-induced luminol chemiluminescence. Biochem. J. 1993, 290, 51–57. [Google Scholar] [CrossRef]
- Prolo, C.; Piacenza, L.; Radi, R. Peroxynitrite: A multifaceted oxidizing and nitrating metabolite. Curr. Opin. Chem. Biol. 2024, 80, 102459. [Google Scholar] [CrossRef]
- Lai, W.W.-P.; Hsu, M.-H.; Lin, A.Y.-C. The role of bicarbonate anions in methotrexate degradation via UV/TiO2: Mechanisms, reactivity and increased toxicity. Water Res. 2017, 112, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Chen, J.; Ischiropoulos, H.; Crow, J.P. Oxidative chemistry of peroxynitrite. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1994; Volume 233, pp. 229–240. [Google Scholar] [CrossRef]
- Wang, W.-L.; Wu, Q.-Y.; Du, Y.; Huang, N.; Hu, H.-Y. Elimination of chlorine-refractory carbamazepine by breakpoint chlorination: Reactive species and oxidation byproducts. Water Res. 2018, 129, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Du, Y.; Zhou, Y.; Wu, Q.; Zheng, S.; Fang, J. Formation of nitro(so) and chlorinated products and toxicity alteration during the UV/monochloramine treatment of phenol. Water Res. 2021, 194, 116914. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/O•− in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Huang, Y.; Kong, M.; Westerman, D.; Xu, E.G.; Coffin, S.; Cochran, K.H.; Liu, Y.; Richardson, S.D.; Schlenk, D.; Dionysiou, D.D. Effects of HCO3– on degradation of toxic contaminants of emerging concern by UV/NO3–. Environ. Sci. Technol. 2018, 52, 12697–12707. [Google Scholar] [CrossRef]
- Goldstein, S.; Saha, A.; Lymar, S.V.; Czapski, G. Oxidation of peroxynitrite by inorganic radicals: A pulse radiolysis study. J. Am. Chem. Soc. 1998, 120, 5549–5554. [Google Scholar] [CrossRef]
- Brienza, M.; Manasfi, R.; Chiron, S. Relevance of N-nitrosation reactions for secondary amines in nitrate-rich wastewater under UV-C treatment. Water Res. 2019, 162, 22–29. [Google Scholar] [CrossRef]
- Kissner, R.; Nauser, T.; Bugnon, P.; Lye, P.G.; Koppenol, W.H. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem. Res. Toxicol. 1997, 10, 1285–1292. [Google Scholar] [CrossRef]
- Squadrito, G.L.; Pryor, W.A. Oxidative chemistry of nitric oxide: The roles of superoxide, peroxynitrite, and carbon dioxide. Free Radic. Biol. Med. 1998, 25, 392–403. [Google Scholar] [CrossRef]
- Denicola, A.; Freeman, B.A.; Trujillo, M.; Radi, R. Peroxynitrite reaction with carbon dioxide/bicarbonate: Kinetics and influence on peroxynitrite-mediated oxidations. Arch. Biochem. Biophys. 1996, 333, 49–58. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Serrano-Luginbuehl, S.; Nauser, T.; Kissner, R. Thinking outside the cage: A new hypothesis that accounts for variable yields of radicals from the reaction of CO2 with ONOO–. Chem. Res. Toxicol. 2020, 33, 1516–1527. [Google Scholar] [CrossRef]
- Petersson, F.; Jonsson, M. The reactivity of hydroxyl radicals toward boric acid as a function of pH. J. Phys. Chem. A 2024, 128, 7593–7600. [Google Scholar] [CrossRef]
- Buxton, G.V.; Sellers, R.M. Reactivity of the hydrated electron and the hydroxyl radical with boric acid in aqueous solutions. Int. J. Radiat. Appl. Instrum. Part C Radiat. Phys. Chem. 1987, 29, 137–140. [Google Scholar] [CrossRef]
- Sikora, A.; Zielonka, J.; Lopez, M.; Dybala-Defratyka, A.; Joseph, J.; Marcinek, A.; Kalyanaraman, B. Reaction between peroxynitrite and boronates: EPR spin-trapping, HPLC analyses, and quantum mechanical study of the free radical pathway. Chem. Res. Toxicol. 2011, 24, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Frassati, S.; Carena, L.; Barbaro, E.; Roman, M.; Feltracco, M.; Minella, M.; Sordello, F.; Minero, C.; Spolaor, A.; Scalabrin, E.; et al. Photodegradation of bisphenol A and identification of photoproducts in artificial snow under UVA radiation. Environ. Pollut. 2025, 381, 126503. [Google Scholar] [CrossRef]
- Zhou, S.; Li, L.; Wu, Y.; Zhu, S.; Zhu, N.; Bu, L.; Dionysiou, D.D. UV365 induced elimination of contaminants of emerging concern in the presence of residual nitrite: Roles of reactive nitrogen species. Water Res. 2020, 178, 115829. [Google Scholar] [CrossRef]
- Fu, Z.; Lin, Z.; Jian, J.; Zhang, S.; Zhang, F.; Wang, Y.; He, Y.; Liu, L.; Wang, Y.; Zhan, H.; et al. Synergistic enhancement of bisphenol A photodegradation by BiOCl via nitrite-induced charge separation and reactive oxygen species generation: A dual role for nitrite in pollutant degradation and transformation. J. Environ. Chem. Eng. 2026, 14, 120680. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, M.; Deng, Y.; Liu, M.; Chen, Y.; Gao, N.; Du, E.; Chu, W.; Guo, H. Generality and diversity on the kinetics, toxicity and DFT studies of sulfate radical-induced transformation of BPA and its analogues. Water Res. 2022, 219, 118506. [Google Scholar] [CrossRef]
- Vione, D.; Maurino, V.; Minero, C.; Pelizzetti, E. Nitration and photonitration of naphthalene in aqueous systems. Environ. Sci. Technol. 2005, 39, 1101–1110. [Google Scholar] [CrossRef]
- Schreiber, I.M.; Mitch, W.A. Enhanced Nitrogenous Disinfection By Product Formation near the Breakpoint: Implications for Nitrification Control. Environ. Sci. Technol. 2007, 41, 7039–7046. [Google Scholar] [CrossRef]
- Schreiber, I.M.; Mitch, W.A. Influence of the Order of Reagent Addition on NDMA Formation during Chloramination. Environ. Sci. Technol. 2005, 39, 3811–3818. [Google Scholar] [CrossRef]
- Charrois, J.W.A.; Hrudey, S.E. Breakpoint chlorination and free-chlorine contact time: Implications for drinking water N-nitrosodimethylamine concentrations. Water Res. 2007, 41, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Mower, H.F.; Pignatelli, B.; Celan, I.; Friesen, M.D.; Nishino, H.; Ohshima, H. Formation of N-nitrosamines and N-nitramines by the reaction of secondary amines with peroxynitrite and other reactive nitrogen species: Comparison with nitrotyrosine formation. Chem. Res. Toxicol. 2000, 13, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Yang, J.; Zhang, C.; Song, Y.; Jiang, J.; Ma, J. The role of biochar in algal source water treatment: Algal cells integrity and N-nitrosodimethylamine (NDMA) formation potential. J. Hazard. Mater. 2025, 492, 138292. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.