

Degradation of 1,4-Dioxane by Au/TiO2 Janus Nanoparticles Under Ultraviolet Light: Experiments and Modeling

 , , , , , , and
, , , , , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Synthesis of Au/TiO2 JNPs
2.3. Characterization Methods
2.4. Photocatalytic Degradation Experiments
2.5. Computational Modeling
3. Results
3.1. Characterization
3.1.1. Morphology and Elemental Composition of JNPs
3.1.2. TiO2 Crystalline Phase
3.1.3. Particle Size Distribution
3.1.4. Light Absorption
3.1.5. XPS Analysis
3.2. Factors Affecting Degradation Kinetics
3.2.1. Effect of Wavelength
3.2.2. Effect of Light Intensity
3.2.3. Effects of Solution Salinity and Radical Scavengers
3.2.4. Role of Self-Propulsion
3.3. Simulations of Light Absorption
4. Discussion
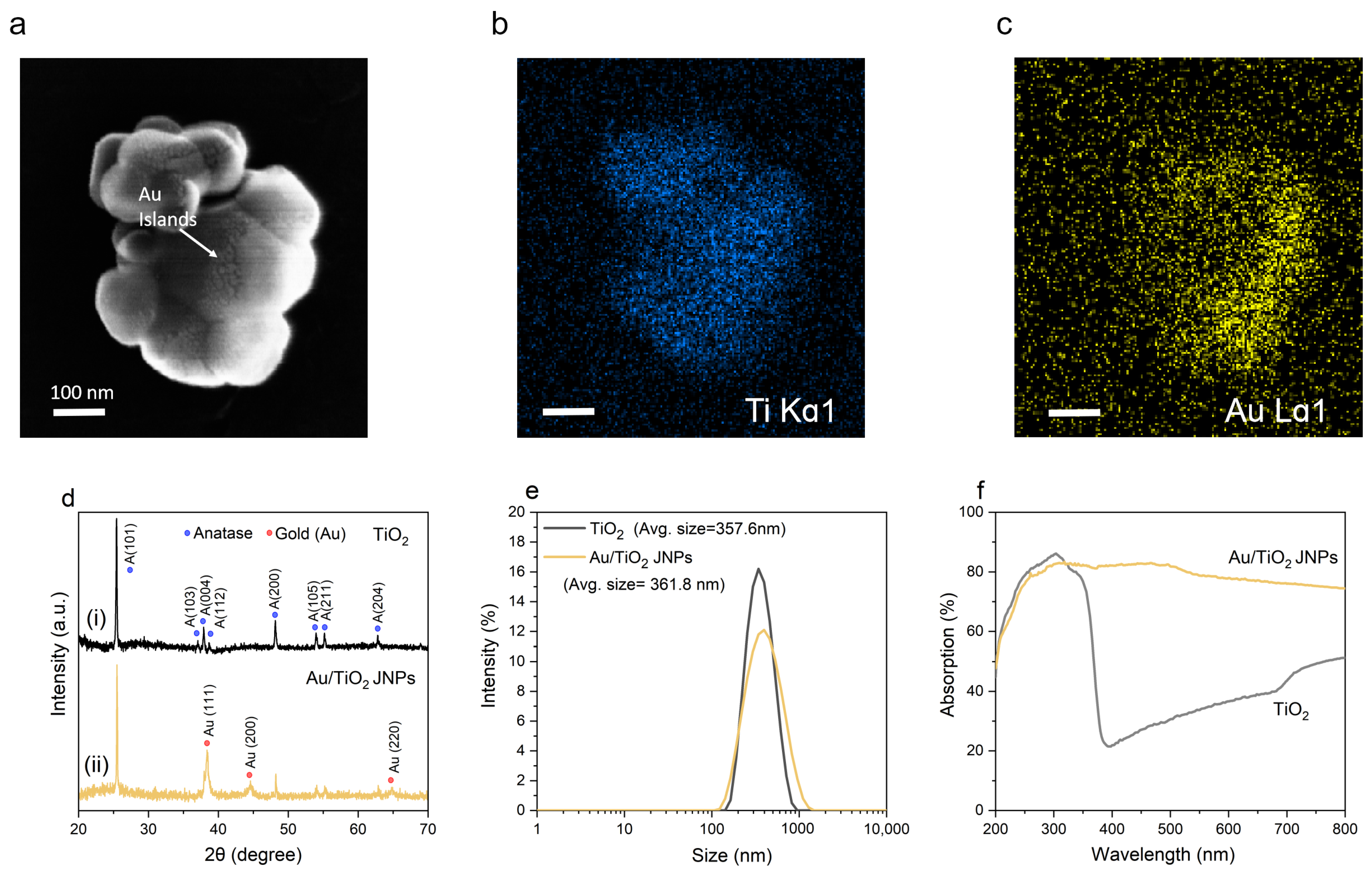
- In the case of 254 nm light, light was preferentially absorbed close to the water/TiO2 interface, whereas 365 nm light was mostly absorbed near the particle’s core. This suggests that even though roughly the same amount of light was absorbed by the particles overall at the two wavelengths (gold data in Figure 1f, black data in Figure 4e), a significantly larger proportion of the electron–hole pairs created by 254 nm light resulted in viable ●OH radicals to degrade pollutants. This could explain why the reaction rate constant was nearly 15 times larger for 254 nm light than for 365 nm light for the Au/TiO2 JNPs at the same light intensity (see Table S1 in the Supplementary Information).
- For 254 nm light, more light was absorbed by the TiO2 overall, as evidenced by Figure 4d,e. The data in Figure 1f are consistent with this observation, since the absorbance for pure TiO2 was roughly 80% at 254 nm and roughly 62% at 365 nm. This observation suggests that more electron–hole pairs were created overall for 254 nm light, but the difference was not necessarily sufficient to explain the vast enhancement in the degradation rate that was observed between the two wavelengths.
- Although the total amount of light absorbed was similar for 254 nm and 365 nm light, a significantly greater portion was absorbed by the TiO2 rather than by the Au. This could explain why, even though the total absorption was similar for the Au/TiO2 nanostructures (Figure 1f), the rate of reaction was significantly higher (rate constant of 0.138 min−1 for 254 nm light and 0.009 min−1 for 365 nm light).
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SPR | Surface plasmon resonance |
| JNP | Janus nanoparticle |
| AOP | Advanced oxidation process |
| RSM | Response surface methodology |
| DLS | Dynamic light scattering |
| XRD | X-ray diffraction |
| XPS | X-ray photoelectron spectroscopy |
| EDS | Energy-dispersive X-ray spectroscopy |
| UV/Vis | Ultraviolet–visible spectroscopy |
| CEC | Contaminant of emerging concern |
| ROS | Reactive oxygen species |
| DI | Deionized (water) |
| HPLC | High-performance liquid chromatography |
| DAD | Diode-array detection |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
| NaCl | Sodium chloride |
| MeOH | Methanol |
| UPS | Ultraviolet photoelectron spectroscopy |
| ●OH | Hydroxyl radical |
| CB | Conduction band |
| VB | Valence band |
References
- Stepanov, M.G.; Arutiunian, A.V.; Ailamazian, E.K. Disruption of Central Regulation of Reproductive Function under the Effect of Unfavorable Environmental Factors. Vopr. Med. Khimii 1995, 41, 33–35. [Google Scholar] [PubMed]
- 1,4-Dioxane Priority Existing Chemical No. 7 Full Public Report. 1998. Available online: https://www.industrialchemicals.gov.au/sites/default/files/PEC7-1-4-Dioxane.pdf (accessed on 19 May 2025).
- Lam, S.W.; Hermawan, M.; Coleman, H.M.; Fisher, K.; Amal, R. The Role of Copper(II) Ions in the Photocatalytic Oxidation of 1,4-Dioxane. J. Mol. Catal. A Chem. 2007, 278, 152–159. [Google Scholar] [CrossRef]
- Sittig, M. Handbook of Toxic and Hazardous Chemicals and Carcinogens: 1991—Third Edition; Elsevier Inc.: Amsterdam, The Netherlands, 2009; ISBN -13-978-0815512868. [Google Scholar]
- Coleman, H.M.; Vimonses, V.; Leslie, G.; Amal, R. Degradation of 1,4-Dioxane in Water Using TiO2 Based Photocatalytic and H2O2/UV Processes. J. Hazard. Mater. 2007, 146, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Aǐlamazian, E.K. Effect of Ecological Factors on the Course of Pregnancy. Vestn. Akad. Med. Nauk SSSR 1990, 23–25. Available online: https://pubmed.ncbi.nlm.nih.gov/2220046/ (accessed on 19 May 2025). [PubMed]
- Godri Pollitt, K.J.; Kim, J.H.; Peccia, J.; Elimelech, M.; Zhang, Y.; Charkoftaki, G.; Hodges, B.; Zucker, I.; Huang, H.; Deziel, N.C.; et al. 1,4-Dioxane as an Emerging Water Contaminant: State of the Science and Evaluation of Research Needs. Sci. Total Environ. 2019, 690, 853–866. [Google Scholar] [CrossRef]
- Zhang, S.; Gedalanga, P.B.; Mahendra, S. Advances in Bioremediation of 1,4-Dioxane-Contaminated Waters. J. Environ. Manag. 2017, 204, 765–774. [Google Scholar] [CrossRef]
- Mohr, T.K.; Stickney, J.A.; DiGuiseppi, W.H.; Anderson, J. Environmental Investigation and Remediation 1,4-Dioxane and Other Solvent Stabilizers, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- Stepien, D.K.; Diehl, P.; Helm, J.; Thoms, A.; Püttmann, W. Fate of 1,4-Dioxane in the Aquatic Environment: From Sewage to Drinking Water. Water Res. 2014, 48, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Adamson, D.T.; Mahendra, S.; Walker, K.L.; Rauch, S.R.; Sengupta, S.; Newell, C.J. A Multisite Survey to Identify the Scale of the 1,4-Dioxane Problem at Contaminated Groundwater Sites. Environ. Sci. Technol. Lett. 2014, 1, 254–258. [Google Scholar] [CrossRef]
- Reviews of environmental contamination and toxicology: Vol. 98 (Continuation of Residue Reviews). Edited by G.W. Ware, Springer-Verlag, Berlin, 1987. Pp. 166, ISBN 3 540 96448 7. Price: DM 92·00. Environ. Pollut. 1988, 50, 253–254. [CrossRef]
- Xu, X.; Liu, S.; Sun, P.; Guo, Z.; Smith, K.; Zhang, D.; Li, H.; Bedia, J.; Belver, C. Iron Tungstate on Nano-γ-Alumina as Photocatalyst for 1,4-Dioxane Solar Degradation in Water. J. Clean. Prod. 2022, 377, 134232. [Google Scholar] [CrossRef]
- Maurino, V.; Calza, P.; Minero, C.; Pelizzetti, E.; Vincenti, M. Light-Assisted 1,4-Dioxane Degradation. Chemosphere 1997, 35, 2675–2688. [Google Scholar] [CrossRef]
- Klečka, G.M.; Gonsior, S.J. Removal of 1,4-Dioxane from Wastewater. J. Hazard. Mater. 1986, 13, 161–168. [Google Scholar] [CrossRef]
- Adams, C.D.; Scanlan, P.A.; Secrist, N.D. Oxidation and Biodegradability Enhancement of 1,4-Dioxane Using Hydrogen Peroxide and Ozone. Environ. Sci. Technol. 1994, 28, 1812–1818. [Google Scholar] [CrossRef]
- Hoigné, J.; Bader, H. Rate Constants of Reactions of Ozone with Organic and Inorganic Compounds in Water-I. Non-Dissociating Organic Compounds. Water Res. 1983, 17, 173–183. [Google Scholar] [CrossRef]
- Howard, P.H. Handbook of Environmental Fate and Exposure Data for Organic Chemicals; Taylor & Francis: New York, NY, USA, 1991. [Google Scholar]
- Sauvé, S.; Desrosiers, M. A Review of What Is an Emerging Contaminant. Chem. Cent. J. 2014, 8, 15. [Google Scholar] [CrossRef]
- Kamat, P.V.; Meisel, D. Nanoparticles in Advanced Oxidation Processes. Curr. Opin. Colloid Interface Sci. 2002, 7, 282–287. [Google Scholar] [CrossRef]
- Hodges, B.C.; Cates, E.L.; Kim, J.H. Challenges and Prospects of Advanced Oxidation Water Treatment Processes Using Catalytic Nanomaterials. Nat. Nanotechnol. 2018, 13, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Araki, S.; Yamamoto, H. Evaluation of Advanced Oxidation Processes (AOP) Using O3, UV, and TiO2 for the Degradation of Phenol in Water. J. Water Process Eng. 2015, 7, 54–60. [Google Scholar] [CrossRef]
- Fernandes, A.; Makoś, P.; Wang, Z.; Boczkaj, G. Synergistic Effect of TiO2 Photocatalytic Advanced Oxidation Processes in the Treatment of Refinery Effluents. Chem. Eng. J. 2020, 391, 123488. [Google Scholar] [CrossRef]
- Dewil, R.; Mantzavinos, D.; Poulios, I.; Rodrigo, M.A. New Perspectives for Advanced Oxidation Processes. J. Environ. Manag. 2017, 195, 93–99. [Google Scholar] [CrossRef]
- Venkatachalam, N.; Palanichamy, M.; Murugesan, V. Sol-Gel Preparation and Characterization of Alkaline Earth Metal Doped Nano TiO2: Efficient Photocatalytic Degradation of 4-Chlorophenol. J. Mol. Catal. A Chem. 2007, 273, 177–185. [Google Scholar] [CrossRef]
- Prakash, J.; Sun, S.; Swart, H.C.; Gupta, R.K. Noble Metals-TiO2 Nanocomposites: From Fundamental Mechanisms to Photocatalysis, Surface Enhanced Raman Scattering and Antibacterial Applications. Appl. Mater. Today 2018, 11, 82–135. [Google Scholar] [CrossRef]
- Chen, Y.; Bian, J.; Qi, L.; Liu, E.; Fan, J. Efficient Degradation of Methylene Blue over Two-Dimensional Au/TiO2 Nanosheet Films with Overlapped Light Harvesting Nanostructures. J. Nanomater. 2015, 2015, 905259. [Google Scholar] [CrossRef]
- Pradhan, S.; Ghosh, D.; Chen, S. Janus Nanostructures Based on Au-TiO2 Heterodimers and Their Photocatalytic Activity in the Oxidation of Methanol. ACS Appl. Mater. Interfaces 2009, 1, 2060–2065. [Google Scholar] [CrossRef]
- Mrowetz, M.; Villa, A.; Prati, L.; Selli, E. Effect of Au Nanoparticles on TiO2 in the Photocatalytic Degradation of an Azo Dye. Gold Bull. 2007, 40, 154–160. [Google Scholar] [CrossRef]
- Orlov, A.; Jefferson, D.A.; Tikhov, M.; Lambert, R.M. Enhancement of MTBE Photocatalytic Degradation by Modification of TiO2 with Gold Nanoparticles. Catal. Commun. 2007, 8, 821–824. [Google Scholar] [CrossRef]
- Bumajdad, A.; Madkour, M.; Abdel-Moneam, Y.; El-Kemary, M. Nanostructured Mesoporous Au/TiO2 for Photocatalytic Degradation of a Textile Dye: The Effect of Size Similarity of the Deposited Au with That of TiO2 Pores. J. Mater. Sci. 2014, 49, 1743–1754. [Google Scholar] [CrossRef]
- Dang, X.; Zhang, X.; Lu, Z.; Yang, Z.; Dong, X.; Zhang, X.; Ma, C.; Ma, H.; Xue, M.; Shi, F. Construction of Au@TiO2/Graphene Nanocomposites with Plasmonic Effect and Super Adsorption Ability for Enhanced Visible-Light-Driven Photocatalytic Organic Pollutant Degradation. J. Nanopartic. Res. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Do, T.C.M.V.; Nguyen, D.Q.; Nguyen, K.T.; Le, P.H. TiO2 and Au-TiO2 Nanomaterials for Rapid Photocatalytic Degradation of Antibiotic Residues in Aquaculture Wastewater. Materials 2019, 12, 2434. [Google Scholar] [CrossRef]
- Seh, Z.W.; Liu, S.; Low, M.; Zhang, S.Y.; Liu, Z.; Mlayah, A.; Han, M.Y. Janus Au-TiO2 Photocatalysts with Strong Localization of Plasmonic near-Fields for Efficient Visible-Light Hydrogen Generation. Adv. Mater. 2012, 24, 2310–2314. [Google Scholar] [CrossRef]
- Tseng, Y.H.; Chang, I.G.; Tai, Y.; Wu, K.W. Effect of Surface Plasmon Resonance on the Photocatalytic Activity of Au/TiO2 under UV/Visible Illumination. J. Nanosci. Nanotechnol. 2012, 12, 416–422. [Google Scholar] [CrossRef]
- Yasmeen, H.; Zada, A.; Ali, S.; Khan, I.; Ali, W.; Khan, W.; Khan, M.; Anwar, N.; Ali, A.; Huerta-Flores, A.M.; et al. Visible Light-Excited Surface Plasmon Resonance Charge Transfer Significantly Improves the Photocatalytic Activities of ZnO Semiconductor for Pollutants Degradation. J. Chin. Chem. Soc. 2020, 67, 1611–1617. [Google Scholar] [CrossRef]
- Yu, Y.; Wen, W.; Qian, X.Y.; Liu, J.B.; Wu, J.M. UV and Visible Light Photocatalytic Activity of Au/TiO2 Nanoforests with Anatase/Rutile Phase Junctions and Controlled Au Locations. Sci. Rep. 2017, 7, 41253. [Google Scholar] [CrossRef] [PubMed]
- Jose, D.; Sorensen, C.M.; Rayalu, S.S.; Shrestha, K.M.; Klabunde, K.J. Au-TiO2 Nanocomposites and Efficient Photocatalytic Hydrogen Production under UV-Visible and Visible Light Illuminations: A Comparison of Different Crystalline Forms of TiO2. Int. J. Photoenergy 2013, 2013, 685614. [Google Scholar] [CrossRef]
- Chauhan, A.; Rastogi, M.; Scheier, P.; Bowen, C.; Kumar, R.V.; Vaish, R. Janus Nanostructures for Heterogeneous Photocatalysis. Appl. Phys. Rev. 2018, 5, 041111. [Google Scholar] [CrossRef]
- Tian, Y.; Tatsuma, T. Mechanisms and Applications of Plasmon-Induced Charge Separation at TiO2 Films Loaded with Gold Nanoparticles. J. Am. Chem. Soc. 2005, 127, 7632–7637. [Google Scholar] [CrossRef]
- Soler, L.; Magdanz, V.; Fomin, V.M.; Sanchez, S.; Schmidt, O.G. Self-Propelled Micromotors for Cleaning Polluted Water. ACS Nano 2013, 7, 9611–9620. [Google Scholar] [CrossRef]
- Min, B.K.; Heo, J.E.; Youn, N.K.; Joo, O.S.; Lee, H.; Kim, J.H.; Kim, H.S. Tuning of the Photocatalytic 1,4-Dioxane Degradation with Surface Plasmon Resonance of Gold Nanoparticles on Titania. Catal. Commun. 2009, 10, 712–715. [Google Scholar] [CrossRef]
- Tsukamoto, D.; Shiraishi, Y.; Sugano, Y.; Ichikawa, S.; Tanaka, S.; Hirai, T. Gold Nanoparticles Located at the Interface of Anatase/Rutile TiO2 Particles as Active Plasmonic Photocatalysts for Aerobic Oxidation. J. Am. Chem. Soc. 2012, 134, 6309–6315. [Google Scholar] [CrossRef]
- Li, H.; Wang, S.; Hong, F.; Gao, Y.; Zeng, B.; Haider, R.S.; Fan, F.; Huang, J.; Li, C. Effects of the Interfacial Defects in Au/TiO2on Plasmon-Induced Water Oxidation. J. Chem. Phys. 2020, 152, 194702. [Google Scholar] [CrossRef]
- Primo, A.; Corma, A.; García, H. Titania Supported Gold Nanoparticles as Photocatalyst. Phys. Chem. Chem. Phys. 2011, 13, 886–910. [Google Scholar] [CrossRef] [PubMed]
- Lattuada, M.; Hatton, T.A. Synthesis, Properties and Applications of Janus Nanoparticles. Nano Today 2011, 6, 286–308. [Google Scholar] [CrossRef]
- Yaou Balarabe, B.; Maity, P. A Polymer-Au/TiO2 Nano-Composite Based Floating Catalyst for Photocatalytic Dye Degradation under Natural Sunlight. J. Photochem. Photobiol. A Chem. 2024, 449, 115405. [Google Scholar] [CrossRef]
- Youn, N.K.; Heo, J.E.; Joo, O.S.; Lee, H.; Kim, J.; Min, B.K. The effect of dissolved oxygen on the 1,4-dioxane degradation with TiO2 and Au-TiO2 photocatalysts. J. Hazard. Mater. 2010, 177, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.; Ganguly, P.; Maccioni, M.B.; Nolan, M.; Hermosilla, D.; Merayo, N.; Blanco, A.; Hinder, S.; Pillai, S.C. Impact of Au on the transition temperature and photocatalytic activity of TiO2. J. Photochem. Photobiol. A 2024, 456, 115848. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Toi, S.; Ichikawa, S.; Hirai, T. Photocatalytic NH3 Splitting on TiO2 Particles Decorated with Pt-Au Bimetallic Alloy Nanoparticles. ACS Appl. Nano Mater. 2020, 3, 1612–1620. [Google Scholar] [CrossRef]
- Collado, L.; Reynal, A.; Coronado, J.M.; Serrano, D.P.; Durrant, J.R.; De la Peña O’Shea, V.A. Effect of Au Surface Plasmon Nanoparticles on the Selective CO2 Photoreduction to CH4. Appl. Catal. B Environ. 2015, 178, 177–185. [Google Scholar] [CrossRef]
- Collado, L.; Reynal, A.; Fresno, F.; Barawi, M.; Escudero, C.; Perez-Dieste, V.; Coronado, J.M.; Serrano, D.P.; Durrant, J.R.; de la Peña O’Shea, V.A. Unravelling the Effect of Charge Dynamics at the Plasmonic Metal/Semiconductor Interface for CO2 Photoreduction. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Fontelles-Carceller, O.; Muñoz-Batista, M.J.; Rodríguez-Castellón, E.; Conesa, J.C.; Fernández-García, M.; Kubacka, A. Measuring and Interpreting Quantum Efficiency for Hydrogen Photo-Production Using Pt-Titania Catalysts. J. Catal. 2017, 347, 157–169. [Google Scholar] [CrossRef]
- Hernández Rodríguez, M.J.; Pulido Melián, E.; García Santiago, D.; González Díaz, O.; Navío, J.A.; Doña Rodríguez, J.M. NO Photooxidation with TiO2 Photocatalysts Modified with Gold and Platinum. Appl. Catal. B Environ. 2017, 205, 148–157. [Google Scholar] [CrossRef]
- Kowalska, E.; Abe, R.; Ohtani, B. Visible Light-Induced Photocatalytic Reaction of Gold-Modified Titanium(IV) Oxide Particles: Action Spectrum Analysis. Chem. Commun. 2009, 9, 241–243. [Google Scholar] [CrossRef]
- Ghasemi, S.; Hashemian, S.J.; Alamolhoda, A.A.; Gocheva, I.; Rahman Setayesh, S. Plasmon Enhanced Photocatalytic Activity of Au@TiO2-Graphene Nanocomposite under Visible Light for Degradation of Pollutants. Mater. Res. Bull. 2017, 87, 40–47. [Google Scholar] [CrossRef]
- Xu, L.; Mou, F.; Gong, H.; Luo, M.; Guan, J. Light-Driven Micro/Nanomotors: From Fundamentals to Applications. Chem. Soc. Rev. 2017, 46, 6905–6926. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Cai, Y.; Yang, Y.; Gao, W.; Ren, B. Photocatalytic Micro/Nanomotors: From Construction to Applications. Acc. Chem. Res. 2018, 51, 1940–1947. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Zhang, Q.; Gao, W.; Pei, A.; Ren, B. Highly Efficient Light-Driven TiO2-Au Janus Micromotors. ACS Nano 2016, 10, 839–844. [Google Scholar] [CrossRef]
- Maric, T.; Nasir, M.Z.M.; Webster, R.D.; Pumera, M. Tailoring Metal/TiO2 Interface to Influence Motion of Light-Activated Janus Micromotors. Adv. Funct. Mater. 2020, 30, 1908614. [Google Scholar] [CrossRef]
- Moran, J.L.; Wheat, P.M.; Posner, J.D. Locomotion of Electrocatalytic Nanomotors Due to Reaction Induced Charge Autoelectrophoresis. Phys. Rev. E Stat. Nonlinear, Soft Matter Phys. 2010, 81, 065302. [Google Scholar] [CrossRef]
- Xiao, Z.; Chen, J.; Duan, S.; Lv, X.; Wang, J.; Ma, X.; Tang, J.; Wang, W. Bimetallic Coatings Synergistically Enhance the Speeds of Photocatalytic TiO2 Micromotors. Chem. Commun. 2020, 56, 4728–4731. [Google Scholar] [CrossRef]
- Paxton, W.F.; Sen, A.; Mallouk, T.E. Motility of Catalytic Nanoparticles through Self-Generated Forces. Chem. Eur. J. 2005, 11, 6462–6470. [Google Scholar] [CrossRef]
- Mitchell, P. Self-Electrophoretic Locomotion in Microorganisms: Bacterial Flagella as Giant Ionophores. FEBS Lett. 1972, 28, 1–4. [Google Scholar] [CrossRef]
- Paxton, W.F.; Baker, P.T.; Kline, T.R.; Wang, Y.; Mallouk, T.E.; Sen, A. Catalytically Induced Electrokinetics for Motors and Micropumps. J. Am. Chem. Soc. 2006, 128, 14881–14888. [Google Scholar] [CrossRef]
- Cui, X.; Li, J.; Ng, D.H.L.; Liu, J.; Liu, Y.; Yang, W. 3D Hierarchical ACFs-Based Micromotors as Efficient Photo-Fenton-like Catalysts. Carbon 2020, 158, 738–748. [Google Scholar] [CrossRef]
- Vilela, D.; Parmar, J.; Zeng, Y.; Zhao, Y.; Sánchez, S. Graphene-Based Microbots for Toxic Heavy Metal Removal and Recovery from Water. Nano Lett. 2016, 16, 2860–2866. [Google Scholar] [CrossRef]
- Wang, H.; Gu, X.; Wang, C. Self-Propelling Hydrogel/Emulsion-Hydrogel Soft Motors for Water Purification. ACS Appl. Mater. Interfaces 2016, 8, 9413–9422. [Google Scholar] [CrossRef]
- Soler, L.; Sánchez, S. Catalytic Nanomotors for Environmental Monitoring and Water Remediation. Nanoscale 2014, 6, 7175–7182. [Google Scholar] [CrossRef]
- Eskandarloo, H.; Kierulf, A.; Abbaspourrad, A. Nano- and Micromotors for Cleaning Polluted Waters: Focused Review on Pollutant Removal Mechanisms. Nanoscale 2017, 9, 13850–13863. [Google Scholar] [CrossRef] [PubMed]
- Parmar, J.; Vilela, D.; Villa, K.; Wang, J.; Sánchez, S. Micro- and Nanomotors as Active Environmental Microcleaners and Sensors. J. Am. Chem. Soc. 2018, 140, 9317–9331. [Google Scholar] [CrossRef]
- Wu, Y.; Dong, R.; Zhang, Q.; Ren, B. Dye-Enhanced Self-Electrophoretic Propulsion of Light-Driven TiO2-Au Janus Micromotors. Nano-Micro Lett. 2017, 9, 30. [Google Scholar] [CrossRef]
- Luna, M.; Barawi, M.; Gómez-Moñivas, S.; Colchero, J.; Rodríguez-Peña, M.; Yang, S.; Zhao, X.; Lu, Y.H.; Chintala, R.; Reñones, P.; et al. Photoinduced Charge Transfer and Trapping on Single Gold Metal Nanoparticles on TiO2. ACS Appl. Mater. Interfaces 2021, 13, 50531–50538. [Google Scholar] [CrossRef] [PubMed]
- Pelaez, M.; Falaras, P.; Likodimos, V.; O’Shea, K.; de la Cruz, A.A.; Dunlop, P.S.M.; Byrne, J.A.; Dionysiou, D.D. Use of Selected Scavengers for the Determination of NF-TiO2 Reactive Oxygen Species during the Degradation of Microcystin-LR under Visible Light Irradiation. J. Mol. Catal. A Chem. 2016, 425, 183. [Google Scholar] [CrossRef]
- Granick, S.; Jiang, S.; Chen, Q. Janus Particles. Phys. Today 2009, 62, 68. [Google Scholar] [CrossRef]
- Wang, J.; Yu, J.; Zhu, X.; Kong, X.Z. Preparation of Hollow TiO2 Nanoparticles through TiO2 Deposition on Polystyrene Latex Particles and Characterizations of Their Structure and Photocatalytic Activity. Nanoscale Res. Lett. 2012, 7, 646. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Ahmad, M.; Nisar, A.; Sun, H.; Karim, S.; Khan, M.; Khan, S.D.; Iqbal, M.; Hussain, S.Z. Enhanced Photocatalytic and Electrochemical Properties of Au Nanoparticles Supported TiO2 Microspheres. New J. Chem. 2014, 38, 1424–1432. [Google Scholar] [CrossRef]
- Vert, M.; Doi, Y.; Hellwich, K.-H.; Hess, M.; Hodge, P.; Kubisa, P.; Rinaudo, M.; Schué, F. Terminology for biorelated polymers and applications (IUPAC Recommendations 2012). Pure Appl. Chem. 2012, 84, 377–410. [Google Scholar] [CrossRef]
- Bharti, B.; Kumar, S.; Lee, H.N.; Kumar, R. Formation of Oxygen Vacancies and Ti3+ State in TiO2 Thin Film and Enhanced Optical Properties by Air Plasma Treatment. Sci. Rep. 2016, 6, 32355. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, R.; Kait, C.F.; Chia, H.Y.; Isa, M.H.; Huei, L.W. Glycerol-Mediated Facile Synthesis of Colored Titania Nanoparticles for Visible Light Photodegradation of Phenolic Compounds. Nanomaterials 2019, 9, 1586. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, S.; Wan, P.; Sun, J.; Hood, Z.D. Introducing Ti3+ Defects Based on Lattice Distortion for Enhanced Visible Light Photoreactivity in TiO2 Microspheres. RSC Adv. 2017, 7, 32461–32467. [Google Scholar] [CrossRef]
- Veziroglu, S.; Ullrich, M.; Hussain, M.; Drewes, J.; Shondo, J.; Strunskus, T.; Adam, J.; Faupel, F.; Aktas, O.C. Plasmonic and Non-Plasmonic Contributions on Photocatalytic Activity of Au-TiO2 Thin Film under Mixed UV–Visible Light. Surf. Coatings Technol. 2020, 389, 125613. [Google Scholar] [CrossRef]
- Bertóti, I.; Mohai, M.; Sullivan, J.L.; Saied, S.O.; Bertóti, I.; Mohai, M.; Sullivan, J.L.; Saied, S.O. Surface Characterisation of Plasma-Nitrided Titanium: An XPS Study. Appl. Surf. Sci. 1995, 84, 357–371. [Google Scholar] [CrossRef]
- Zhao, S.; Liu, H.X.; Qiu, Y.; Liu, S.Q.; Diao, J.X.; Chang, C.R.; Si, R.; Guo, X.H. An Oxygen Vacancy-Rich Two-Dimensional Au/TiO2 Hybrid for Synergistically Enhanced Electrochemical N2 Activation and Reduction. J. Mater. Chem. A 2020, 8, 6586–6596. [Google Scholar] [CrossRef]
- Liu, X.; Gao, S.; Xu, H.; Lou, Z.; Wang, W.; Huang, B.; Dai, Y. Green Synthetic Approach for Ti3+ Self-Doped TiO2-x Nanoparticles with Efficient Visible Light Photocatalytic Activity. Nanoscale 2013, 5, 1870–1875. [Google Scholar] [CrossRef]
- Ullattil, S.G.; Pumera, M. Light-Powered Self-Adaptive Mesostructured Microrobots for Simultaneous Microplastics Trapping and Fragmentation via in Situ Surface Morphing. Small 2023, 19, 2301467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Dong, R.; Wu, Y.; Gao, W.; He, Z.; Ren, B. Light-Driven Au-WO3@C Janus Micromotors for Rapid Photodegradation of Dye Pollutants. ACS Appl. Mater. Interfaces 2017, 9, 4674–4683. [Google Scholar] [CrossRef] [PubMed]
- Manion, J.A.; Huie, R.E.; Levin, R.D.; Burgess, D.R., Jr.; Orkin, V.L.; Tsang, W.; McGivern, W.S.; Hudgens, J.W.; Knyazev, V.D.; Atkinson, D.B.; et al. NIST Chemical Kinetics Database (Web Version). Available online: http://kinetics.nist.gov/ (accessed on 25 April 2025).
- Yan, N.; Liu, F.; Liu, B.; Brusseau, M.L. Treatment of 1,4-Dioxane and Trichloroethene Co-Contamination by an Activated Binary Persulfate-Peroxide Oxidation Process. Environ. Sci. Pollut. Res. 2018, 25, 32088–32095. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.M.; Chiesa, M. EPR of Paramagnetic Centres on Solid Surfaces. In Electron Paramagnetic Resonance; Royal Society of Chemistry: London, UK, 2009; Volume 21, pp. 105–130. [Google Scholar]
- Bockris, J.O.M.; Oldfield, L.F. The Oxidation-Reduction Reactions of Hydrogen Peroxide at Inert Metal Electrodes and Mercury Cathodes. Trans. Faraday Soc. 1955, 51, 249–259. [Google Scholar] [CrossRef]
- Buettner, G.R. The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, α-Tocopherol, and Ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Heck, K.N.; Wang, Y.; Wu, G.; Wang, F.; Tsai, A.L.; Adamson, D.T.; Wong, M.S. Effectiveness of Metal Oxide Catalysts for the Degradation of 1,4-Dioxane. RSC Adv. 2019, 9, 27042–27049. [Google Scholar] [CrossRef]
- Feng, C.; Yu, Z.; Liu, H.; Yuan, K.; Wang, X.; Zhu, L.; Zhang, G.; Xu, D. Enhanced Photocatalytic Performance of Au/TiO2 Nanofibers by Precisely Manipulating the Dosage of Uniform-Sized Au Nanoparticles. Appl. Phys. A Mater. Sci. Process. 2017, 123, 519. [Google Scholar] [CrossRef]
- Winkelmann, J. Diffusion Coefficient of 1,4-Dioxane in Water. In Diffusion in Gases, Liquids and Electrolytes; Springer: Berlin/Heidelberg, Germany, 2018; pp. 547–549. [Google Scholar] [CrossRef]
- Amendola, V.; Pilot, R.; Frasconi, M.; Maragò, O.M.; Iatì, M.A. Surface Plasmon Resonance in Gold Nanoparticles: A Review. J. Phys. Condens. Matter 2017, 29, 203002. [Google Scholar] [CrossRef]
- Majeed, I.; Ali, H.; Idrees, A.; Arif, A.; Ashraf, W.; Rasul, S.; Khan, M.A.; Nadeem, M.A.; Nadeem, M.A. Understanding the Role of Metal Supported on TiO2 in Photoreforming of Oxygenates. Energy Adv. 2022, 1, 842–867. [Google Scholar] [CrossRef]
- Zhang, Z.; Yates, J.T. Band Bending in Semiconductors: Chemical and Physical Consequences at Surfaces and Interfaces. Chem. Rev. 2012, 112, 5520–5551. [Google Scholar] [CrossRef]
- Subramanian, V.; Wolf, E.E.; Kamat, P.V. Catalysis with TiO2/Gold Nanocomposites. Effect of Metal Particle Size on the Fermi Level Equilibration. J. Am. Chem. Soc. 2004, 126, 4943–4950. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.H.; Cheng, W.H.; Crumlin, E.J.; Drisdell, W.S.; Atwater, H.A.; Schmeißer, D.; Lewis, N.S.; Brunschwig, B.S. X-Ray Photoelectron Spectroscopy and Resonant X-Ray Spectroscopy Investigations of Interactions between Thin Metal Catalyst Films and Amorphous Titanium Dioxide Photoelectrode Protection Layers. Chem. Mater. 2021, 33, 1265–1275. [Google Scholar] [CrossRef]
- Kumar, S.G.; Devi, L.G. Review on Modified TiO2 Photocatalysis under UV/Visible Light: Selected Results and Related Mechanisms on Interfacial Charge Carrier Transfer Dynamics. J. Phys. Chem. A 2011, 115, 13211–13241. [Google Scholar] [CrossRef]
- Pan, X.; Yang, M.Q.; Fu, X.; Zhang, N.; Xu, Y.J. Defective TiO2 with Oxygen Vacancies: Synthesis, Properties and Photocatalytic Applications. Nanoscale 2013, 5, 3601–3614. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, A.C.; Beglitis, N.S.; Pang, C.L.; Teobaldi, G.; Cabailh, G.; Chen, Q.; Fisher, A.J.; Hofer, W.A.; Thornton, G. Electron Traps and Their Effect on the Surface Chemistry of TiO2(110). Proc. Natl. Acad. Sci. USA 2010, 107, 2391–2396. [Google Scholar] [CrossRef]
- Sun, H.; Yu, M.; Wang, G.; Sun, X.; Lian, J. Temperature-Dependent Morphology Evolution and Surface Plasmon Absorption of Ultrathin Gold Island Films. J. Phys. Chem. C 2012, 116, 9000–9008. [Google Scholar] [CrossRef]
- Juvé, V.; Cardinal, M.F.; Lombardi, A.; Crut, A.; Maioli, P.; Pérez-Juste, J.; Liz-Marzán, L.M.; Del Fatti, N.; Vallée, F. Size-Dependent Surface Plasmon Resonance Broadening in Nonspherical Nanoparticles: Single Gold Nanorods. Nano Lett. 2013, 13, 2234–2240. [Google Scholar] [CrossRef]
- Hill, R.R.; Jeffs, G.E.; Roberts, D.R. Photocatalytic Degradation of 1,4-Dioxane in Aqueous Solution. J. Photochem. Photobiol. A Chem. 1997, 108, 55–88. [Google Scholar] [CrossRef]
- Yamazaki, S.; Yamabe, N.; Nagano, S.; Fukuda, A. Adsorption and Photocatalytic Degradation of 1,4-Dioxane on TiO2. J. Photochem. Photobiol. A Chem. 2007, 185, 150–155. [Google Scholar] [CrossRef]
- Tawfik, A. Degradation Pathways of 1,4-Dioxane in Biological and Advanced Oxidation Processes. Desalin. Water Treat. 2020, 178, 360–386. [Google Scholar] [CrossRef]
- Padikkaparambil, S.; Narayanan, B.; Yaakob, Z.; Viswanathan, S.; Tasirin, S.M. Au/TiO2 Reusable Photocatalysts for Dye Degradation. Int. J. Photoenergy 2013, 2013, 752605. [Google Scholar] [CrossRef]
- Dorfman, L.E.; Adams, G.E. Reactivity of the Hydroxyl Radical in Aqueous Solutions; National Bureau of Standards: Gaithersburg, MD, USA, 1972.
- Schmid, J.; Hoenes, K.; Rath, M.; Vatter, P.; Hessling, M. Antimicrobial efficacy of 222 nm UVC light in the hospital environment. GMS Hyg. Infect. Control 2017, 12, Doc06. [Google Scholar] [PubMed]
- Chitra, S.; Paramasivan, K.; Cheralathan, M.; Sinha, P.K. Degradation of 1,4-dioxane using advanced oxidation processes. Environ. Sci. Pollut. Res. 2012, 19, 871–878. [Google Scholar] [CrossRef]
- Barndõk, H.; Hermosilla, D.; Han, C.; Dionysiou, D.D.; Negro, C.; Blanco, Á. Degradation of pharmaceuticals in water by advanced oxidation processes: A review. Appl. Catal. B Environ. 2016, 180, 44–52. [Google Scholar] [CrossRef]
- Seshadri, H.; Cheralathan, M.; Sinha, P.K. Photocatalytic performance of combustion-synthesized β and γ-Ga2O3 in the degradation of 1,4-dioxane in aqueous solution. Res. Chem. Intermed. 2013, 39, 991–1001. [Google Scholar] [CrossRef]
- Wang, W.; Qiao, Z.; Lee, G.J.; Chen, H.; Ding, L.; Zhu, M.; Liu, N.; Wu, J.J. In-situ anodic precipitation process for highly efficient separation of aluminium from impurities. Sep. Purif. Technol. 2020, 235, 116194. [Google Scholar] [CrossRef]
- Wang, W.M.; Zhang, L.; Wang, W.L.; Huang, J.Y.; Wu, Q.Y.; Wu, J.J. Photocatalytic Degradation of 1,4-Dioxane by Heterostructured Bi2O3/Cu-MOF with Oxygen Vacancies. Catalysts 2023, 13, 1211. [Google Scholar] [CrossRef]
- Son, H.S.; Im, J.K.; Zoh, K.D. A Fenton-like degradation mechanism for 1,4-dioxane using zero-valent iron (Fe⁰) and UV light. Water Res. 2009, 43, 1457–1463. [Google Scholar] [CrossRef]
- Xu, X.; Liu, S.; Smith, K.; Wang, Y.; Hu, H. Photocatalytic degradation of 1,4-dioxane using TiO2 modified with W: Influence of W content and calcination temperature. Chem. Eng. J. 2019, 373, 508. [Google Scholar]
- Mehrvar, M.; Anderson, W.A.; Moo-Young, M. Photocatalytic degradation of aqueous organic contaminants by UV/H2O2: Process optimization using response surface methodology. Int. J. Photoenergy 2000, 2, 67–80. [Google Scholar] [CrossRef]
- Mehrvar, M.; Anderson, W.A.; Moo-Young, M. Photocatalytic degradation of aqueous organic contaminants by UV/H2O2: A kinetic approach. Int. J. Photoenergy 2002, 4, 141–146. [Google Scholar] [CrossRef]
- Park, Y.K.; Chung, K.H.; Park, I.S.; Kim, S.C.; Kim, S.J.; Jung, S.C. Photocatalytic degradation of 1,4-dioxane using TiO2 under UV irradiation: Kinetics and mechanism. J. Hazard. Mater. 2020, 399, 123087. [Google Scholar] [CrossRef]
- Byrne, C.; Dervin, S.; Hermosilla, D.; Merayo, N.; Blanco, Á.; Hinder, S.; Harb, M.; Dionysiou, D.D.; Pillai, S.C. A review of heterogeneous photocatalysis for water and wastewater treatment: Fundamentals, mechanisms, and applications. Catal. Today 2021, 380, 199–208. [Google Scholar] [CrossRef]
- Ajmal, A.; Majeed, I.; Malik, R.N.; Idriss, H.; Nadeem, M.A. Principles and mechanisms of photocatalytic dye degradation on TiO2 based photocatalysts: A comparative overview. RSC Adv. 2014, 4, 37003–37026. [Google Scholar] [CrossRef]
- Lee, K.M.; Lai, C.W.; Ngai, K.S.; Juan, J.C. Recent developments of zinc oxide based photocatalyst in water treatment technology: A review. Water Res. 2016, 88, 428–448. [Google Scholar] [CrossRef]
- Makuła, P.; Pacia, M.; Macyk, W. How to correctly determine the band gap energy of modified semiconductor photocatalysts based on UV–Vis spectra. J. Phys. Chem. Lett. 2018, 9, 6814–6817. [Google Scholar] [CrossRef] [PubMed]
- Coulter, J.B.; Birnie, D.P. Determining direct and indirect optical band gap energies from UV–Vis absorption spectra. Phys. Status Solidi Basic Res. 2018, 255, 1700393. [Google Scholar] [CrossRef]
- López, R.; Gómez, R. Band-gap energy estimation from diffuse reflectance measurements on sol–gel and commercial TiO2: A comparative study. J. Sol-Gel Sci. Technol. 2012, 61, 1–7. [Google Scholar] [CrossRef]
- Thermo Fisher Scientific. Band Gap Analysis Through UV-Visible Spectroscopy; Thermo Fisher Scientific: Waltham, MA, USA, 2023. [Google Scholar]
- Zhao, Y.; Jia, X.; Waterhouse, G.I.N.; Wu, L.Z.; Tung, C.H.; O’Hare, D.; Zhang, T. Layered double hydroxide nanostructured photocatalysts for water splitting: A review. Adv. Energy Mater. 2016, 6, 1501974. [Google Scholar] [CrossRef]
- Miao, L.; Jin, P.; Kaneko, K.; Terai, A.; Nabatova-Gabain, N.; Tanemura, S. Preparation and characterization of TiO2 thin films on Si substrates by sol–gel method. Appl. Surf. Sci. 2003. [Google Scholar] [CrossRef]
- Nagaraj, G.; Brundha, D.; Chandraleka, C.; Arulpriya, M.; Kowsalya, V.; Sangavi, S.; Jayalakshmi, R.; Tamilarasu, S.; Murugan, R. Green synthesis of zinc oxide nanoparticles using plant extracts and their antimicrobial activity: A review. SN Appl. Sci. 2020, 2, 1–9. [Google Scholar] [CrossRef]
- Jiang, L.; Gao, X.; Chen, S.; Ashok, J.; Kawi, S. Oxygen-Deficient WO3/TiO2/CC Nanorod Arrays for Visible-Light Photocatalytic Degradation of Methylene Blue. Catalysts 2021, 11, 1349. [Google Scholar] [CrossRef]
- Senthil, R.A.; Theerthagiri, J.; Selvi, A.; Madhavan, J. Synthesis and characterization of low-cost g-C3N4/TiO2 composite with enhanced photocatalytic performance under visible-light irradiation. Opt. Mater. 2017, 64, 533–539. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.; Tao, M.J.; Mair, L.O.; Singh, A.K.; Fang, Y.; Rajendran, S.; Beechem, T.E.; Warsinger, D.M.; Moran, J.L. Degradation of 1,4-Dioxane by Au/TiO2 Janus Nanoparticles Under Ultraviolet Light: Experiments and Modeling. Water 2025, 17, 1708. https://doi.org/10.3390/w17111708
Ji Y, Tao MJ, Mair LO, Singh AK, Fang Y, Rajendran S, Beechem TE, Warsinger DM, Moran JL. Degradation of 1,4-Dioxane by Au/TiO2 Janus Nanoparticles Under Ultraviolet Light: Experiments and Modeling. Water. 2025; 17(11):1708. https://doi.org/10.3390/w17111708
Chicago/Turabian StyleJi, Yangyuan, Matthew J. Tao, Lamar O. Mair, Amit Kumar Singh, Yuhang Fang, Sathish Rajendran, Thomas E. Beechem, David M. Warsinger, and Jeffrey L. Moran. 2025. "Degradation of 1,4-Dioxane by Au/TiO2 Janus Nanoparticles Under Ultraviolet Light: Experiments and Modeling" Water 17, no. 11: 1708. https://doi.org/10.3390/w17111708
APA StyleJi, Y., Tao, M. J., Mair, L. O., Singh, A. K., Fang, Y., Rajendran, S., Beechem, T. E., Warsinger, D. M., & Moran, J. L. (2025). Degradation of 1,4-Dioxane by Au/TiO2 Janus Nanoparticles Under Ultraviolet Light: Experiments and Modeling. Water, 17(11), 1708. https://doi.org/10.3390/w17111708