Abstract
The discharge of ammonia-rich wastewater poses significant threats to water quality and ecosystem health, driving the need for efficient and sustainable treatment technologies. The electrochemical ammonia oxidation reaction (eAOR) has emerged as a promising alternative to conventional biological and physicochemical methods, offering advantages such as in situ oxidant generation, tunable product selectivity, and applicability under challenging water matrices. This comprehensive review systematically examines the mechanisms, catalyst design, and environmental factors influencing eAOR performance. Two primary pathways are detailed: direct eAOR, involving stepwise dehydrogenation of NH3 on the electrode surface, and indirect eAOR, mediated by electrogenerated reactive chlorine species (RCS). The mechanisms—including the Oswin-Salomon and Gerischer-Mauerer pathways for direct oxidation, as well as breakpoint chlorination and radical-mediated routes for indirect oxidation—are critically discussed alongside experimental and theoretical evidence. Recent advances in electrocatalyst development are highlighted, covering noble metals, non-noble transition metal oxides, alloys, and hybrid materials, with an emphasis on enhancing activity, selectivity toward N2, and durability. Key operational parameters such as pH, chloride concentration, and coexisting ions are analyzed for their impact on reaction kinetics and byproduct formation. Finally, the review identifies current challenges—including catalyst poisoning, toxic byproduct generation, and scalability—and outlines future research directions aimed at advancing eAOR toward energy-efficient, resource-recovering water treatment systems.
1. Introduction
The escalating discharge of ammonia-laden wastewater from industrial sectors—including chemical synthesis, fertilizer production, and semiconductor manufacturing—has intensified pressure on global water security [1]. As a pervasive aquatic pollutant, ammonia nitrogen (NH3/NH4+) degrades water quality by driving eutrophication and depleting dissolved oxygen, while also serving as a precursor for the formation of carcinogenic byproducts (e.g., nitrosamines) through environmental transformations [2]. With regulatory frameworks such as the EU Water Framework Directive and US EPA effluent guidelines imposing stringent ammonia discharge limits (<1–2 mg-N L−1), the development of robust, energy-efficient treatment technologies has become imperative. Conventional biological nitrification-denitrification processes, though widely adopted, exhibit critical limitations when treating wastewater with extreme salinity (>3% NaCl), low carbon-to-nitrogen ratios (C/N < 2), or inhibitory compounds (e.g., heavy metals) [3,4]. Physicochemical methods, including breakpoint chlorination and air stripping, face operational bottlenecks such as hazardous chlorine gas handling, excessive chemical consumption, and secondary contamination risks from chlorinated byproducts or ammonia-laden air emissions [5,6].
Electrochemical ammonia oxidation reaction (eAOR) has emerged as a disruptive technology that synergizes the merits of biological and physicochemical approaches while circumventing their inherent drawbacks. Unlike conventional methods requiring external oxidant dosing or microbial acclimatization, eAOR enables in situ generation of reactive species through interfacial electron transfer, achieving tunable selectivity toward harmless nitrogen gas (N2) or nitrate (NO3−) based on operational parameters [7,8]. The process operates via two distinct pathways: direct electrochemical oxidation, where NH3 undergoes stepwise dehydrogenation and oxidation at the anode surface [9,10,11], and indirect oxidation, mediated by electrochemically generated reactive chlorine species (RCS) such as free chlorine (HClO/ClO−) or chlorine radicals (Cl•) [12]. Direct eAOR excels in low-chloride environments but demands electrocatalysts with high intrinsic activity to overcome sluggish reaction kinetics and intermediate poisoning [13]. Indirect eAOR, leveraging chloride ions (Cl−) ubiquitously present in industrial effluents, converts Cl− to oxidizing agents via the chlorine evolution reaction (CER), enabling efficient ammonia degradation even in complex matrices. However, both pathways contend with competing side reactions—notably the oxygen evolution reaction (OER)—and undesirable byproduct formation (e.g., chloramines, perchlorates), necessitating strategic optimization of catalysts and operating conditions [14].
Recent advancements in electrocatalyst engineering have revitalized interest in eAOR. For direct oxidation, research focuses on tailoring electronic structures to enhance NH3 adsorption and weaken N≡N bond formation barriers, while indirect oxidation catalysts prioritize selective CER activation over OER. Despite progress, fundamental challenges persist at the nexus of materials science and process engineering. These include reconciling the trade-off between catalyst durability and activity under harsh electrochemical conditions, suppressing toxic byproduct accumulation across variable water chemistries, and scaling laboratory breakthroughs to industrial systems with fluctuating contaminant loads.
This review systematically synthesizes mechanistic insights, material innovations, and operational considerations shaping the evolution of electrochemical ammonia oxidation. In this review, we first give the systematic comparison and integration of direct and indirect eAOR pathways with a focus on N2 selectivity; (2) the critical analysis of catalyst design strategies across noble and non-noble metals; and (3) the synthesis of operational challenges and future research directions aimed at practical application. Accordingly, providing a critical knowledge base for the rational design of efficient and scalable electrochemical reactors for ammonia removal and resource recovery in real-world water treatment.
2. Direct eAOR
Direct eAOR is highly dependent on the solution pH [15]. Under acidic conditions, ammonia primarily exists in the form of ammonium ions (NH4+), which are prone to indirect oxidation by external oxidants such as hypochlorous acid (HOCl). This indirect oxidation route tends to suffer from low efficiency and sluggish kinetics. In contrast, alkaline electrolytes facilitate the direct electrochemical oxidation of NH3, where hydroxide ions (OH−) participate as reactants. As a result, most investigations into direct eAOR have been conducted under alkaline aqueous environments, as shown in Figure 1a.
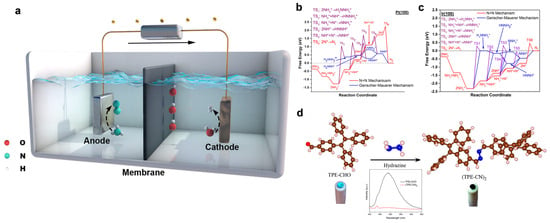
Figure 1.
(a) Schematic of an NH3 electrolysis cell (AEC) under aqueous alkaline electrolytes. (b) Comparison of the free-energy diagrams for NH3 electro-oxidation on Ir(100) at 0 V using the N + N mechanism and the G-M mechanism. (c) Comparison of the free-energy diagrams for NH3 electro-oxidation on Ir(100) at 0 V using N + N mechanism and G-M mechanism. Reprinted with permission from Ref. [16]. Copyright 2021, Elsevier. (d) Schematic illustration of the reaction between TPE-CHO and N2H4 to form (TPE-CN)2 for probing the reaction intermediates in the AOR process. NH3. Reprinted with permission from Ref. [17]. Copyright 2021, American Chemical Society.
In alkaline media, the anodic and cathodic half-reactions, along with the overall electrochemical process, are represented by the following equations:
Anode: 2NH3 + 6OH− → N2 + 6H2O + 6e− (E0 = −0.77 V vs. SHE)
Cathode: 6H2O + 6e− → 3H2 + 6OH− (E0 = −0.83 V vs. SHE)
Overall: 2NH3 → N2 + 3H2 (Ecell = 0.06 V)
The direct eAOR process involves a six-electron transfer, indicating its complex, multi-step nature. Consequently, several mechanistic studies have been conducted to identify the key steps and intermediates involved in this oxidation pathway. Two major mechanisms have been proposed over the years: the Oswin-Salomon (O-S) mechanism and the Gerischer–Mauerer (G-M) mechanism.
The O-S mechanism was initially proposed by Oswin and Salomon in 1963, suggesting that ammonia undergoes stepwise dehydrogenation on the catalyst surface, ultimately forming adsorbed nitrogen atoms (Nads), which couple to produce N2. Their analysis, based on Tafel slope comparisons using Langmuir and Temkin adsorption models, indicated that the conversion of *NH2 (* denoted as adsorption intermediate) to *NH is the rate-determining step (RDS) at low current densities [18]. The reaction sequence is as follows:
NH3 (aq) → *NH3
*NH3 + OH− → *NH2 + H2O + e−
*NH2 + OH− → *NH + H2O + e−
*NH + OH− → *N + H2O + e−
2*N → N2
On the other hand, the G-M mechanism, introduced by Gerischer and Mauerer in 1970, proposes that N–N coupling occurs at the intermediate stage rather than after full dehydrogenation. In this pathway, *NH2 and *NH species can dimerize to form N2Hx+y-type intermediates (e.g., hydrazine), which subsequently undergo electrochemical oxidation to generate N2 [19]. The reaction proceeds as follows:
NH3 (aq) → *NH3
*NH3 + OH− → *NH2 + H2O + e−
*NH2 + OH− → *NH + H2O + e−
*NHx + *NHy → *N2Hx+y
*N2Hx+y + (x + y)OH− → N2 + (x + y)H2O + (x + y)e−
*NHads + OH− → *N + H2O + e−
One notable feature of the G-M pathway is that strongly adsorbed nitrogen atoms (*N) can poison the electrode surface by occupying active sites, thereby diminishing the long-term catalytic performance. To evaluate and distinguish between these two mechanisms, density functional theory (DFT) calculations have been carried out for various metal surfaces [16]. On platinum Pt(100), for instance, the O-S mechanism suggests that the deprotonation of *NH2 to *NH is the potential-determining step (PDS), requiring a free energy of 0.87 eV. The subsequent *N dimerization to form N2 is kinetically favorable with an activation barrier of 0.53 eV and is thermodynamically downhill (−1.69 eV). In contrast, the G-M mechanism identifies the deprotonation of HNNH* to NNH* as the RDS, with a lower onset potential of 0.29 V (Figure 1b). For Ir(100), the onset potential under the O-S mechanism is 0.47 V, and the barrier for N* dimerization increases to 1.24 eV. The G-M mechanism for Ir(100) involves NNH* deprotonation as the PDS with an onset potential of 0.46 V (Figure 1c).
Although both mechanisms involve proton-coupled electron transfer (PCET) steps and appear pH-independent on the reversible hydrogen electrode (RHE) scale, experimental studies by Katsounaros et al. [20] introduced a revised viewpoint. They observed that increasing the pH of the electrolyte reduces the onset potential for Pt(100) and suppresses the formation of *NO. They proposed that OH− ions (not *OH) directly participate in a decoupled proton–electron transfer step, forming negatively charged intermediates that exhibit pH sensitivity:
This modification implies that the electrochemical step does not occur concurrently with proton removal, leading to distinct mechanistic behavior compared to classical PCET assumptions.
*NxHy + OH− ⇌ *(NxHy−1)− + H2O
*(NxHy−1)− ⇌ *NxHy−1 + e−
To validate the proposed mechanisms, several experimental techniques have been employed, including surface-enhanced Raman spectroscopy (SERS), differential electrochemical mass spectrometry (DEMS), and rotating ring-disk electrode analysis. For instance, De Vooys et al. [21] used SERS to identify *N species on gold and palladium electrodes, supporting the existence of poisoning intermediates consistent with the G-M model. Hydrazine (N2H4), one of the key intermediates, has been detected using aggregation-induced emission, further confirming its role in the N2 formation pathway (Figure 1d) [17].
Moreover, a reaction involving azide ions (N3−) has also been proposed as a potential intermediate route:
The presence of N3− was confirmed by SERS, offering the first experimental evidence for its involvement in eAOR [22].
NH3 + N2H4 + 7OH− ⇌ N3− + 7H2O + 6e−
Pt + N3− ⇌ PtN3−
PtN3− + 2H2O + e− ⇌ PtNH2 + 2OH− + N2
Further analysis by Vidal-Iglesias et al. [23] using DEMS revealed that ammonia oxidation on different Pt single crystal facets ((100), (110), and (111)) produces various products including N2, NO, and N2O, with Pt(100) showing the highest activity. Wasmus et al. [24] and Gootzen [25] et al. also demonstrated that nitrogen gas can be produced at potentials below 0.7 V vs. RHE, while nitrogen oxides dominate at higher anodic potentials (>0.8 V). Under such conditions, further oxidation can lead to the formation of nitrite (NO2−) and nitrate (NO3−) via [26,27]:
2NO2 + 2OH− → NO3− + NO2− + H2O
NO + NO2 + 2OH− → 2NO2− + H2O
In conclusion, Figure 2 integrates the O-S and G-M mechanisms with supporting experimental evidence, offering a comprehensive picture of the reaction landscape for electrochemical ammonia oxidation.
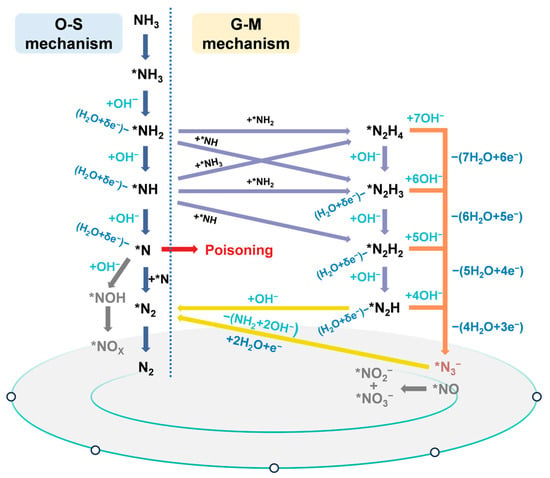
Figure 2.
Schematic diagram of possible eAOR mechanisms based on previous reports.
3. Indirect eAOR via RCS
Indirect eAOR exploits the in situ generation of RCS from chloride ions, which are ubiquitous in many industrial and municipal wastewaters. Unlike direct eAOR, where NH3 is oxidized through surface-mediated dehydrogenation, the indirect pathway relies on chlorine-derived oxidants that can rapidly and non-selectively attack ammonia, leading to the formation of nitrogen gas or oxidized nitrogen products. Two main mechanistic routes dominate this process: the well-established breakpoint chlorination pathway and radical-driven pathways where Cl• and ClO• species play decisive roles. While the indirect ammonia oxidation benefits from high reactivity and effectiveness in saline matrices, it introduces challenges associated with chloramine accumulation, halogenated byproducts, and the balance between molecular chlorine and radical pathways [28]. Understanding these dual routes is essential for developing selective and environmentally benign indirect eAOR systems.
3.1. Breakpoint Chlorination Oxidation Mechanism
Breakpoint chlorination has long been recognized as a classical pathway for chemical ammonia removal and is also operative in electrochemical chlorine-mediated oxidation systems. In this process, chloride ions (Cl−) are oxidized at the anode to generate free chlorine species such as Cl2, HOCl, and ClO−. These active chlorine species subsequently react with ammonia to form chloramines (NH2Cl, NHCl2, and NCl3), which undergo further oxidation and decomposition, ultimately leading to the production of nitrogen gas (N2) [29]. The reaction sequence is often divided into three main stages: (i) rapid chloramine formation, (ii) stepwise conversion and oxidation of chloramines, and (iii) final destruction of residual chloramines at the so-called “breakpoint,” yielding nitrogenous products (Figure 3a).
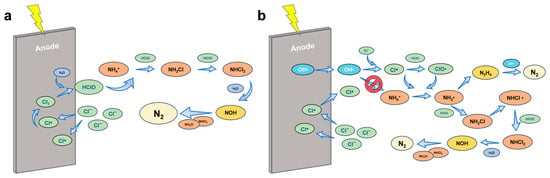
Figure 3.
Proposed reaction mechanisms and product transformation for ammonia oxidation via (a) breakpoint chlorination and (b) chlorine radicals.
The stoichiometric requirement for complete conversion of ammonia to nitrogen gas is typically achieved when the molar ratio of chlorine to nitrogen (Cl/N) exceeds 1.6 [5,30,31]. At this ratio, chloramines are no longer stable intermediates and are further oxidized to nitrogen gas or nitrate [32]. Despite its efficiency, the breakpoint chlorination pathway suffers from several drawbacks: (i) the demand for high concentrations of active chlorine, (ii) the accumulation of intermediate chloramines, which may exert ecological and toxicological risks, and (iii) the potential formation of halogenated byproducts such as trihalomethanes and haloacetic acids. In electrochemical systems, these challenges are partially mitigated by the in situ generation of active chlorine, yet they remain significant hurdles for practical water treatment applications.
3.2. Chlorine Radical Oxidation Mechanism
Beyond molecular chlorine species, chlorine radicals (Cl• and ClO•) generated under anodic polarization have been shown to play a decisive role in indirect electrochemical ammonia oxidation [33,34,35]. These highly reactive transient species possess higher oxidation potentials and exhibit faster reaction kinetics than HOCl or ClO− [33]. Experimental and computational studies suggest that Cl• can abstract hydrogen atoms directly from NH3, forming amidogen radicals (•NH2), which subsequently couple or react with chlorine-derived oxidants to yield N2. Similarly, ClO• can act through single-electron transfer pathways to facilitate ammonia dehydrogenation and radical chain propagation [12,36,37,38] (Figure 3b).
A key distinction between the breakpoint chlorination and chlorine radical mechanisms lies in the onset conditions and product distributions. Whereas breakpoint chlorination generally requires bulk Cl/N ratios >1.6, chlorine radical pathways can initiate ammonia oxidation even at much lower Cl/N ratios, often within the diffusion boundary layer of the anode, where radical concentrations are transiently high [39]. This helps explain why chloramines are sometimes undetectable during electrolysis, as the reactions occur within micrometer-scale regions before intermediates can diffuse into the bulk solution [37]. Nevertheless, the high reactivity of chlorine radicals can also trigger side reactions with coexisting organic matter or inorganic anions, increasing the risk of halogenated disinfection byproducts (DBPs) [40]. Balancing efficiency with selectivity, therefore, remains a critical challenge.
4. Fundamentals of the Design of Catalysts in eAOR
The efficiency and selectivity of eAOR are intrinsically governed by the properties of the electrocatalyst. In direct eAOR, catalysts must overcome sluggish multi-electron transfer kinetics, suppress electrode poisoning by nitrogen intermediates, and steer product distribution toward N2 rather than undesired NOₓ species. Noble metals such as Pt and Ir exhibit excellent intrinsic activity but suffer from high costs and stability issues, motivating the development of alloyed systems and earth-abundant transition metal alternatives. In contrast, indirect eAOR catalysts focus on promoting the CER over competing OER, stabilizing reactive chlorine species, and resisting chlorine-induced corrosion. Both pathways increasingly rely on advanced design concepts such as single-atom dispersion, heterostructure engineering, and operando-guided tuning of electronic structures. These principles form the foundation for translating laboratory insights into scalable catalytic systems capable of addressing the dual goals of pollutant removal and sustainable nitrogen management. To this end, Table 1 provides a performance comparison of representative metal-based catalysts.

Table 1.
Performance comparison of metal-based catalysts for direct eAOR.
4.1. Noble Metal-Based Catalysts for Direct eAOR
Noble metals have long been regarded as highly active electrocatalysts due to their favorable electronic structures, which enable effective adsorption and activation of reaction intermediates. In the context of direct eAOR, noble metal-based catalysts—especially those involving Pt, Ir, and their alloys—exhibit relatively high activity toward N2 formation, attributed to their strong affinity for adsorbed nitrogen species [48]. In contrast, coinage metals such as Cu, Ag, and Au exhibit weak binding energy for nitrogen species, rendering them catalytically inactive for NH3 dehydrogenation [49].
De Vooys et al. [50] systematically evaluated the performance of noble metals and demonstrated that 5d metals (Pt, Ir) possess relatively high steady-state activity for selective N2 production, whereas 4d metals (Ru, Rh, Pd) show limited catalytic ability toward this reaction. The coinage metals, though thermodynamically capable, are kinetically hindered and generally fail to initiate the dehydrogenation of ammonia. This difference is rooted in their respective binding energies toward nitrogen species, which dictate the desorption or further transformation of NHx intermediates on the catalyst surface.
Among noble metals, Pt has been widely recognized as one of the most efficient catalysts for direct eAOR. sts reveal that the Pt surface is prone to deactivation at this potential due to the accumulation of poisoning intermediates, which block active sites and hinder continuous catalytic turnover.
To gain deeper mechanistic insights, Ir-based catalysts have been investigated through in situ surface-sensitive techniques. Wei et al. [51] employed attenuated total reflection-surface enhanced infrared absorption spectroscopy (ATR-SEIRAS), a powerful tool capable of probing interfacial species at the molecular level [52,53]. The cyclic voltammetry (CV) profiles of Ir electrodes show two prominent oxidation peaks at approximately 0.2 V and 0.62 V vs. RHE (Figure 4a).
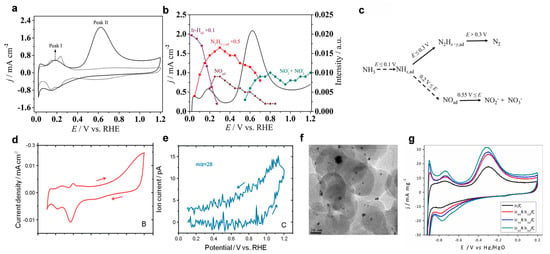
Figure 4.
(a) CV collected on the Ir film electrode in 1 M NH3 + 0.1 M NaOH at a scan rate of 20 mV s−1. (b) Potential-dependent intensity variation in adsorbed species (Ir-Hads, N2Hx+y,ads, NOads) and soluble products (NO2−, NO3−) on an Ir surface. (c) Proposed mechanism of the electrochemical AOR (eAOR) on an Ir electrode in alkaline electrolyte. Solid arrows indicate processes established by the spectral evidence; dotted arrows indicate those inferred from the Gerischer-Mauerer mechanism on Pt electrodes. Reprinted with permission from Ref. [51]. Copyright 2021, Elsevier. (d) CV curve and (e) MSCV signal for N2 (m/z = 28) of a Pd electrode in 1 M KOH + 0.1 M NH3. Reprinted with permission from Ref. [37]. Copyright 2011, American Chemical Society. (f) TEM image of the Ir50Rh50/C catalyst. (g) Comparative CV curves of Ir/C and IrxRh1−x/C catalysts in 1 M KOH + 2 M NH4OH. Reprinted with permission from Ref. [54]. Copyright 2017, Wiley-VCH.
In situ spectral analysis revealed that the low-potential peak (0.2 V) corresponds to the initial dehydrogenation of NH3, generating *H and NHₓ intermediates. These species can dimerize on the Ir surface to form *N2Hx+y. The higher-potential peak (0.6 V) is attributed to the further oxidation of these dimerized intermediates to form molecular N2 (Figure 4b). At potentials beyond 0.55 V vs. RHE, a competing pathway emerges, wherein *NHx is partially oxidized to NO2− or NO3−, reducing the N2 selectivity. The proposed eAOR mechanism on Ir surfaces involves dual potential-dependent routes: (1) the N2 formation pathway through NH3 dehydrogenation and N–N coupling at lower potentials, and (2) the oxygenated byproduct formation at higher potentials (Figure 4c).
Palladium (Pd), another 4d metal with strong *N adsorption, has also been evaluated for direct eAOR. However, both theoretical predictions and experimental data suggest low overall activity on Pd surfaces. CV measurements in 1 mol L−1 KOH (with and without 0.1 mol L−1 NH3) show weak anodic features (Figure 4d), consistent with its limited catalytic performance. Surprisingly, DEMS detected an N2 signal at m/z = 28 (Figure 4e), suggesting that N2 may still be formed Via surface-mediated reactions. Furthermore, when Pd electrodes were examined in a non-aqueous NH3-saturated acetonitrile solution, a noticeable increase in anodic current and N2 generation was observed. [55] These results suggest that surface oxygenated species may play a dual role: they can deactivate active Pd sites but may also facilitate partial NH3 oxidation under specific conditions, such as the surface coverage of oxygen.
To address the activity–stability trade-offs of single noble metals, alloying strategies have emerged as a promising approach. Silva et al. [54] reported the synthesis of IrRh nanoparticles supported on carbon substrates to optimize catalytic performance while reducing the reliance on expensive Ir. Ir, although catalytically competent and relatively resistant to poisoning of adsorbed nitrogen species, suffers from scarcity and cost issues. By forming IrRh alloys with tunable compositions (Figure 4f), the authors were able to adjust the electronic properties of the active sites. Electrochemical evaluation in 1 mol L−1 KOH + 2 mol L−1 NH4OH revealed that certain Ir/Rh ratios led to a pronounced enhancement in peak current density compared to monometallic Ir/C catalysts (Figure 4g). This performance enhancement is likely due to synergistic effects between Ir and Rh atoms, which modulate the binding strength for nitrogen species and facilitate the desorption of final products.
In summary, noble metal-based catalysts, particularly Pt and Ir, exhibit superior catalytic activity toward direct eAOR owing to their favorable energetics for nitrogen species and their ability to mediate multi-electron transfer processes. However, issues such as intermediate poisoning, limited selectivity, and high material cost constrain their large-scale application. Strategies including alloy engineering (e.g., IrRh) and the utilization of in situ diagnostic techniques (e.g., ATR-SEIRAS, DEMS) provide valuable insights and design directions for developing next-generation direct eAOR catalysts with enhanced activity, selectivity, and stability.
4.2. Non-Noble Metal in Direct eAOR
Direct eAOR holds significant promise for environmental remediation and energy applications, but the widespread use of noble metal catalysts such as Pt is severely limited by their high cost and susceptibility to rapid deactivation, especially due to poisoning by nitrogen intermediates [41]. Consequently, the development of low-cost, stable, and efficient non-noble metal electrocatalysts has become a critical research frontier. Among these, nickel (Ni)-based catalysts have attracted considerable attention due to their earth abundance, low cost, chemical stability, and versatility in surface modification, making them highly promising candidates for eAOR (Figure 5a) [43,56,57,58,59].
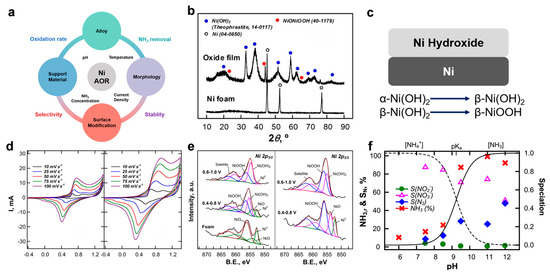
Figure 5.
(a) Key parameters and strategies for designing efficient Ni-based catalysts toward eAOR. (b) XRD patterns of Ni foam and electrochemically oxidized Ni(OH)2/NiOOH. (c) Proposed mechanism of in situ-generated Ni hydroxide for enhanced eAOR. (d) Voltammetric responses of Ni foam in 0.1 M Na2SO4 without and with 3 mM NH3. (e) Ni 2p3/2 XPS spectra of pristine Ni foam and after CV cycling (100 cycles at 50 mV/s, pH 11) in 0.1 M Na2SO4 with and without 10 mM NH3. Reprinted with permission from Ref. [43]. Copyright 2018, Elsevier. (f) Ammonia removal efficiency and product selectivity (SN2, NO2−, NO3−) as a function of pH after 7 h of electrolysis on Ni foam. Reprinted with permission from Ref. [42]. Copyright 2018, Elsevier.
Despite the advantageous properties of Ni, pristine Ni metal exhibits inherently low catalytic activity for ammonia oxidation in alkaline media. This low activity primarily arises from the spontaneous formation of a surface nickel oxide (NiO) or hydroxide layer, which adversely affects ammonia adsorption and activation, as explained by valence bond orbital theory. The presence of NiO reduces the binding affinity for NH3 molecules, thus limiting their effective oxidation. Moreover, the oxidation potential of Ni overlaps with that of NH3, further hindering direct catalytic activity [60,61]. Initial experimental studies, such as those employing Raney Ni electrodes, confirmed these intrinsic limitations of pure Ni under typical eAOR conditions [62].
Recent mechanistic studies have demonstrated that the true active catalytic species are not the metallic Ni itself but rather the electrochemically generated nickel hydroxides and oxyhydroxides, specifically Ni(OH)2 and NiOOH, which form on the electrode surface under anodic polarization (Figure 5b). The electrochemical cycling induces a phase transformation sequence: metallic Ni → NiOx → α-Ni(OH)2 → β-Ni(OH)2 → β-NiOOH (Figure 5c). These oxyhydroxide phases possess favorable electronic and structural properties that facilitate the adsorption and oxidation of ammonia, promoting its conversion into nitrogen (N2) and oxidized nitrogen species such as nitrite (NO2−) and nitrate (NO3−).
The catalytic activity enhancement is attributed to the redox cycling between Ni(II) and Ni(III) states in NiOOH, which mediates electron transfer processes and enables the formation of key reaction intermediates such as NHx monolayers. Electrochemical characterization, including CV, confirms that the formation of NHx intermediates occurs prominently within the potential range of 0.6–0.8 V vs. Hg/HgO, where N2 selectivity is maximized, whereas higher potentials tend to favor the generation of NO2− and NO3− (Figure 5d). Advanced spectroscopic methods such as X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy have corroborated the presence and dynamic transformation of these nickel oxyhydroxide species during the reaction (Figure 5e).
The efficiency and selectivity of Ni-based catalysts for eAOR are highly sensitive to reaction parameters such as pH, temperature, and applied current density [42,43]. Experimental evidence indicates that alkaline conditions (pH > 9.5) favor ammonia oxidation with enhanced N2 selectivity due to the predominance of un-ionized NH3 species (Figure 5f). Elevated temperatures generally promote higher N2 selectivity while reducing nitrite and nitrate yields, likely due to thermodynamic and kinetic effects on the oxygen evolution and nitrogen evolution reactions. Moreover, lower current densities tend to favor N2 formation, whereas higher current densities increase NO3− selectivity but suppress N2 generation. These trends reflect the complex interplay between electrochemical potential, surface adsorbates, and competing reaction pathways.
To overcome the intrinsic activity limitations of pure Ni catalysts, alloying with other transition metals has emerged as an effective strategy to tailor the electronic structure, surface adsorption properties, and morphology of Ni-based catalysts. Among various candidates, copper (Cu) has been identified as an optimal alloying element due to its complementary electronic effects and catalytic behavior [63]. NiCu alloys, particularly in the form of layered double hydroxides (LDHs) or double hydroxides (DHs), have been synthesized with diverse morphologies such as nanoflowers, nanonets, and nanotyres [64,65,66,67,68]. The incorporation of Cu enhances catalytic activity by facilitating charge transfer, increasing the electrochemically active surface area (ECSA), and modulating the adsorption energies of reaction intermediates. The synergistic effect arises because NiOOH serves as a conductive backbone enabling rapid electron transport, while Cu(OH)2 sites modify the reaction pathways, favoring the formation of intermediates such as *NO2 and ultimately leading to higher selectivity toward NO2− and NO3− products.
While NiCu alloys are desirable in producing NO2− and NO3−, their ability to selectively generate N2 remains limited [44,45,69,70]. To address this, the incorporation of a third element, such as cobalt (Co), has been explored. Co exhibits strong bonding with nitrogen but weak interaction with hydrazine-like intermediates, facilitating the dimerization of NHx species into N2 rather than partial oxidation to NOx species [71]. NiCo and NiCoCu ternary alloys, including layered double hydroxides and metal nitrides (e.g., NiCo2N), have demonstrated remarkable improvements in N2 selectivity, often exceeding 60–90%. These materials balance electronic structure to optimize NH3 adsorption, NHx radical formation, and final N2 desorption [13,46,72]. DFT calculations and advanced spectroscopic characterizations reveal that moderate contraction of the d-band center enhances metal-NH3 bonding and stabilizes key cation radical intermediates essential for N2 formation, while suppressing side reactions that lead to soluble metal-amine complexes [73,74].
Furthermore, high-entropy alloys and spinel oxides containing multiple transition metals (Mn, Fe, Co, Ni, Cu) have shown promise due to their unique electronic structures, which provide abundant active sites and optimal binding energies for efficient and selective ammonia oxidation to N2 [47].
4.3. Fundamentals of the Design of Catalysts in Indirect eAOR
The design of electrocatalysts for indirect eAOR must address two often competing objectives: maximizing the generation of RCS for efficient ammonia degradation, while minimizing energy consumption and the formation of toxic byproducts. Unlike direct eAOR, which relies on the intrinsic activity of the catalyst surface toward NH3 adsorption and dehydrogenation, indirect eAOR is mediated by chlorine-containing oxidants electrogenerated at the anode. This distinction necessitates a unique set of material design principles, emphasizing stability under harsh oxidative conditions, selective chlorine evolution, and resistance to corrosion. To evaluate the efficiency of different anodic materials in meeting these design criteria, Table 2 summarizes the performance of various anodes for ammonia oxidation reported in different studies.
4.3.1. Noble Metal and Metal Oxide Anodes
Active chlorine and chlorine radicals are produced at high anodic potentials, requiring materials with high oxidation resistance. Noble metal-based anodes, such as Pt, Ir, Ru, and their oxides, have been widely adopted due to their excellent conductivity, catalytic activity, and corrosion stability [75,76,77,78]. For instance, Ti/RuO2 and Ti/IrO2–RuO2 are among the most commonly used dimensionally stable anodes (DSAs) in chloride-mediated eAOR systems. Zhang et al. [79] demonstrated that a Ti/IrO2–RuO2 anode could remove 50 mg L−1 of ammonia within 30 min (Figure 6a). Similarly, PbO2/Ti anodes have shown promise, achieving over 85% removal of both COD and ammonia within 6 h [36]. Recent studies suggest that Ti-, Ru-, and Ir-based anodes may exhibit higher active chlorine production efficiencies compared to Pt or Pb anodes [80]. Novel non-noble metal anodes, such as WO3–Sb/SnO2 and 3D Co3O4 nanowires, have also been developed, demonstrating the complete degradation of ammonia and phenolic compounds within 45–60 min (Figure 6b) [81,82]. To improve the electron transfer performance in indirect eAOR, Xu et al. [83] used the MXene substrate to support the Co3O4 catalysts, and the obtained Co3O4/MXene anode exhibited 97.8% ammonium removal within 5 h. In addition, the Co3O4/MXene electrode exhibited exceptional stability and achieved desirable degradation efficiency for actual tannery wastewater, with the desirable Faradaic Efficiency (24.12%) and energy consumption (85 kWh/kg-N). Wu et al. [84] demonstrated that micro-doping Co3O4 with Sn and Bi effectively enhances its CER performance by modulating the electronic structure. This synergistic doping creates electron-deficient Co(III) sites, enriches surface Cl−, and ultimately leads to superior catalytic activity, selectivity, and long-term stability.
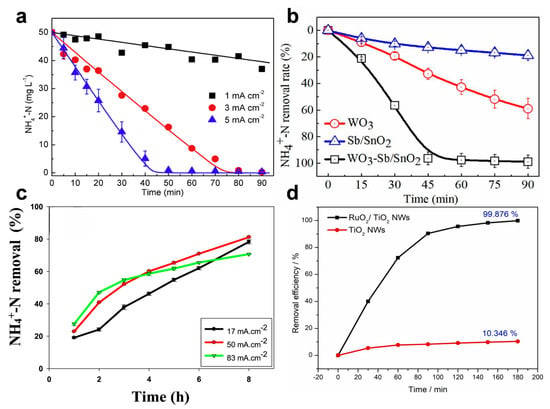
Figure 6.
(a) NH4+-N removal under different current densities with an initial Cl− concentration of 10 mM and pH = 6.0. Reprinted with permission from Ref. [79]. Copyright 2018, Elsevier. (b) Ammonia removal rates under the following conditions: pH = 5.0, 50 mM NaCl, and 50 mM Na2SO4, 2.0 V vs. Ag/AgCl. Reprinted with permission from Ref. [82]. Copyright 2020, Elsevier. (c) Ammonium (NH4+-N) elimination efficiency using Ti/BDD anodes. Reprinted with permission from Ref. [85]. Copyright 2020, Elsevier. (d) Comparison of ammonia nitrogen degradation by RuO2/TiO2 NWs and pure TiO2 NWs electrodes at 10 mA cm−2 for 60 min. Reprinted with permission from Ref. [80]. Copyright 2022, Elsevier.
4.3.2. Non-Metal and Diamond-Based Electrodes
Non-metal anodes, particularly boron-doped diamond (BDD), offer a wide potential window (up to 4.5 V), high oxygen evolution potential, and exceptional electrochemical stability. These properties make BDD highly suitable for indirect eAOR, especially in complex water matrices. Luu [85] reported that a Ti/BDD anode achieved over 80% ammonia removal from landfill leachate (initial concentration 1006–1197 mg L−1) after 8 h of treatment (Figure 6c). Although BDD may show slightly lower ammonia removal efficiency compared to Ti/RuO2, it often outperforms metal oxides in COD removal. Díaz et al. [86] demonstrated complete removal of 8 mg L−1 ammonia within 45 min using a BDD anode. Other non-metal materials like graphite have been less widely adopted due to higher wear rates and lower efficiency in chlorine production [87].
4.3.3. Membrane Electrodes and Hybrid Materials
Membrane electrodes have gained attention for their large surface area and efficient contact with wastewater, enabling enhanced mass transfer and faster ammonia oxidation rates. These electrodes function similarly to filter membranes and are often used in electrochemical filtration systems. Sun et al. [80] developed a RuO2/TiO2 nanowire (NW) membrane anode that achieved 99.88% ammonia oxidation within 3 h under optimized conditions (200 mg L−1 NH3, 2000 mg L−1 Cl−, 20 mA cm−2, pH 9) (Figure 6d). The electrode also exhibited remarkable stability over 100 h of continuous operation. Pan et al. [88] constructed an electrochemical membrane reactor (EMR) featuring a coal-based carbon membrane as a flow-through anode for ammonia nitrogen wastewater decontamination. Under optimized conditions, the system achieved complete ammonia nitrogen removal with a low energy consumption of 0.0380 kWh/g-NH4-N, demonstrating significant potential for practical application. To balance corrosion resistance and plasticity, recent studies have explored hybrid structures incorporating noble metals into low-cost substrates. For example, Chi et al. [89] electrodeposited RuO2 nanospheres on lignin-derived carbon, achieving a low resistance of 0.91 Ω and a high current density of 129 mA cm−2 at 1.2 V. Li et al. [90] fabricated a Ti/Ir-coated ceramic membrane anode that removed 93.5% of ammonia and 69.4% of dissolved organic nitrogen within 60 min, demonstrating stability over 14 days of continuous operation.
In summary, the indirect eAOR catalysts must (i) favor the chlorine evolution reaction (CER) over the OER, (ii) anode materials must withstand prolonged exposure to corrosive RCS, (iii) the balance between molecular chlorine (HOCl/ClO−) and radicals (Cl•, ClO•) should be considered for modulate the ammonia oxidation efficiency and byproduct formation, (iv) high-surface-area structures (e.g., nanowires, nanospheres, porous membranes) are important for enhancing mass transport and active site availability, improving overall catalytic performance. That is, the design of catalysts for indirect eAOR requires a multidisciplinary approach that integrates materials science, electrochemistry, and environmental engineering. Future efforts should focus on developing low-cost, high-efficiency, and durable anodes that can operate effectively across diverse water quality conditions while minimizing energy input and secondary pollution.
Table 2.
Different anodes for ammonia oxidation and their effect on ammonia oxidation in different studies.
Table 2.
Different anodes for ammonia oxidation and their effect on ammonia oxidation in different studies.
| Anode Material | Rate Constant or Removal Efficiency | Concentration of Ammonia | Concentration of Chlorine | Current Density/Potential | References |
|---|---|---|---|---|---|
| Ti/IrO2–RuO2 | 100%/25 min | 50 mg L−1 | 20 mM | 5 mA cm−2 | [79] |
| PbO2/Ti | 0.306 h−1 | 78.2 mg L−1 | 40 mM | 37.5 mA cm−2 | [36] |
| WO3-Sb/SnO2 | 3.516 h−1 | 30 mg L−1 | 50 mM | 2.0 V (VS Ag/AgCl) | [82] |
| 3D Co3O4 NWs | 4.836 h−1 | 56 mg L−1 | 50 mM | 1.8 V (VS Ag/AgCl) | [81] |
| Co3O4/MXene | 0.6059 h−1 | 200 mg L−1 | 50 mM | 20 mA cm−2 | [83] |
| Sn, Bi co-doped Co3O4 | 0.0055 min−1 | 100 mg L−1 | 600 mM | 10 mA cm−2 | [84] |
| Ti/BDD | >80%/480 min | 1006–1197 mg L−1 | 1880–2700 mg L−1 | 17–83 mA cm−2 | [85] |
| BDD | 100%/45 min | 8 mg L−1 | 26167 mg L−1 | 50 A m−2 | [86] |
| RuO2/TiO2 NW membrane | 99.876%/180 min | 200 mg L−1 | 2000 mg L−1 | 20 mA cm−2 | [80] |
| coal-based carbon membrane | 100%/120 min | 30 mg L−1 | 100 mM | 2.8 V | [88] |
| Ceramic membrane | 93.5%/60 min | 8.8 mg L−1 | 1781.9 mg L−1 | 25 mA cm−2 | [90] |
5. Key Environmental Factors in Practical Water Treatment
The practical application of electrochemical ammonia oxidation (eAOR) is governed not only by catalyst properties but also by environmental parameters that dictate the kinetics, selectivity, and byproduct profiles of both direct eAOR and indirect, chlorine-mediated eAOR. Understanding these environmental influences is essential to bridging the gap between laboratory-scale studies and real-world water treatment systems.
5.1. pH Dependence and Speciation Effects
In direct pathways, ammonia oxidation strongly depends on the acid–base equilibrium of ammonia (pKa ≈ 9.25). At alkaline pH, NH3 is the dominant species, which adsorbs readily onto electrode surfaces and undergoes dehydrogenation according to the Oswin-Salomon or Gerischer–Mauerer mechanisms [18,19]. Alkaline conditions also favor OH−-assisted proton–electron transfer steps, lowering the onset potential for ammonia oxidation and improving selectivity toward N2. Conversely, in acidic or neutral environments, ammonia exists mainly as NH4+, which adsorbs poorly on metal surfaces, suppressing direct eAOR efficiency.
For chlorine-mediated oxidation, pH controls both the speciation of chlorine (HOCl vs. ClO−) and ammonia (NH3 vs. NH4+). HOCl, predominant at near-neutral pH, is a much stronger oxidant than ClO− and react sefficiently with NH4+ to form chloramines [91]. Acidic conditions, therefore, favor breakpoint chlorination. In contrast, alkaline solutions promote the formation of ClO• radicals and direct NH3 attack by RCS, enhancing radical-driven pathways [92]. However, too high alkalinity lowers the reactivity of active chlorine and shifts selectivity toward nitrate formation [93]. Hence, optimal performance often requires pH tuning to balance reactivity, selectivity, and byproduct control.
5.2. Formation of Nitrogenous and Halogenated Byproducts
The principal challenge for direct oxidation is the generation of nitrogen oxides (NO2−, NO3−, N2O) at elevated anodic potentials. While noble metal catalysts such as Pt and Ir can selectively yield N2 at moderate potentials, higher bias leads to overoxidation of intermediates and accumulation of undesired NOx species [51,94]. These not only reduce nitrogen selectivity but also pose environmental risks. Maintaining potential windows and designing catalysts that stabilize N–N coupling intermediates are critical for minimizing NOx formation.
In chlorine-mediated pathways, the most concerning byproducts are chloramines (NH2Cl, NHCl2, NCl3), oxychlorine species (ClO3−, ClO4−), and halogenated organics (e.g., trihalomethanes, haloacetic acids). Their formation is influenced by chlorine dosage (Cl/N ratio), current density, NOM content, and electrolysis duration. For example, exceeding the breakpoint Cl/N ratio significantly increases DBP generation [95]. Advanced catalysts that favor radical oxidation over molecular chlorine pathways and integrated treatment trains (e.g., adsorption, biological polishing) are being explored to mitigate these risks.
5.3. Background Matrix Effects and Coexisting Constituents
Inorganic ions such as sulfate, carbonate, and phosphate can influence direct eAOR by modifying the electrolyte conductivity, buffering capacity, and electrode surface chemistry. Carbonate/bicarbonate ions may alter local pH near the electrode, indirectly affecting NH3 adsorption. Transition metal ions or heavy metals present in industrial effluents can poison the active sites of catalysts, further reducing their stability and efficiency.
In chlorine-mediated systems, background constituents play a more pronounced role because many species can scavenge reactive chlorine. Bicarbonate (HCO3−) reacts with chlorine radicals to form CO3•−, which has much lower reactivity toward ammonia [96]. Nitrite (NO2−) competes strongly with NH3 for HOCl, reducing ammonia oxidation rates while forming secondary nitrogenous DBPs [97]. Natural organic matter (NOM) is a particularly strong competitor, consuming HOCl and radicals and acting as a precursor to halogenated DBPs [98]. Consequently, in natural waters with high NOM or alkalinity, chlorine-mediated eAOR efficiency is often lower than in synthetic laboratory solutions. In real water treatment scenarios, the selection between direct and indirect eAOR—or a hybrid strategy—should be guided by the specific water matrix. Direct eAOR is more suitable for chloride-deficient streams (e.g., inland industrial effluents), whereas indirect eAOR is advantageous in saline or chloride-rich wastewaters (e.g., aquaculture, coking wastewater, seawater-based systems). However, both are strongly conditioned by environmental parameters such as pH, coexisting ions, and NOM. The integration of pretreatment (e.g., NOM removal), operational control (pH and potential optimization), and post-treatment (adsorption or biological polishing) is necessary to ensure high ammonia removal efficiency with minimal byproduct risks.
6. Conclusions and Outlook
Electrochemical ammonia oxidation (eAOR) has emerged as a versatile and promising strategy for nitrogen removal and transformation in water treatment, benefiting from its flexibility, environmental compatibility, and capacity for integration with renewable electricity. Both direct eAOR and indirect, chlorine-mediated eAOR play pivotal roles depending on water chemistry, catalyst design, and treatment goals. Direct eAOR operates primarily via surface adsorption and stepwise dehydrogenation of NH3 under alkaline conditions, and Pt and Ir offer high intrinsic activity but suffer from cost and poisoning, while Ni (in its NiOOH form) and NiCo alloys are promising non-noble alternatives due to their optimal d-band center for facilitating N–N coupling. This pathway offers the advantage of avoiding halogenated byproducts but is challenged by sluggish kinetics, poisoning of electrode surfaces by *N intermediates, and competing side reactions that yield NO2−, NO3−, or N2O. Recent advances in catalyst design—including noble metal alloys, transition metal oxides, and single-atom catalysts—have improved activity and nitrogen selectivity, yet durability and cost remain pressing concerns.
Indirect eAOR, mediated by free chlorine and reactive chlorine species (RCS), leverages the abundance of chloride in many wastewaters. And the crucial properties are high CER selectivity and corrosion resistance. RuO2 and IrO2-based DSAs are benchmarks, while non-noble options like Co3O4 and doped diamond (BDD) are emerging for their stability and radical-generation capabilities. Through mechanisms of breakpoint chlorination and radical oxidation, indirect pathways provide rapid and effective ammonia degradation, even at low concentrations. However, their reliance on reactive chlorine introduces environmental challenges: chloramine accumulation, oxychlorine formation (ClO3−, ClO4−), and halogenated organic byproducts (e.g., THMs, HAAs). Catalyst innovations that tune the balance between free chlorine and chlorine radicals, suppress side reactions, and enhance corrosion resistance are central to improving the sustainability of this approach.
Looking forward, several key research directions can be identified:
Mechanistic Elucidation and In Situ Characterization. Despite progress, the detailed interplay between direct electron transfer, free chlorine, and chlorine radicals remains insufficiently understood. Advanced operando spectroscopies (e.g., Raman, X-ray absorption, EPR) and computational studies are needed to capture short-lived intermediates and map out the competition between nitrogen–nitrogen coupling and overoxidation pathways.
Catalyst Development for Activity, Selectivity, and Stability. Rationally designed catalysts that stabilize key intermediates (*NH2, *N–N) in direct eAOR and precisely control radical flux in indirect eAOR are critical. Strategies such as single-atom anchoring, defect engineering, and heterostructure interfaces hold promise for achieving both efficiency and longevity, while minimizing noble metal dependence.
Byproduct Minimization and Process Integration. Addressing the generation of NOx (in direct eAOR) and halogenated DBPs (in indirect eAOR) remains a major bottleneck. Coupling eAOR with downstream adsorption, ion exchange, advanced oxidation, or biological polishing could provide synergistic mitigation. Integrated processes must be tailored to the water matrix to ensure compliance with water quality standards. In addition, the future of eAOR lies not only in pollutant removal but also in resource recovery. Coupling ammonia oxidation with value-added product generation could transform eAOR from a remediation strategy into a sustainable resource management platform, such as two prominent examples of co-production of hydrogen and the recovery of nitrate.
System-Level and Engineering Considerations. Beyond electrode materials, the scalability of eAOR depends on reactor design, energy efficiency, and operational robustness. Three-dimensional porous electrodes, flow-through architectures, and modular electrochemical cells can improve mass transfer and reduce energy input. Life-cycle assessments and techno-economic analyses will be essential to evaluate the feasibility of real-world deployment.
Toward Sustainable and Resource-Oriented Treatment. The future of eAOR lies not only in pollutant removal but also in resource recovery. Coupling ammonia oxidation with hydrogen production, nitrogen fixation to value-added chemicals, or integration into circular nitrogen cycles could transform eAOR from a remediation strategy into a sustainable resource management platform.
In conclusion, electrochemical ammonia oxidation offers a powerful toolbox for addressing nitrogen pollution under diverse water treatment scenarios. The complementary roles of direct and indirect eAOR—one offering halogen-free selectivity, the other leveraging chloride-mediated reactivity—underscore the need for adaptive and context-specific approaches. Through advances in mechanistic understanding, materials innovation, process integration, and system-level optimization, eAOR has the potential to become a cornerstone technology for sustainable nitrogen management in the era of global water challenges.
Author Contributions
X.S., conceptualization, formal analysis, validation, writing—original draft; F.M., funding acquisition, review, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (No. 52070054) and the State Key Laboratory of Urban-rural Water Resources and Environment (Harbin Institute of Technology) (No. 2025DX17). The APC was funded by (No. 52070054).
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Acknowledgments
The authors sincerely appreciate the anonymous reviewer and editors’ valuable comments and suggestions to improve the manuscript.
Conflicts of Interest
The authors declare no competing financial interests.
References
- Wen, Z.; Huang, B.; Wang, Y.; Wang, K.; Tu, X.; Xie, P.; Fu, X. Ammonia as a renewable energy carrier from synthesis to utilization. Nat. Rev. Clean Technol. 2025, 1–16. [Google Scholar] [CrossRef]
- Edwards, T.M.; Puglis, H.J.; Kent, D.B.; Durán, J.L.; Bradshaw, L.M.; Farag, A.M. Ammonia and aquatic ecosystems—A review of global sources, biogeochemical cycling, and effects on fish. Sci. Total Environ. 2024, 907, 167911. [Google Scholar] [CrossRef]
- Mishra, S.; Singh, V.; Cheng, L.; Hussain, A.; Ormeci, B. Nitrogen removal from wastewater: A comprehensive review of biological nitrogen removal processes, critical operation parameters and bioreactor design. J. Environ. Chem. Eng. 2022, 10, 107387. [Google Scholar] [CrossRef]
- He, Y.; Liu, Y.; Yan, M.; Zhao, T.; Liu, Y.; Zhu, T.; Ni, B.-J. Insights into N2O turnovers under polyethylene terephthalate microplastics stress in mainstream biological nitrogen removal process. Water Res. 2022, 224, 119037. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, S.; Li, H.; Liu, J.; Li, S.; Zhang, L. Treatment of ammonia-nitrogen wastewater by the ultrasonic strengthened break point chlorination method. J. Water Process Eng. 2022, 45, 102501. [Google Scholar] [CrossRef]
- Zhang, X.; Ren, P.; Zhou, J.; Li, J.; Li, Z.; Wang, D. Formation of disinfection byproducts in an ammonia-polluted source water with UV/chlorine treatment followed by post-chlorination: A pilot-scale study. Environ. Technol. Innov. 2022, 26, 102266. [Google Scholar] [CrossRef]
- Han, B.; Butterly, C.; Zhang, W.; He, J.-Z.; Chen, D. Adsorbent materials for ammonium and ammonia removal: A review. J. Clean. Prod. 2021, 283, 124611. [Google Scholar] [CrossRef]
- Almomani, F.; Bhosale, R.; Khraisheh, M.; Kumar, A.; Tawalbeh, M. Electrochemical oxidation of ammonia on nickel oxide nanoparticles. Int. J. Hydrogen Energy 2020, 45, 10398–10408. [Google Scholar] [CrossRef]
- Zöllig, H.; Fritzsche, C.; Morgenroth, E.; Udert, K.M. Direct electrochemical oxidation of ammonia on graphite as a treatment option for stored source-separated urine. Water Res. 2015, 69, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Rahardjo, S.S.P.; Shih, Y.-J. Electrochemical characteristics of silver/nickel oxide (Ag/Ni) for direct ammonia oxidation and nitrogen selectivity in paired electrode system. Chem. Eng. J. 2023, 452, 139370. [Google Scholar] [CrossRef]
- Chauhan, R.; Srivastava, V.C. Mechanistic kinetic modeling of simultaneous electrochemical nitrate reduction and ammonium ion oxidation in wastewater. Chem. Eng. Sci. 2022, 247, 117025. [Google Scholar] [CrossRef]
- Yan, C.; Liu, L. Oxidation of gas phase ammonia via accelerated generation of radical species and synergy of photo electrochemical catalysis with persulfate activation by CuO-Co3O4 on cathode electrode. J. Hazard. Mater. 2020, 388, 121793. [Google Scholar] [CrossRef]
- Shih, Y.-J.; Hsu, C.-H. Kinetics and highly selective N2 conversion of direct electrochemical ammonia oxidation in an undivided cell using NiCo oxide nanoparticle as the anode and metallic Cu/Ni foam as the cathode. Chem. Eng. J. 2021, 409, 128024. [Google Scholar] [CrossRef]
- Hansen, H.A.; Man, I.C.; Studt, F.; Abild-Pedersen, F.; Bligaard, T.; Rossmeisl, J. Electrochemical chlorine evolution at rutile oxide (110) surfaces. Phys. Chem. Chem. Phys. 2010, 12, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Candido, L.; Gomes, J.A.C.P. Evaluation of anode materials for the electro-oxidation of ammonia and ammonium ions. Mater. Chem. Phys. 2011, 129, 1146–1151. [Google Scholar] [CrossRef]
- Elnabawy, A.O.; Herron, J.A.; Karraker, S.; Mavrikakis, M. Structure sensitivity of ammonia electro-oxidation on transition metal surfaces: A first-principles study. J. Catal. 2021, 397, 137–147. [Google Scholar] [CrossRef]
- Siddharth, K.; Alam, P.; Hossain, M.D.; Xie, N.; Nambafu, G.S.; Rehman, F.; Lam, J.W.Y.; Chen, G.; Cheng, J.; Luo, Z.; et al. Hydrazine Detection during Ammonia Electro-oxidation Using an Aggregation-Induced Emission Dye. J. Am. Chem. Soc. 2021, 143, 2433–2440. [Google Scholar] [CrossRef] [PubMed]
- Oswin, H.G.; Salomon, M. The Anodic Oxidation of Ammonia at Platinum Black Electrodes in Aqueous Koh Electrolyte. Can. J. Chem. 1963, 41, 1686–1694. [Google Scholar] [CrossRef]
- Gerischer, H.; Mauerer, A. Untersuchungen Zur anodischen Oxidation von Ammoniak an Platin-Elektroden. J. Electroanal. Chem. Interfacial Electrochem. 1970, 25, 421–433. [Google Scholar] [CrossRef]
- Katsounaros, I.; Chen, T.; Gewirth, A.A.; Markovic, N.M.; Koper, M.T.M. Evidence for Decoupled Electron and Proton Transfer in the Electrochemical Oxidation of Ammonia on Pt(100). J. Phys. Chem. Lett. 2016, 7, 387–392. [Google Scholar] [CrossRef]
- de Vooys, A.C.A.; Mrozek, M.F.; Koper, M.T.M.; van Santen, R.A.; van Veen, J.A.R.; Weaver, M.J. The nature of chemisorbates formed from ammonia on gold and palladium electrodes as discerned from surface-enhanced Raman spectroscopy. Electrochem. Commun. 2001, 3, 293–298. [Google Scholar] [CrossRef]
- Kim, H.; Yang, W.; Lee, W.H.; Han, M.H.; Moon, J.; Jeon, C.; Kim, D.; Ji, S.G.; Chae, K.H.; Lee, K.-S.; et al. Operando Stability of Platinum Electrocatalysts in Ammonia Oxidation Reactions. ACS Catal. 2020, 10, 11674–11684. [Google Scholar] [CrossRef]
- Vidal-Iglesias, F.J.; Solla-Gullón, J.; Feliu, J.M.; Baltruschat, H.; Aldaz, A. DEMS study of ammonia oxidation on platinum basal planes. J. Electroanal. Chem. 2006, 588, 331–338. [Google Scholar] [CrossRef]
- Wasmus, S.; Vasini, E.J.; Krausa, M.; Mishima, H.T.; Vielstich, W. DEMS-cyclic voltammetry investigation of the electrochemistry of nitrogen compounds in 0.5 M potassium hydroxide. Electrochim. Acta 1994, 39, 23–31. [Google Scholar] [CrossRef]
- Gootzen, J.F.E.; Wonders, A.H.; Visscher, W.; van Santen, R.A.; van Veen, J.A.R. A DEMS and cyclic voltammetry study of NH3 oxidation on platinized platinum. Electrochim. Acta 1998, 43, 1851–1861. [Google Scholar] [CrossRef]
- Siddharth, K.; Chan, Y.; Wang, L.; Shao, M. Ammonia electro-oxidation reaction: Recent development in mechanistic understanding and electrocatalyst design. Curr. Opin. Electrochem. 2018, 9, 151–157. [Google Scholar] [CrossRef]
- Bunce, N.J.; Bejan, D. Mechanism of electrochemical oxidation of ammonia. Electrochim. Acta 2011, 56, 8085–8093. [Google Scholar] [CrossRef]
- He, J.; Zhang, C.; Yang, Y.; Kang, J.; Zhang, C.; He, D.; Ma, J. Chlorine-Mediated Ammonia and Organics Transformation during Electrochemical Ammonia Recovery from Human Urine. Environ. Sci. Technol. 2025, 59, 13096–13107. [Google Scholar] [CrossRef]
- Qiang, Z.; Adams, C.D. Determination of Monochloramine Formation Rate Constants with Stopped-Flow Spectrophotometry. Environ. Sci. Technol. 2004, 38, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Chen, X.; Zhang, S.; Wu, D. Treatment of high chlorine-containing composting leachate biochemical effluent by Ti/RuO2-IrO2 anodic electrochemical oxidation: Optimization and evolution of pollutants. J. Environ. Chem. Eng. 2023, 11, 109674. [Google Scholar] [CrossRef]
- Deng, Y.; Chen, N.; Feng, C.; Chen, F.; Wang, H.; Kuang, P.; Feng, Z.; Liu, T.; Gao, Y.; Hu, W. Treatment of organic wastewater containing nitrogen and chlorine by combinatorial electrochemical system: Taking biologically treated landfill leachate treatment as an example. Chem. Eng. J. 2019, 364, 349–360. [Google Scholar] [CrossRef]
- Liu, Z.; Tao, Y.; Zhang, Z.; He, J.; Yang, K.; Ma, J. Active chlorine mediated ammonia oxidation in an electrified SnO2–Sb filter: Reactivity, mechanisms and response to matrix effects. Sep. Purif. Technol. 2023, 312, 123369. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Bai, J.; Li, L.; Chen, S.; Zhou, T.; Wang, J.; Xia, L.; Xu, Q.; Zhou, B. Extremely Efficient Decomposition of Ammonia N to N2 Using ClO• from Reactions of HO• and HOCl Generated in Situ on a Novel Bifacial Photoelectroanode. Environ. Sci. Technol. 2019, 53, 6945–6953. [Google Scholar] [CrossRef]
- Yan, Z.; Dai, Z.; Zheng, W.; Lei, Z.; Qiu, J.; Kuang, W.; Huang, W.; Feng, C. Facile ammonium oxidation to nitrogen gas in acid wastewater by in situ photogenerated chlorine radicals. Water Res. 2021, 205, 117678. [Google Scholar] [CrossRef]
- Lu, S.; Li, X.; Liao, Y.; Zhang, Z.; Luo, H.; Zhang, G. Boosting generation of reactive oxygen and chlorine species on TNT photoanode and Ni/graphite fiber cathode towards efficient oxidation of ammonia wastewater. Chemosphere 2023, 313, 137363. [Google Scholar] [CrossRef]
- Zheng, W.; Zhu, L.; Liang, S.; Ye, J.; Yang, X.; Lei, Z.; Yan, Z.; Li, Y.; Wei, C.; Feng, C. Discovering the Importance of ClO• in a Coupled Electrochemical System for the Simultaneous Removal of Carbon and Nitrogen from Secondary Coking Wastewater Effluent. Environ. Sci. Technol. 2020, 54, 9015–9024. [Google Scholar] [CrossRef]
- Li, F.; Peng, X.; Liu, Y.; Mei, J.; Sun, L.; Shen, C.; Ma, C.; Huang, M.; Wang, Z.; Sand, W. A chloride-radical-mediated electrochemical filtration system for rapid and effective transformation of ammonia to nitrogen. Chemosphere 2019, 229, 383–391. [Google Scholar] [CrossRef]
- Chen, X.; Dai, T.; Yin, M.-Y.; Xia, X.-Y.; Xing, Q.-J.; Tian, L.; Zou, J.-P. Enhanced anodic mass transfer enables interfacial Cl• for efficient ammonia oxidation. Chin. Chem. Lett. 2025, 111445. [Google Scholar] [CrossRef]
- Yuan, K.-X.; Wu, Q.; Hu, K.; Liu, Y.-L.; Wang, W.; Feng, H.; Liu, Y.; Bao, X.; Ma, J. Harnessing Electrochemistry Synergy in Reverse Osmosis: Modulating Ammonium Localized Oxidation and Restricted Transport. Environ. Sci. Technol. 2025, 59, 4188–4198. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, H.; Wang, R.; Hu, Q.; Zhang, Y.; Wang, Z.; Zhou, J.; Qu, G.; Wang, T.; Jia, H.; et al. Underlying mechanisms of promoted formation of haloacetic acids disinfection byproducts after indometacin degradation by non-thermal discharge plasma. Water Res. 2022, 220, 118701. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, X.; Pillai, H.S.; Lattimer, J.; Mohd Adli, N.; Karakalos, S.; Chen, M.; Guo, L.; Xu, H.; Yang, J.; et al. Ternary PtIrNi Catalysts for Efficient Electrochemical Ammonia Oxidation. ACS Catal. 2020, 10, 3945–3957. [Google Scholar] [CrossRef]
- Shih, Y.-J.; Huang, Y.-H.; Huang, C.P. Electrocatalytic ammonia oxidation over a nickel foam electrode: Role of Ni(OH)2(s)-NiOOH(s) nanocatalysts. Electrochim. Acta 2018, 263, 261–271. [Google Scholar] [CrossRef]
- Shih, Y.-J.; Huang, Y.-H.; Huang, C.P. In-situ electrochemical formation of nickel oxyhydroxide (NiOOH) on metallic nickel foam electrode for the direct oxidation of ammonia in aqueous solution. Electrochim. Acta 2018, 281, 410–419. [Google Scholar] [CrossRef]
- Jiang, X.; Ying, D.; Liu, X.; Liu, M.; Zhou, S.; Guo, C.; Zhao, G.; Wang, Y.; Jia, J. Identification of the role of Cu site in Ni-Cu hydroxide for robust and high selective electrochemical ammonia oxidation to nitrite. Electrochim. Acta 2020, 345, 136157. [Google Scholar] [CrossRef]
- Xu, W.; Du, D.; Lan, R.; Humphreys, J.; Miller, D.N.; Walker, M.; Wu, Z.; Irvine, J.T.S.; Tao, S. Electrodeposited NiCu bimetal on carbon paper as stable non-noble anode for efficient electrooxidation of ammonia. Appl. Catal. B Environ. 2018, 237, 1101–1109. [Google Scholar] [CrossRef]
- He, S.; Chen, Y.; Wang, M.; Nuomin, H.; Novello, P.; Li, X.; Zhu, S.; Liu, J. Metal nitride nanosheets enable highly efficient electrochemical oxidation of ammonia. Nano Energy 2021, 80, 105528. [Google Scholar] [CrossRef]
- He, S.; Somayaji, V.; Wang, M.; Lee, S.-H.; Geng, Z.; Zhu, S.; Novello, P.; Varanasi, C.V.; Liu, J. High entropy spinel oxide for efficient electrochemical oxidation of ammonia. Nano Res. 2022, 15, 4785–4791. [Google Scholar] [CrossRef]
- Li, L.; Xu, L.; Wang, H.; Wei, H.; Tang, C.; Li, G.; Dou, Y.; Liu, H.K.; Dou, S.X. Electrocatalytic nitrogen cycle: Mechanism, materials, and momentum. Energy Environ. Sci. 2024, 17, 9027–9050. [Google Scholar] [CrossRef]
- Agharezaei, P.; Ghuman, K.K. Designing Trimetallic Single-Doped Alloy Catalysts for Sustainable Ammonia Production: The Role of Dopants in Active Site Engineering. ACS Catal. 2025, 15, 7853–7866. [Google Scholar] [CrossRef]
- de Vooys, A.C.A.; Koper, M.T.M.; van Santen, R.A.; van Veen, J.A.R. The role of adsorbates in the electrochemical oxidation of ammonia on noble and transition metal electrodes. J. Electroanal. Chem. 2001, 506, 127–137. [Google Scholar] [CrossRef]
- Wei, R.-L.; Liu, Y.; Chen, Z.; Jia, W.-S.; Yang, Y.-Y.; Cai, W.-B. Ammonia oxidation on iridium electrode in alkaline media: An in situ ATR-SEIRAS study. J. Electroanal. Chem. 2021, 896, 115254. [Google Scholar] [CrossRef]
- Zhu, C.; Lan, B.; Wei, R.-L.; Wang, C.-N.; Yang, Y.-Y. Potential-Dependent Selectivity of Ethanol Complete Oxidation on Rh Electrode in Alkaline Media: A Synergistic Study of Electrochemical ATR-SEIRAS and IRAS. ACS Catal. 2019, 9, 4046–4053. [Google Scholar] [CrossRef]
- Yang, Y.-Y.; Ren, J.; Li, Q.-X.; Zhou, Z.-Y.; Sun, S.-G.; Cai, W.-B. Electrocatalysis of Ethanol on a Pd Electrode in Alkaline Media: An in Situ Attenuated Total Reflection Surface-Enhanced Infrared Absorption Spectroscopy Study. ACS Catal. 2014, 4, 798–803. [Google Scholar] [CrossRef]
- Silva, J.C.M.; Assumpção, M.H.M.T.; Hammer, P.; Neto, A.O.; Spinacé, E.V.; Baranova, E.A. Iridium−Rhodium Nanoparticles for Ammonia Oxidation: Electrochemical and Fuel Cell Studies. ChemElectroChem 2017, 4, 1101–1107. [Google Scholar] [CrossRef]
- Peng, W.; Xiao, L.; Huang, B.; Zhuang, L.; Lu, J. Inhibition Effect of Surface Oxygenated Species on Ammonia Oxidation Reaction. J. Phys. Chem. C 2011, 115, 23050–23056. [Google Scholar] [CrossRef]
- Shih, Y.-J.; Wu, Z.-L.; Huang, Y.-H.; Huang, C.-P. Electrochemical nitrate reduction as affected by the crystal morphology and facet of copper nanoparticles supported on nickel foam electrodes (Cu/Ni). Chem. Eng. J. 2020, 383, 123157. [Google Scholar] [CrossRef]
- Lan, R.; Irvine, J.T.S.; Tao, S. Ammonia and related chemicals as potential indirect hydrogen storage materials. Int. J. Hydrogen Energy 2012, 37, 1482–1494. [Google Scholar] [CrossRef]
- Kapałka, A.; Cally, A.; Neodo, S.; Comninellis, C.; Wächter, M.; Udert, K.M. Electrochemical behavior of ammonia at Ni/Ni(OH)2 electrode. Electrochem. Commun. 2010, 12, 18–21. [Google Scholar] [CrossRef]
- Wang, M.; Liu, M.; Zou, H.; Liu, G. Efficient Removal of Ni-Edta Complexes Utilizing 3d Ni-Rm Electro-Fenton System. Sep. Purif. Technol. 2025, 369, 133067. [Google Scholar] [CrossRef]
- Yao, K.; Cheng, Y.F. Investigation of the electrocatalytic activity of nickel for ammonia oxidation. Mater. Chem. Phys. 2008, 108, 247–250. [Google Scholar] [CrossRef]
- Yao, K.; Cheng, Y.F. Electrodeposited Ni–Pt binary alloys as electrocatalysts for oxidation of ammonia. J. Power Sources 2007, 173, 96–101. [Google Scholar] [CrossRef]
- Despić, A.R.; Dražić, D.M.; Rakin, P.M. Kinetics of electrochemical oxidation of ammonia in alkaline solution. Electrochim. Acta 1966, 11, 997–1005. [Google Scholar] [CrossRef]
- Medford, A.J.; Vojvodic, A.; Hummelshøj, J.S.; Voss, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Nørskov, J.K. From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 2015, 328, 36–42. [Google Scholar] [CrossRef]
- Xu, M.; Wei, M. Layered Double Hydroxide-Based Catalysts: Recent Advances in Preparation, Structure, and Applications. Adv. Funct. Mater. 2018, 28, 1802943. [Google Scholar] [CrossRef]
- Xie, J.; Gao, L.; Cao, S.; Liu, W.; Lei, F.; Hao, P.; Xia, X.; Tang, B. Copper-incorporated hierarchical wire-on-sheet α-Ni(OH)2 nanoarrays as robust trifunctional catalysts for synergistic hydrogen generation and urea oxidation. J. Mater. Chem. A 2019, 7, 13577–13584. [Google Scholar] [CrossRef]
- Wei, C.; Sun, Y.; Scherer, G.G.; Fisher, A.C.; Sherburne, M.; Ager, J.W.; Xu, Z.J. Surface Composition Dependent Ligand Effect in Tuning the Activity of Nickel–Copper Bimetallic Electrocatalysts toward Hydrogen Evolution in Alkaline. J. Am. Chem. Soc. 2020, 142, 7765–7775. [Google Scholar] [CrossRef]
- Sonia Theres, G.; Velayutham, G.; Santhana Krishnan, P.; Shanthi, K. Synergistic impact of Ni–Cu hybrid oxides deposited on ordered mesoporous carbon scaffolds as non-noble catalyst for methanol oxidation. J. Mater. Sci. 2019, 54, 1502–1519. [Google Scholar] [CrossRef]
- Jabłońska, M.; Beale, A.M.; Nocuń, M.; Palkovits, R. Ag-Cu based catalysts for the selective ammonia oxidation into nitrogen and water vapour. Appl. Catal. B Environ. 2018, 232, 275–287. [Google Scholar] [CrossRef]
- Yang, A.; Wang, J.; Su, K.; Lei, W.; Qiu, X.; Tang, Y. Modulating Hydroxyl-Rich Interfaces on Nickel–Copper Double Hydroxide Nanotyres to Pre-activate Alkaline Ammonia Oxidation Reactivity. Chem.—A Eur. J. 2021, 27, 4869–4875. [Google Scholar] [CrossRef]
- Xu, W.; Lan, R.; Du, D.; Humphreys, J.; Walker, M.; Wu, Z.; Wang, H.; Tao, S. Directly growing hierarchical nickel-copper hydroxide nanowires on carbon fibre cloth for efficient electrooxidation of ammonia. Appl. Catal. B Environ. 2017, 218, 470–479. [Google Scholar] [CrossRef]
- Tsai, M.-H.; Chen, T.-C.; Juang, Y.; Hua, L.-C.; Huang, C. High catalytic performance of CuCo/nickel foam electrode for ammonia electrooxidation. Electrochem. Commun. 2020, 121, 106875. [Google Scholar] [CrossRef]
- Wang, R.; Liu, H.; Zhang, K.; Zhang, G.; Lan, H.; Qu, J. Ni(II)/Ni(III) redox couple endows Ni foam-supported Ni2P with excellent capability for direct ammonia oxidation. Chem. Eng. J. 2021, 404, 126795. [Google Scholar] [CrossRef]
- Schiffer, Z.J.; Lazouski, N.; Corbin, N.; Manthiram, K. Nature of the First Electron Transfer in Electrochemical Ammonia Activation in a Nonaqueous Medium. J. Phys. Chem. C 2019, 123, 9713–9720. [Google Scholar] [CrossRef]
- Gieshoff, T.; Kehl, A.; Schollmeyer, D.; Moeller, K.D.; Waldvogel, S.R. Insights into the Mechanism of Anodic N–N Bond Formation by Dehydrogenative Coupling. J. Am. Chem. Soc. 2017, 139, 12317–12324. [Google Scholar] [CrossRef]
- Xiao, S.; Qu, J.; Zhao, X.; Liu, H.; Wan, D. Electrochemical process combined with UV light irradiation for synergistic degradation of ammonia in chloride-containing solutions. Water Res. 2009, 43, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, Y. Ammonia removal in electrochemical oxidation: Mechanism and pseudo-kinetics. J. Hazard. Mater. 2009, 161, 1010–1016. [Google Scholar] [CrossRef]
- Kapałka, A.; Katsaounis, A.; Michels, N.-L.; Leonidova, A.; Souentie, S.; Comninellis, C.; Udert, K.M. Ammonia oxidation to nitrogen mediated by electrogenerated active chlorine on Ti/PtOx-IrO2. Electrochem. Commun. 2010, 12, 1203–1205. [Google Scholar] [CrossRef]
- Jung, Y.J.; Baek, K.W.; Oh, B.S.; Kang, J.-W. An investigation of the formation of chlorate and perchlorate during electrolysis using Pt/Ti electrodes: The effects of pH and reactive oxygen species and the results of kinetic studies. Water Res. 2010, 44, 5345–5355. [Google Scholar] [CrossRef]
- Zhang, C.; He, D.; Ma, J.; Waite, T.D. Active chlorine mediated ammonia oxidation revisited: Reaction mechanism, kinetic modelling and implications. Water Res. 2018, 145, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Chen, R.; Wen, Y.; Li, Y.; Zhan, W.; Ma, F.; Jiang, X.; He, W.; Ni, H. Trace amount of RuO2 loaded on TiO2 nanowires for efficient electrocatalytic degradation of ammonia nitrogen in wastewater. J. Alloys Compd. 2022, 928, 167058. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, W.; Bai, J.; Li, J.; Wang, J.; Zhou, T.; Guan, X.; Zhou, B. Highly efficient removal of total nitrogen and dissolved organic compound in waste reverse osmosis concentrate mediated by chlorine radical on 3D Co3O4 nanowires anode. J. Hazard. Mater. 2022, 424, 127662. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, Y.; Bai, J.; Li, J.; Li, L.; Zhou, T.; Chen, S.; Wang, J.; Rahim, M.; Guan, X.; et al. Efficient degradation of N-containing organic wastewater via chlorine oxide radical generated by a photoelectrochemical system. Chem. Eng. J. 2020, 392, 123695. [Google Scholar] [CrossRef]
- Xu, J.; Wang, L.; Mao, X.; Zou, H.; Liu, G. Enhanced electrochlorination for efficient ammonia oxidation facilitated by accelerating electron cycling on Co2+/Co3+. J. Environ. Chem. Eng. 2025, 13, 115415. [Google Scholar] [CrossRef]
- Wu, K.; Cao, J.; Zhang, R.; Pei, Y.; Peng, T.; Chen, G. Micro-doping tin-bismuth on modification of Co3O4 electrocatalyst and degradation of ammonia nitrogen. Environ. Res. 2025, 275, 121366. [Google Scholar] [CrossRef] [PubMed]
- Luu, T.L. Post treatment of ICEAS-biologically landfill leachate using electrochemical oxidation with Ti/BDD and Ti/RuO2 anodes. Environ. Technol. Innov. 2020, 20, 101099. [Google Scholar] [CrossRef]
- Díaz, V.; Ibáñez, R.; Gómez, P.; Urtiaga, A.M.; Ortiz, I. Kinetics of electro-oxidation of ammonia-N, nitrites and COD from a recirculating aquaculture saline water system using BDD anodes. Water Res. 2011, 45, 125–134. [Google Scholar] [CrossRef]
- Zhou, X.; Jin, H.; Ma, Z.; Li, N.; Li, G.; Zhang, T.; Lu, P.; Gong, X. Biochar sacrificial anode assisted water electrolysis for hydrogen production. Int. J. Hydrogen Energy 2022, 47, 36482–36492. [Google Scholar] [CrossRef]
- Pan, Z.; Xu, J.; Zhou, X.; Xu, R.; Yu, H.; Hong, J.; Zhao, S.; Fan, X.; Song, C.; Wang, T. Efficient removal of ammonia from aqueous solution using coal-based carbon membrane via electrochemical oxidation in the present of chloride ion. J. Environ. Chem. Eng. 2024, 12, 114335. [Google Scholar] [CrossRef]
- Chi, M.; Luo, B.; Zhang, Q.; Jiang, H.; Chen, C.; Wang, S.; Min, D. Lignin-based monolithic carbon electrode decorating with RuO2 nanospheres for high-performance chlorine evolution reaction. Ind. Crops Prod. 2021, 159, 113088. [Google Scholar] [CrossRef]
- Li, Y.; Yi, Q.; Wang, D.; Wu, Z.; Wang, Z. Efficient treatment of landfill leachate using an electrochemical ceramic membrane filtration system: Chlorine-mediated oxidation. Chem. Eng. J. 2022, 450, 138102. [Google Scholar] [CrossRef]
- Devkota, L.M.; Williams, D.S.; Matta, J.H.; Albertson, O.E.; Grasso, D.; Fox, P. Variation of oxidation–reduction potential along the breakpoint curves in low-ammonia effluents. Water Environ. Res. 2000, 72, 610–617. [Google Scholar] [CrossRef]
- Guo, K.; Wu, Z.; Chen, C.; Fang, J. UV/Chlorine Process: An Efficient Advanced Oxidation Process with Multiple Radicals and Functions in Water Treatment. Acc. Chem. Res. 2022, 55, 286–297. [Google Scholar] [CrossRef]
- Bagastyo, A.Y.; Radjenovic, J.; Mu, Y.; Rozendal, R.A.; Batstone, D.J.; Rabaey, K. Electrochemical oxidation of reverse osmosis concentrate on mixed metal oxide (MMO) titanium coated electrodes. Water Res. 2011, 45, 4951–4959. [Google Scholar] [CrossRef]
- Kim, H.; Chung, M.W.; Choi, C.H. NOx-induced deactivation of Pt electrocatalysis towards the ammonia oxidation reaction. Electrochem. Commun. 2018, 94, 31–35. [Google Scholar] [CrossRef]
- Lu, S.; Shang, C.; Sun, B.; Xiang, Y. Dominant Dissolved Oxygen-Independent Pathway to Form Hydroxyl Radicals and the Generation of Reactive Chlorine and Nitrogen Species in Breakpoint Chlorination. Environ. Sci. Technol. 2023, 57, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jiang, M.; Su, P.; Lv, Q.; Zeng, G.; An, L.; Cao, J.; Zhou, Y.; Snyder, S.A.; Ma, J.; et al. Refinement of kinetic model and understanding the role of dichloride radical (Cl2•−) in radical transformation in the UV/NH2Cl process. Water Res. 2024, 254, 121440. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, I.M.; Mitch, W.A. Enhanced Nitrogenous Disinfection ByProduct Formation near the Breakpoint: Implications for Nitrification Control. Environ. Sci. Technol. 2007, 41, 7039–7046. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-Y.; Chen, Y.; Wang, W.-L.; Chen, Y.-L.; Wu, Q.-Y. The Impact of Dissolved Organic Matter in Natural Receiving Systems on the Formation Potential and Toxicity of Disinfection By-products: Insights from Origins, Chemical Properties, and Transformations. Curr. Pollut. Rep. 2025, 11, 29. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).