



Distribution of Antibiotic Resistance Genes and Their Association with Microbes in Wastewater Treatment Plants: A Metagenomics Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. Metagenomics Sequencing and Data Analysis
3. Results and Discussion
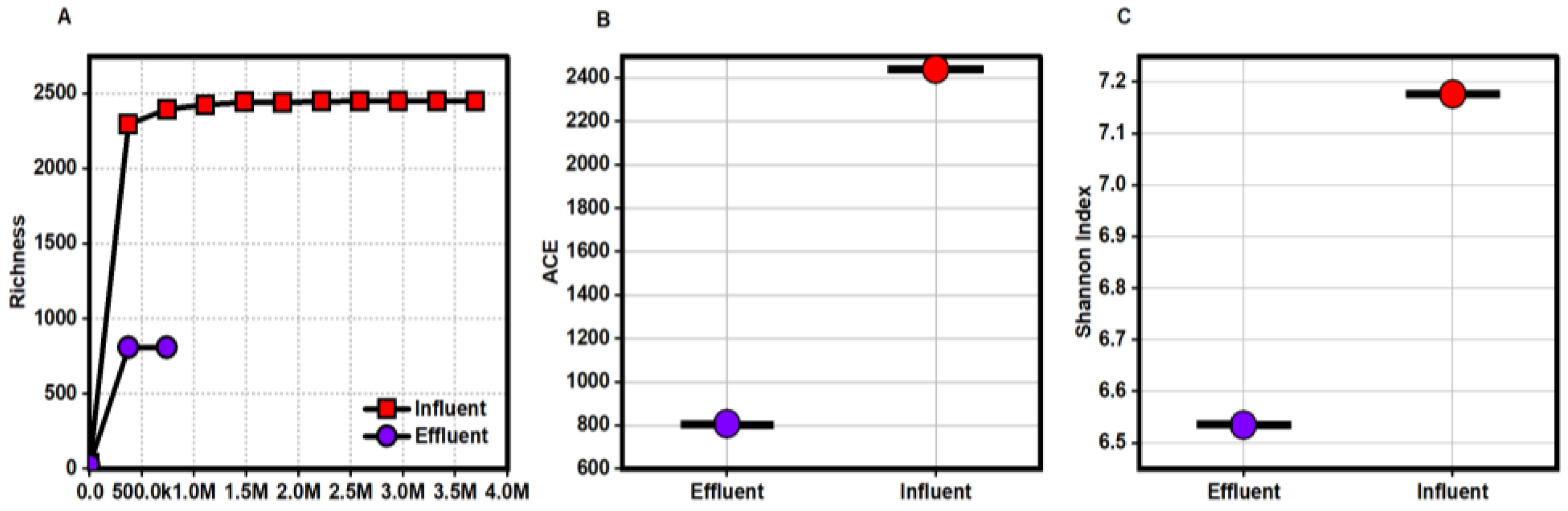
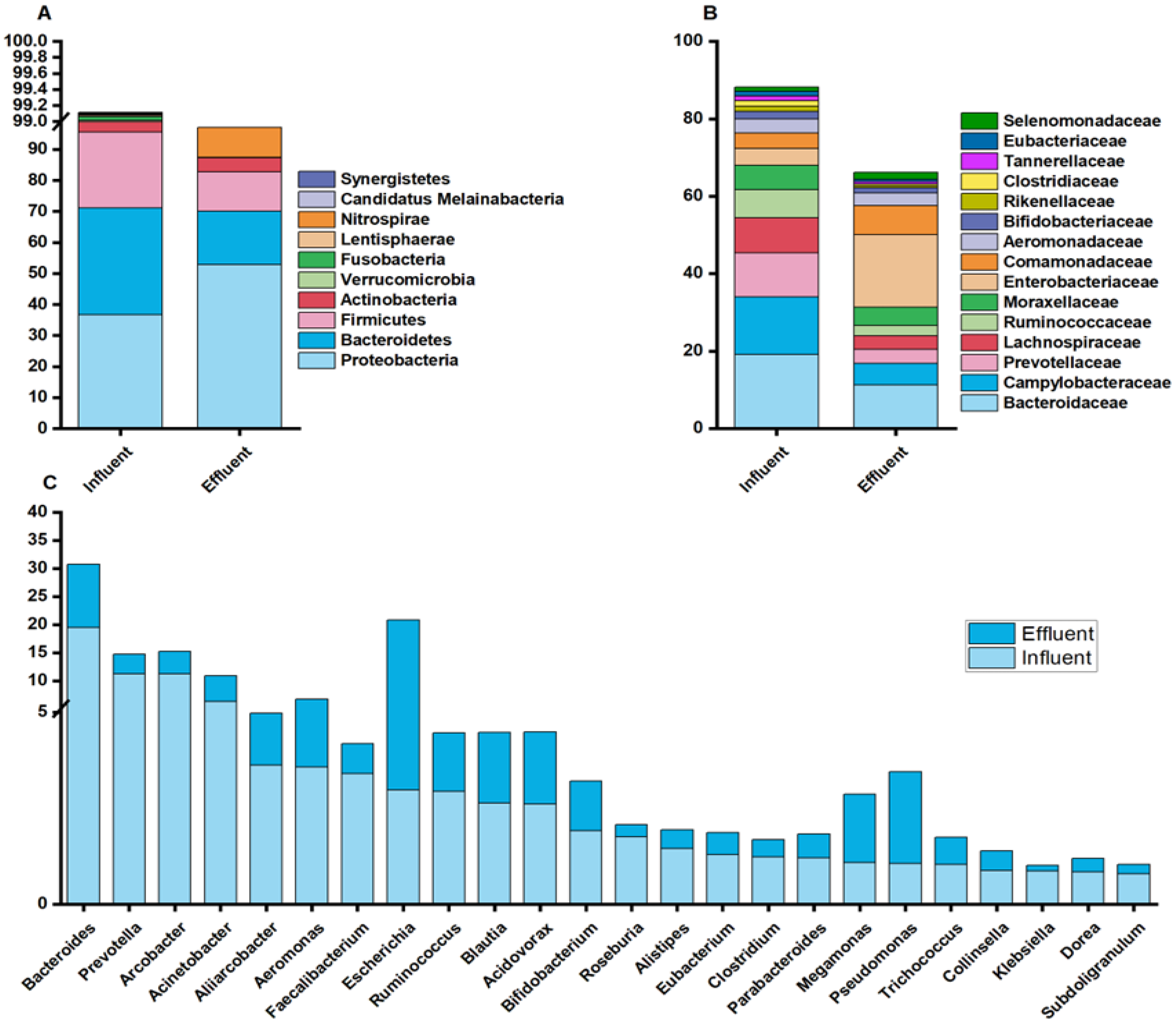
3.1. Diversity and Richness of Microbial Communities
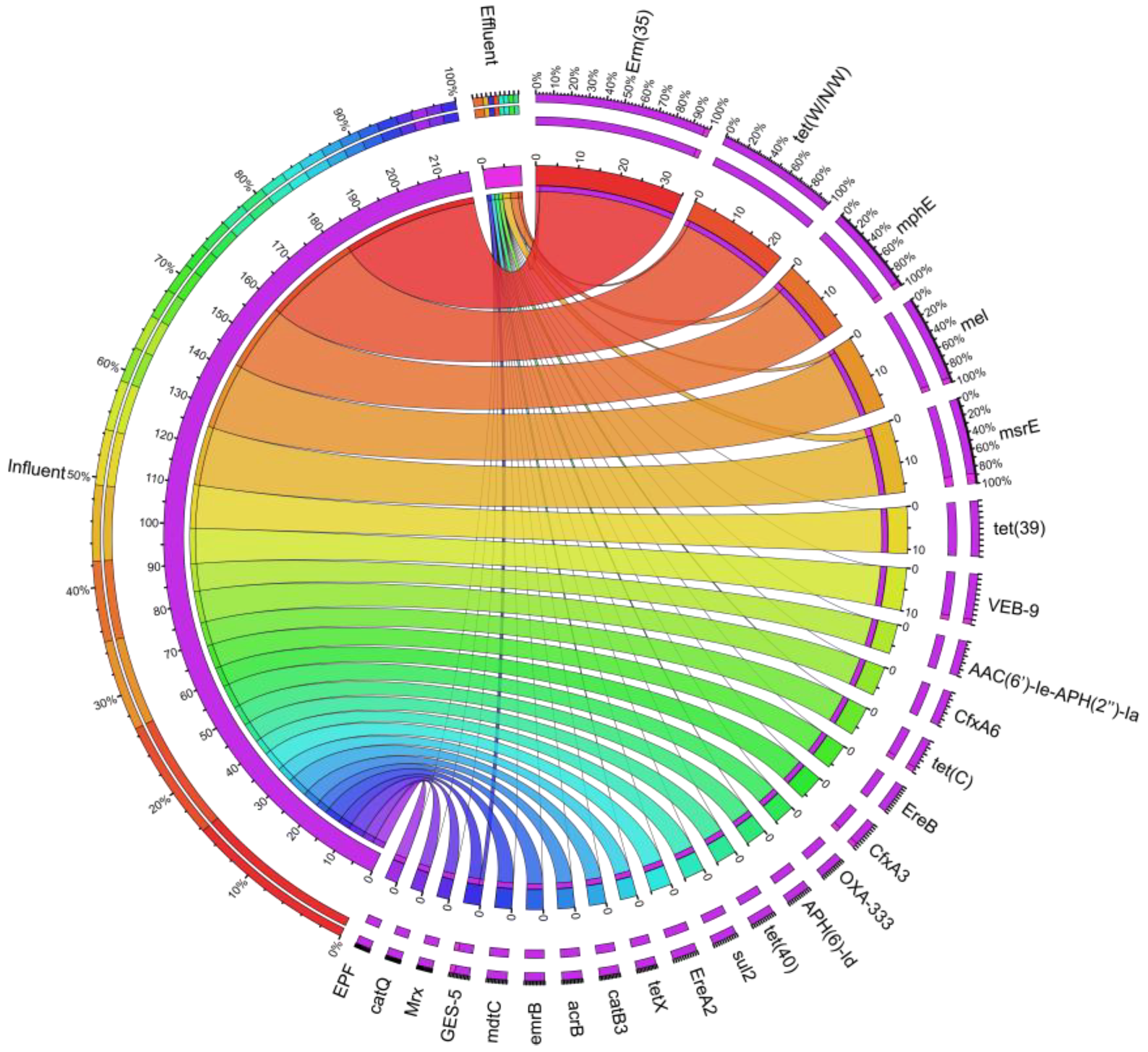
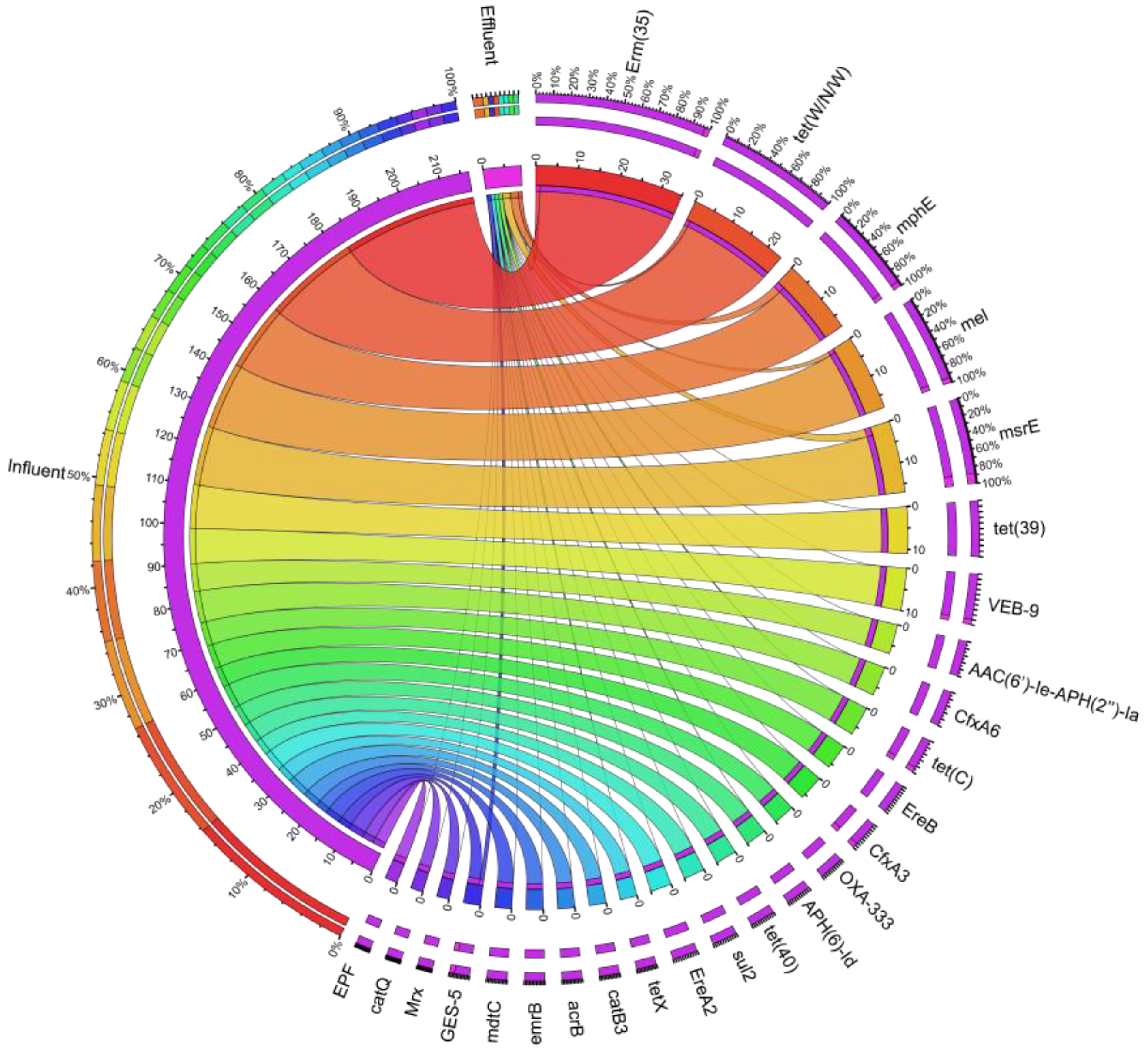
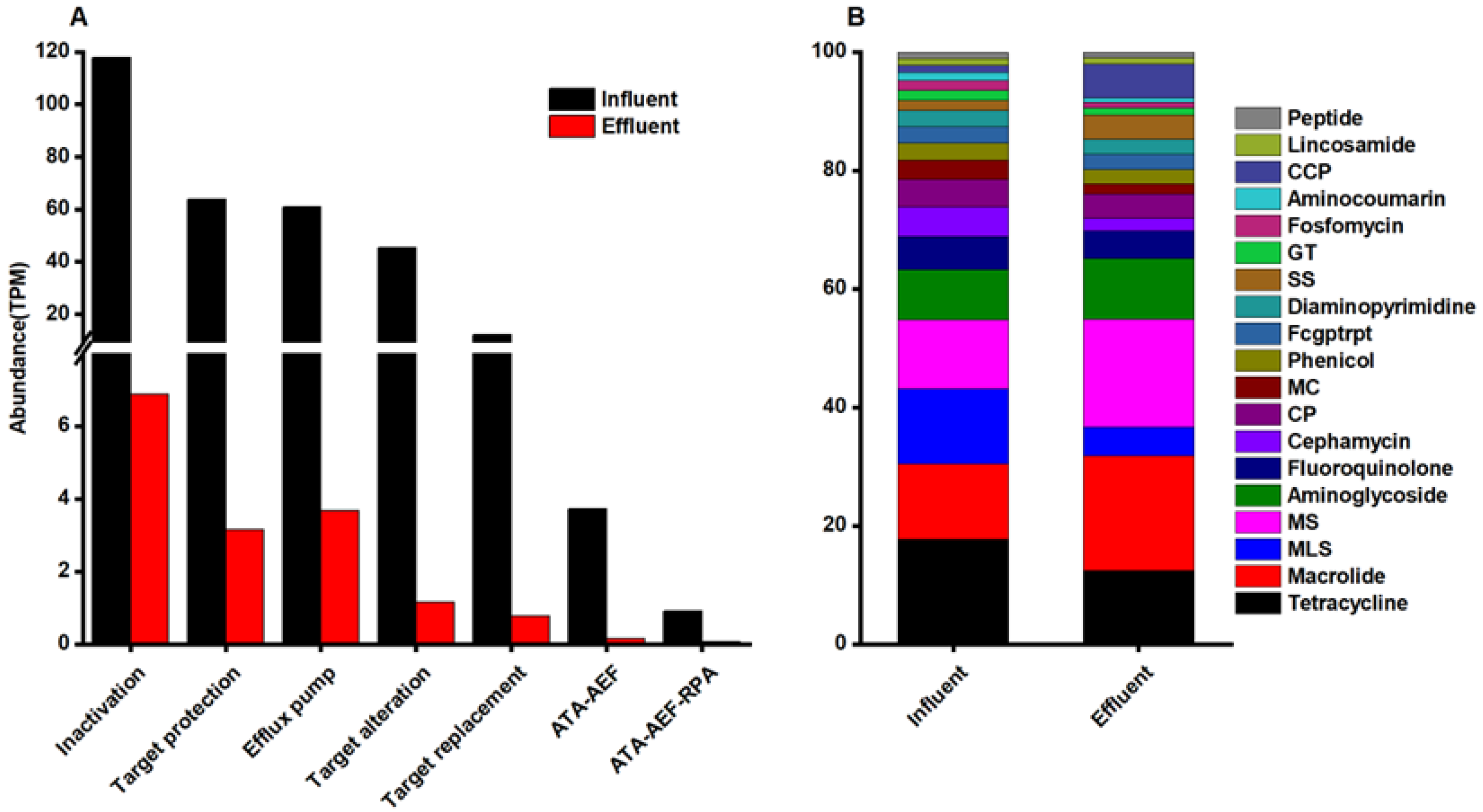
3.2. Characteristics of ARGs
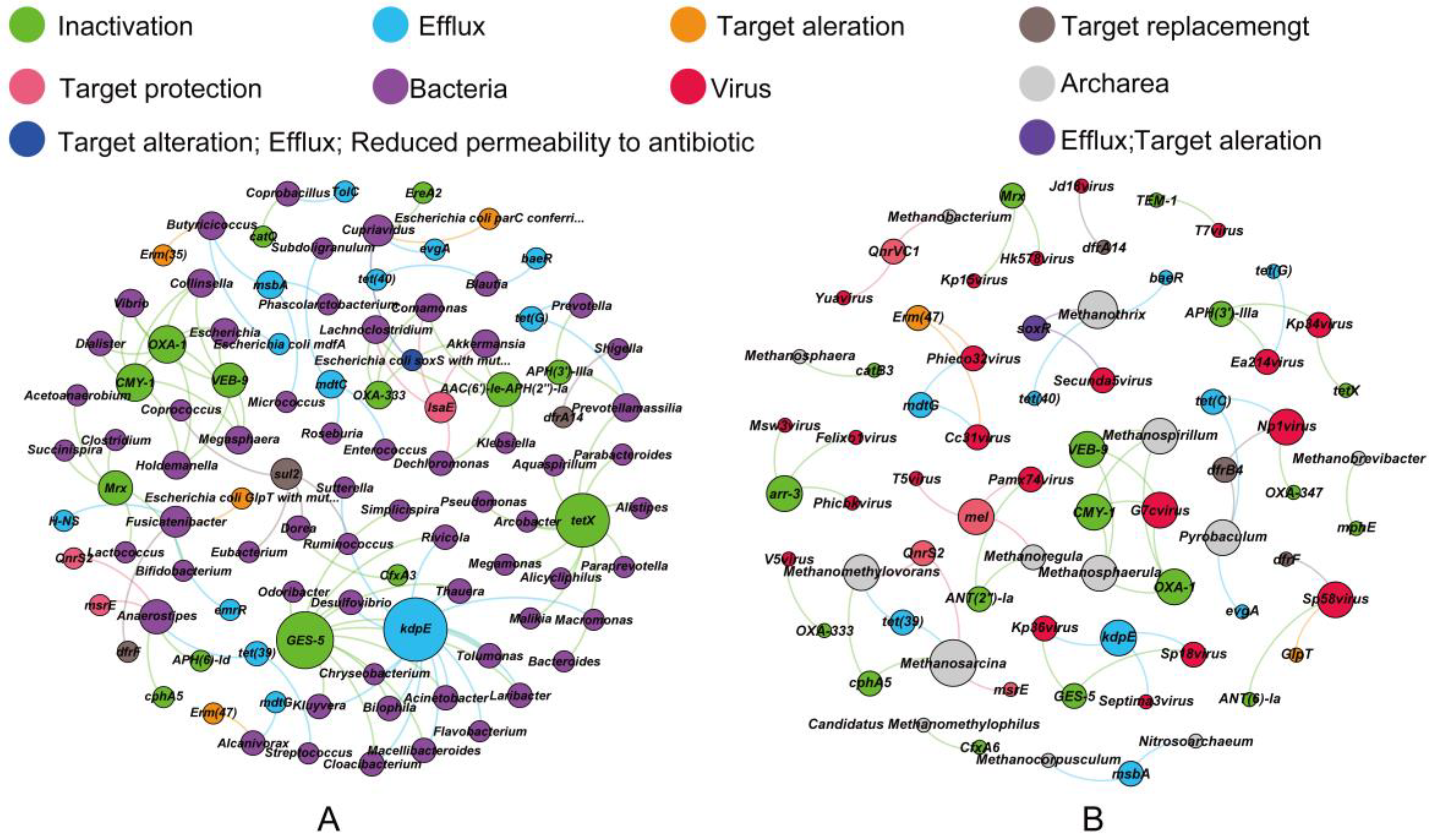
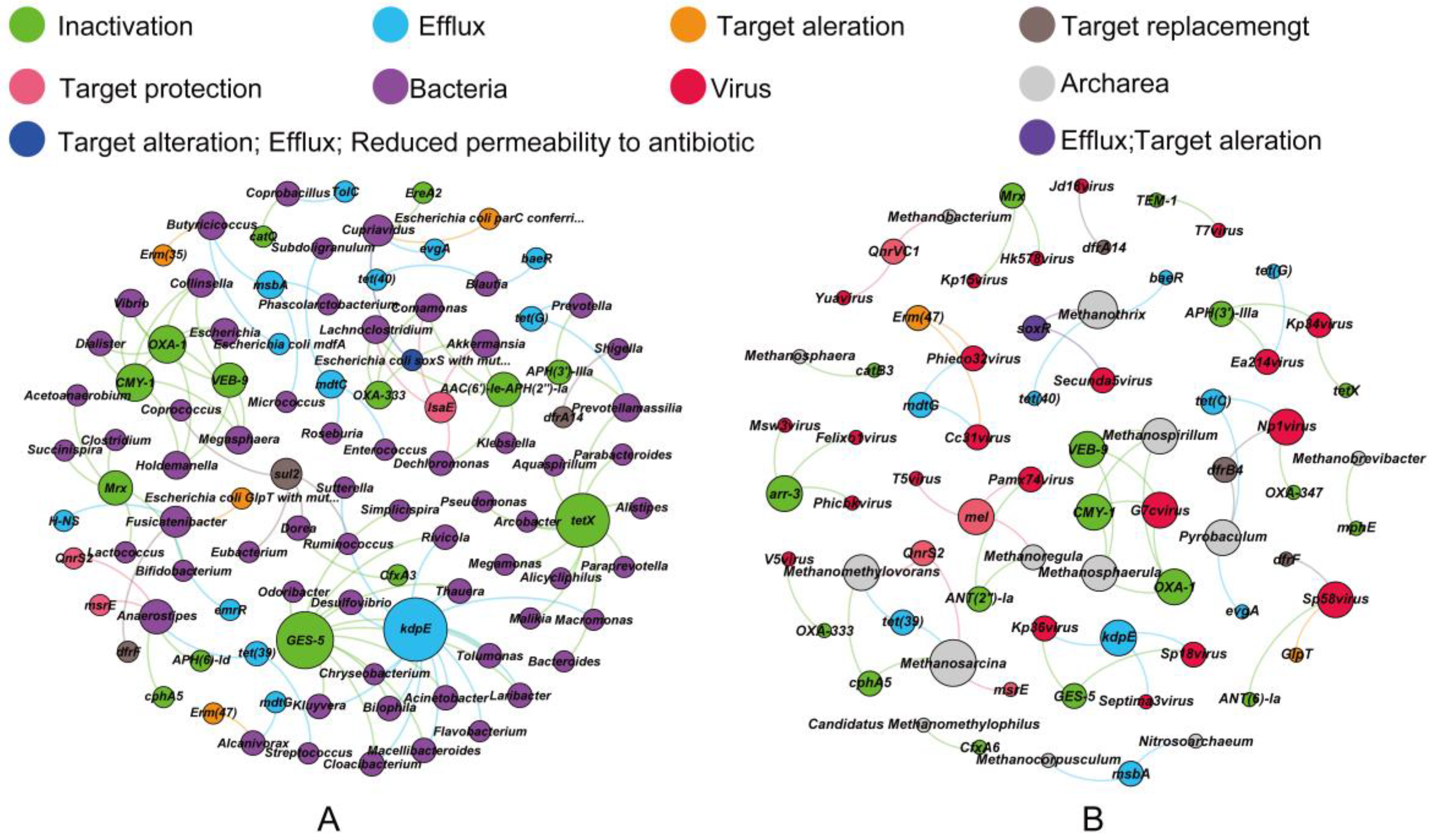
3.3. Co-Occurrence of ARGs and Microbes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviation
| Abbreviation | Full Name |
| MLS | macrolide antibiotic; lincosamide antibiotic; streptogramin antibiotic |
| MS | macrolide antibiotic; streptogramin antibiotic |
| CP | cephalosporin; penam |
| MC | monobactam; cephalosporin |
| FCGPTRPT | fluoroquinolone antibiotic; cephalosporin; glycylcycline; penam; tetracycline antibiotic; rifamycin antibiotic; phenicol antibiotic; triclosan |
| SS | sulfonamide antibiotic; sulfone antibiotic |
| GT | glycylcycline; tetracycline antibiotic |
| CCP | carbapenem; cephalosporin; penam |
References
- Manoharan, R.K.; Ishaque, F.; Ahn, Y.H. Fate of antibiotic resistant genes in wastewater environments and treatment strategies—A review. Chemosphere 2022, 298, 134671. [Google Scholar] [CrossRef]
- van der Donk, C.F.M.; van de Bovenkamp, J.H.B.; De Brauwer, E.I.G.B.; De Mol, P.; Feldhoff, K.-H.; Kalka-Moll, W.M.; Nys, S.; Thoelen, I.; Trienekens, T.A.M.; Stobberingh, E.E. Antimicrobial Resistance and Spread of Multi Drug Resistant Escherichia coli Isolates Collected from Nine Urology Services in the Euregion Meuse-Rhine. PLoS ONE 2012, 7, e47707. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.G.H.; Verner-Jeffreys, D.W.; Baker-Austin, C. Aquatic systems: Maintaining, mixing and mobilising antimicrobial resistance? Trends Ecol. Evol. 2011, 26, 278–284. [Google Scholar] [CrossRef]
- Aminov, R.I. The role of antibiotics and antibiotic resistance in nature. Environ. Microbiol. 2009, 11, 2970–2988. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.D.; Esteve, C.; Alcaide, E. Multiresistant waterborne pathogens isolated from water reservoirs and cooling systems. J. Appl. Microbiol. 2008, 105, 469–475. [Google Scholar] [CrossRef]
- Forsberg, K.J.; Reyes, A.; Wang, B.; Selleck, E.M.; Sommer, M.O.A.; Dantas, G. The Shared Antibiotic Resistome of Soil Bacteria and Human Pathogens. Science 2012, 337, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Bi, X.; Peng, Y.; Bai, M. Research advances of the phosphorus-accumulating organisms of Candidatus Accumulibacter, Dechloromonas and Tetrasphaera: Metabolic mechanisms, applications and influencing factors. Chemosphere 2022, 307, 135675. [Google Scholar] [CrossRef]
- Łuczkiewicz, A.; Jankowska, K.; Fudala-Książek, S.; Olańczuk-Neyman, K. Antimicrobial resistance of fecal indicators in municipal wastewater treatment plant. Water Res. 2010, 44, 5089–5097. [Google Scholar] [CrossRef] [PubMed]
- Novo, A.; Manaia, C.M. Factors influencing antibiotic resistance burden in municipal wastewater treatment plants. Appl. Microbiol. Biot. 2010, 87, 1157–1166. [Google Scholar] [CrossRef]
- Ju, F.; Beck, K.; Yin, X.; Maccagnan, A.; McArdell, C.S.; Singer, H.P.; Johnson, D.R.; Zhang, T.; Bürgmann, H. Wastewater treatment plant resistomes are shaped by bacterial composition, genetic exchange, and upregulated expression in the effluent microbiomes. ISME J. 2019, 13, 346–360. [Google Scholar] [CrossRef]
- Guo, J.; Li, J.; Chen, H.; Bond, P.L.; Yuan, Z. Metagenomic analysis reveals wastewater treatment plants as hotspots of antibiotic resistance genes and mobile genetic elements. Water Res. 2017, 123, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.; Kasprzyk-Hordern, B. Future perspectives of wastewater-based epidemiology: Monitoring infectious disease spread and resistance to the community level. Environ. Int. 2020, 139, 105689. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jeon, J.H.; Shin, J.; Jang, H.M.; Kim, S.; Song, M.S.; Kim, Y.M. Quantitative and qualitative changes in antibiotic resistance genes after passing through treatment processes in municipal wastewater treatment plants. Sci. Total Environ. 2017, 605–606, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, E.A.; Ramirez, D.; Bálcazar J., L.; Jiménez J., N. Metagenomic analysis of urban wastewater resistome and mobilome: A support for antimicrobial resistance surveillance in an endemic country. Environ. Pollut. 2021, 276, 116736. [Google Scholar] [CrossRef]
- Mao, D.; Yu, S.; Rysz, M.; Luo, Y.; Yang, F.; Li, F.; Hou, J.; Mu, Q.; Alvarez, P.J.J. Prevalence and proliferation of antibiotic resistance genes in two municipal wastewater treatment plants. Water Res. 2015, 85, 458–466. [Google Scholar] [CrossRef]
- Yin, X.; Yang, Y.; Deng, Y.; Huang, Y.; Li, L.; Chan, L.Y.L.; Zhang, T. An assessment of resistome and mobilome in wastewater treatment plants through temporal and spatial metagenomic analysis. Water Res. 2022, 209, 117885. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, B.; Lv, P.; Hou, J.; Qiu, Y.; Huang, X. Distribution of antibiotic resistance genes and their association with bacteria and viruses in decentralized sewage treatment facilities. Front. Environ. Sci. Eng. 2021, 16, 35. [Google Scholar] [CrossRef]
- Su, J.-Q.; Wei, B.; Ou-Yang, W.-Y.; Huang, F.-Y.; Zhao, Y.; Xu, H.-J.; Zhu, Y.-G. Antibiotic Resistome and Its Association with Bacterial Communities during Sewage Sludge Composting. Environ. Sci. Technol. 2015, 49, 7356–7363. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Insights into antibiotic resistance through metagenomic approaches. Future Microbiol. 2011, 7, 73–89. [Google Scholar] [CrossRef]
- Yang, Y.; Li, B.; Zou, S.; Fang, H.H.P.; Zhang, T. Fate of antibiotic resistance genes in sewage treatment plant revealed by metagenomic approach. Water Res. 2014, 62, 97–106. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M. Analysis of Microbial Communities and Pathogen Detection in Domestic Sewage Using Metagenomic Sequencing. Diversity 2020, 13, 6. [Google Scholar] [CrossRef]
- Giwa, A.S.; Ali, N.; Athar, M.A.; Wang, K. Dissecting microbial community structure in sewage treatment plant for pathogens’ detection using metagenomic sequencing technology. Arch. Microbiol. 2020, 202, 825–833. [Google Scholar] [CrossRef]
- Sørensen, S.J.; Bailey, M.; Hansen, L.H.; Kroer, N.; Wuertz, S. Studying plasmid horizontal transfer in situ: A critical review. Nat. Rev. Microbiol. 2005, 3, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Guan, Y.; Zhao, R.; Feng, J.; Huang, J.; Ma, L.; Li, B. Metagenomic and network analyses decipher profiles and co-occurrence patterns of antibiotic resistome and bacterial taxa in the reclaimed wastewater distribution system. J. Hazard. Mater. 2020, 400, 123170. [Google Scholar] [CrossRef]
- Ferreira da Silva, M.; Vaz-Moreira, I.; Gonzalez-Pajuelo, M.; Nunes, O.C.; Manaia, C.M. Antimicrobial resistance patterns in Enterobacteriaceae isolated from an urban wastewater treatment plant. FEMS Microbiol. Ecol. 2007, 60, 166–176. [Google Scholar] [CrossRef]
- Tong, J.; Tang, A.; Wang, H.; Liu, X.; Huang, Z.; Wang, Z.; Zhang, J.; Wei, Y.; Su, Y.; Zhang, Y. Microbial community evolution and fate of antibiotic resistance genes along six different full-scale municipal wastewater treatment processes. Bioresource Technol. 2019, 272, 489–500. [Google Scholar] [CrossRef]
- Affek, K.; Muszyński, A.; Załęska-Radziwiłł, M.; Doskocz, N.; Ziętkowska, A.; Widomski, M. Evaluation of ecotoxicity and inactivation of bacteria during ozonation of treated wastewater. Desalin. Water Treat. 2020, 192, 176–184. [Google Scholar] [CrossRef]
- Affek, K.; Muszyński, A.; Doskocz, N.; Załęska-Radziwiłł, M. Ecotoxicological effects of disinfection of treated wastewater. Desalin. Water Treat. 2021, 233, 190–198. [Google Scholar] [CrossRef]
- Li, D.; Jiang, X.; Wang, J.; Wang, K.; Zheng, B. Effect of Sewage and Industrial Effluents on Bacterial and Archaeal Communities of Creek Sediments in the Taihu Basin. Water 2017, 9, 373. [Google Scholar] [CrossRef]
- Xu, R.; Zhang, Y.; Xiong, W.; Sun, W.; Fan, Q.; Zhaohui, Y. Metagenomic approach reveals the fate of antibiotic resistance genes in a temperature-raising anaerobic digester treating municipal sewage sludge. J. Clean. Prod. 2020, 277, 123504. [Google Scholar] [CrossRef]
- Tabatabaei, M.; Rahim, R.A.; Abdullah, N.; Wright, A.-D.G.; Shirai, Y.; Sakai, K.; Sulaiman, A.; Hassan, M.A. Importance of the methanogenic archaea populations in anaerobic wastewater treatments. Process Biochem. 2010, 45, 1214–1225. [Google Scholar] [CrossRef]
- Robertson, C.E.; Harris, J.K.; Spear, J.R.; Pace, N.R. Phylogenetic diversity and ecology of environmental Archaea. Curr. Opin. Microbiol. 2005, 8, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Wojcieszak, M.; Pyzik, A.; Poszytek, K.; Krawczyk, P.S.; Sobczak, A.; Lipinski, L.; Roubinek, O.; Palige, J.; Sklodowska, A.; Drewniak, L. Adaptation of methanogenic inocula to anaerobic digestion of maize silage. Front. Microbiol. 2017, 8, 1881. [Google Scholar] [CrossRef]
- Garlicka, A.; Umiejewska, K.; Nielsen, P.H.; Muszynski, A. Hydrodynamic disintegration of thickened excess sludge and maize silage to intensify methane production: Energy effect and impact on microbial communities. Bioresource Technol. 2023, 376, 128829. [Google Scholar] [CrossRef] [PubMed]
- Gulino, K.; Rahman, J.; Badri, M.; Morton, J.; Bonneau, R.; Ghedin, E. Initial Mapping of the New York City Wastewater Virome. mSystems 2020, 5, e00876-00819. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.-W. Tailed Bacteriophages: The Order Caudovirales. Adv. Virus Res. 1998, 51, 135–201. [Google Scholar] [CrossRef] [PubMed]
- Hultman, J.; Tamminen, M.; Pärnänen, K.; Cairns, J.; Karkman, A.; Virta, M. Host range of antibiotic resistance genes in wastewater treatment plant influent and effluent. FEMS Microbiol. Ecol. 2018, 94, fiy038. [Google Scholar] [CrossRef]
- Pazda, M.; Kumirska, J.; Stepnowski, P.; Mulkiewicz, E. Antibiotic resistance genes identified in wastewater treatment plant systems – A review. Sci. Total Environ. 2019, 697, 134023. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Bu, Y.; Zhang, X.-X.; Huang, K.; He, X.; Ye, L.; Shan, Z.; Ren, H. Metagenomic analysis of bacterial community composition and antibiotic resistance genes in a wastewater treatment plant and its receiving surface water. Ecotoxicol. Environ. Saf. 2016, 132, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Versporten, A.; Zarb, P.; Caniaux, I.; Gros, M.-F.; Drapier, N.; Miller, M.; Jarlier, V.; Nathwani, D.; Goossens, H.; Koraqi, A.; et al. Antimicrobial consumption and resistance in adult hospital inpatients in 53 countries: Results of an internet-based global point prevalence survey. Lancet Global Health 2018, 6, e619–e629. [Google Scholar] [CrossRef] [PubMed]
- Nnadozie, C.F.; Kumari, S.; Bux, F. Status of pathogens, antibiotic resistance genes and antibiotic residues in wastewater treatment systems. Rev. Environ. Sci. Bio/Technol. 2017, 16, 491–515. [Google Scholar] [CrossRef]
- Kumar, N.; Radhakrishnan, A.; Wright, C.C.; Chou, T.-H.; Lei, H.-T.; Bolla, J.R.; Tringides, M.L.; Rajashankar, K.R.; Su, C.-C.; Purdy, G.E.; et al. Crystal structure of the transcriptional regulator Rv1219c of Mycobacterium tuberculosis. Protein Sci. 2014, 23, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Nishino, K.; Roberts, M.C.; Tolmasky, M.; Aminov, R.I.; Zhang, L. Mechanisms of antibiotic resistance. Front. Microbiol. 2015, 6, 34. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria | Archaea | Virus | Eukaryota | Unassigned | ||
|---|---|---|---|---|---|---|
| Influent | Phylum | 99.1243 | 0.00867 | - | 0.0030 | 0.7860 |
| Class | 99.3440 | 0.00867 | - | 0.0023 | 0.5670 | |
| Order | 99.4580 | 0.0867 | 0.0103 | 0.0027 | 0.4423 | |
| Family | 99.4773 | 0.0857 | 0.0020 | 0.0073 | 0.4277 | |
| Genus | 99.6193 | 0.0870 | 0.0023 | 0.0020 | 0.2893 | |
| Species | 99.5957 | 0.0837 | 0.0043 | 0.0020 | 0.3143 | |
| Effluent | Phylum | 97.4540 | 0.7853 | - | 0.1297 | 1.6310 |
| Class | 97.8333 | 0.7803 | - | 0.1293 | 1.2570 | |
| Order | 98.0737 | 0.7827 | 0.0063 | 0.1327 | 1.0047 | |
| Family | 99.1380 | 0.7837 | 0.0050 | 0.1320 | 0.9413 | |
| Genus | 99.3617 | 0.7877 | 0.0000 | 0.1343 | 0.7163 | |
| Species | 98.5607 | 0.7710 | 0.0003 | 0.1897 | 0.4783 |
| Bacteria | Archaea | Virus | Eukaryota | ||
|---|---|---|---|---|---|
| Influent | Phylum | 23 | 3 | - | 3 |
| Class | 40 | 4 | - | 5 | |
| Order | 94 | 6 | 1 | 7 | |
| Family | 195 | 9 | 4 | 11 | |
| Genus | 680 | 14 | 25 | 12 | |
| Species | 2723 | 26 | 15 | 9 | |
| Effluent | Phylum | 19 | 3 | - | 3 |
| Class | 38 | 4 | - | 9 | |
| Order | 88 | 6 | 1 | 11 | |
| Family | 174 | 8 | 3 | 14 | |
| Genus | 410 | 12 | NA | 11 | |
| Species | 971 | 17 | 1 | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, M.; Hu, X.; Li, M.; Lyu, C.; Hu, Y.; Bu, X.; Chen, T.; Cai, H.; Li, C.; Liu, J.; et al. Distribution of Antibiotic Resistance Genes and Their Association with Microbes in Wastewater Treatment Plants: A Metagenomics Analysis. Water 2023, 15, 1587. https://doi.org/10.3390/w15081587
Shen M, Hu X, Li M, Lyu C, Hu Y, Bu X, Chen T, Cai H, Li C, Liu J, et al. Distribution of Antibiotic Resistance Genes and Their Association with Microbes in Wastewater Treatment Plants: A Metagenomics Analysis. Water. 2023; 15(8):1587. https://doi.org/10.3390/w15081587
Chicago/Turabian StyleShen, Mengnan, Xiaowei Hu, Ming Li, Chen Lyu, Yi Hu, Xiaodan Bu, Tao Chen, Hang Cai, Chenyang Li, Jiahong Liu, and et al. 2023. "Distribution of Antibiotic Resistance Genes and Their Association with Microbes in Wastewater Treatment Plants: A Metagenomics Analysis" Water 15, no. 8: 1587. https://doi.org/10.3390/w15081587
APA StyleShen, M., Hu, X., Li, M., Lyu, C., Hu, Y., Bu, X., Chen, T., Cai, H., Li, C., Liu, J., & Fan, K. (2023). Distribution of Antibiotic Resistance Genes and Their Association with Microbes in Wastewater Treatment Plants: A Metagenomics Analysis. Water, 15(8), 1587. https://doi.org/10.3390/w15081587