
Potential Use of Precipitates from Acid Mine Drainage (AMD) as Arsenic Adsorbents

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. OxPFe Precipitate Obtention
2.3. Acid Mine Drainage and Supernatant Characterization
2.4. Adsorbent Preparation
2.5. Adsorbents Characterization
2.6. Theoretical Speciation of Supernatant AMD and Mass Balance of Precipitates
2.7. Batch As(V) Adsorption Experiments
3. Results
3.1. Raw AMD and Treated Supernatant Characterization
3.2. Adsorbents Characterization
3.2.1. Calcium, Iron, Aluminum and Sulfate Content Determination
3.2.2. X-ray Diffraction Analyses
3.2.3. Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray Spectroscopy (EDX) Characterization
3.2.4. Adsorbent Particle Size Distribution and pH Point of Zero Charge (pHPZC) Determination
3.2.5. Theoretical Speciation of Supernatant AMD and Mass Balance of Precipitates
3.2.6. Batch Adsorption Experiments
3.2.7. Adsorption Mechanisms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, H.; Stüben, D.; Berner, Z. Removal of arsenic from aqueous solution by natural siderite and hematite. Appl. Geochem. 2007, 22, 1039–1051. [Google Scholar] [CrossRef]
- Su, C.; Jiang, L.; Zhang, W. A review on heavy metal contamination in the soil worldwide: Situation, impact and remediation techniques. Environ. Skept. Crit. 2014, 3, 24–38. [Google Scholar]
- Casiot, C.; Egal, M.; Elbaz-Poulichet, F.; Bruneel, O.; Bancon-Montigny, C.; Cordier, M.-A.; Gomez, E.; Aliaume, C. Hydrological and geochemical control of metals and arsenic in a Mediterranean river contaminated by acid mine drainage (the Amous River, France); preliminary assessment of impacts on fish (Leuciscus cephalus). Appl. Geochem. 2009, 24, 787–799. [Google Scholar] [CrossRef]
- Sarkar, A.; Paul, B. The global menace of arsenic and its conventional remediation—A critical review. Chemosphere 2016, 158, 37–49. [Google Scholar] [CrossRef]
- Singh, R.; Singh, S.; Parihar, P.; Singh, V.P.; Prasad, S.M. Arsenic contamination, consequences and remediation techniques: A review. Ecotoxicol. Environ. Saf. 2015, 112, 247–270. [Google Scholar] [CrossRef]
- Paikaray, S. Arsenic Geochemistry of Acid Mine Drainage. Mine Water Environ. 2015, 34, 181–196. [Google Scholar] [CrossRef]
- Martí, V.; Jubany, I.; Fernández-Rojo, L.; Ribas, D.; Benito, J.A.; Diéguez, B.; Ginesta, A. Improvement of As(V) Adsorption by Reduction of Granular to Micro-Sized Ferric Hydroxide. Processes 2022, 10, 1029. [Google Scholar] [CrossRef]
- Giles, D.E.; Mohapatra, M.; Issa, T.B.; Anand, S.; Singh, P. Iron and aluminium based adsorption strategies for removing arsenic from water. J. Environ. Manag. 2011, 92, 3011–3022. [Google Scholar] [CrossRef]
- Mamindy-Pajany, Y.; Hurel, C.; Marmier, N.; Roméo, M. Arsenic adsorption onto hematite and goethite. Comptes Rendus Chim. 2009, 12, 876–881. [Google Scholar] [CrossRef]
- Alka, S.; Shahir, S.; Ibrahim, N.; Ndejiko, M.J.; Vo, D.-V.N.; Manan, F.A. Arsenic removal technologies and future trends: A mini review. J. Clean. Prod. 2021, 278, 123805. [Google Scholar] [CrossRef]
- WHO. Guidelines for Drinking-Water Quality, 4th ed.; World Health Organization: Geneva, Switzerland, 2017; ISBN 978-92-4-154995-0.
- The European Union Parliament. Directive (EU) 2020/2184 of the European Parliament. Off. J. Eur. Union 2020, 435, 1–62. [Google Scholar]
- Bissen, M.; Frimmel, F.H. Arsenic— a Review. Part II: Oxidation of Arsenic and its Removal in Water Treatment. Acta Hydrochim. Hydrobiol. 2003, 31, 97–107. [Google Scholar] [CrossRef]
- Liu, R.; Qu, J. Review on heterogeneous oxidation and adsorption for arsenic removal from drinking water. J. Environ. Sci. 2021, 110, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Lopez-Valdivieso, A.; Hernandez-Campos, D.J.; Peng, C.; Monroy-Fernandez, M.G.; Razo-Soto, I. Arsenic removal from high-arsenic water by enhanced coagulation with ferric ions and coarse calcite. Water Res. 2006, 40, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, D.; Clifford, D.A.; Samanta, G. Comparative study of arsenic removal by iron using electrocoagulation and chemical coagulation. Water Res. 2010, 44, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Nur, T.; Loganathan, P.; Ahmed, M.B.; Johir, M.A.H.; Nguyen, T.V.; Vigneswaran, S. Removing arsenic from water by coprecipitation with iron: Effect of arsenic and iron concentrations and adsorbent incorporation. Chemosphere 2019, 226, 431–438. [Google Scholar] [CrossRef]
- Wang, J.W.; Bejan, D.; Bunce, N.J. Removal of Arsenic from Synthetic Acid Mine Drainage by Electrochemical pH Adjustment and Coprecipitation with Iron Hydroxide. Environ. Sci. Technol. 2003, 37, 4500–4506. [Google Scholar] [CrossRef]
- Marino, T.; Figoli, A. Arsenic Removal by Liquid Membranes. Membranes 2015, 5, 150–167. [Google Scholar] [CrossRef]
- He, Y.; Liu, J.; Han, G.; Chung, T.-S. Novel thin-film composite nanofiltration membranes consisting of a zwitterionic co-polymer for selenium and arsenic removal. J. Memb. Sci. 2018, 555, 299–306. [Google Scholar] [CrossRef]
- Akin, I.; Arslan, G.; Tor, A.; Cengeloglu, Y.; Ersoz, M. Removal of arsenate [As(V)] and arsenite [As(III)] from water by SWHR and BW-30 reverse osmosis. Desalination 2011, 281, 88–92. [Google Scholar] [CrossRef]
- Chen, A.S.C.; Wang, L.; Sorg, T.J.; Lytle, D.A. Removing arsenic and co-occurring contaminants from drinking water by full-scale ion exchange and point-of-use/point-of-entry reverse osmosis systems. Water Res. 2020, 172, 115455. [Google Scholar] [CrossRef]
- Aliaskari, M.; Schäfer, A.I. Nitrate, arsenic and fluoride removal by electrodialysis from brackish groundwater. Water Res. 2021, 190, 116683. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.K.; Gutiérrez, C.; Leiva Gonzalez, J.; Lazo, A.; Hansen, M.E.; Lazo, P.; Ottosen, L.M.; Ortiz, R. Combined Electrodialysis and Electrocoagulation as Treatment for Industrial Wastewater Containing Arsenic and Copper. Membranes 2023, 13, 264. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-G.; Alvarez, P.J.J.; Nam, A.; Park, S.-J.; Do, T.; Choi, U.-S.; Lee, S.-H. Arsenic(V) removal using an amine-doped acrylic ion exchange fiber: Kinetic, equilibrium, and regeneration studies. J. Hazard. Mater. 2017, 325, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Rathi, B.S.; Kumar, P.S.; Ponprasath, R.; Rohan, K.; Jahnavi, N. An effective separation of toxic arsenic from aquatic environment using electrochemical ion exchange process. J. Hazard. Mater. 2021, 412, 125240. [Google Scholar] [CrossRef]
- Younger, P.L.; Banwart, S.A.; Hedin, R.S. Mine Water; Environmental Pollution; Springer: Dordrecht, The Netherlands, 2002; Volume 5, ISBN 978-1-4020-0138-3. [Google Scholar]
- Trumm, D. Selection of active and passive treatment systems for AMD—Flow charts for New Zealand conditions. N. Z. J. Geol. Geophys. 2010, 53, 195–210. [Google Scholar] [CrossRef]
- Mohapatra, D.; Mishra, D.; Chaudhury, G.R.; Das, R.P. Arsenic adsorption mechanism on clay minerals and its dependence on temperature. Korean J. Chem. Eng. 2007, 24, 426–430. [Google Scholar] [CrossRef]
- Meez, E.; Tolkou, A.K.; Giannakoudakis, D.A.; Katsoyiannis, I.A.; Kyzas, G.Z. Activated Carbons for Arsenic Removal from Natural Waters and Wastewaters: A Review. Water 2021, 13, 2982. [Google Scholar] [CrossRef]
- Asadullah, M.; Jahan, I.; Ahmed, M.B.; Adawiyah, P.; Malek, N.H.; Rahman, M.S. Preparation of microporous activated carbon and its modification for arsenic removal from water. J. Ind. Eng. Chem. 2014, 20, 887–896. [Google Scholar] [CrossRef]
- Foroutan, R.; Mohammadi, R.; Adeleye, A.S.; Farjadfard, S.; Esvandi, Z.; Arfaeinia, H.; Sorial, G.A.; Ramavandi, B.; Sahebi, S. Efficient arsenic(V) removal from contaminated water using natural clay and clay composite adsorbents. Environ. Sci. Pollut. Res. 2019, 26, 29748–29762. [Google Scholar] [CrossRef]
- Chutia, P.; Kato, S.; Kojima, T.; Satokawa, S. Arsenic adsorption from aqueous solution on synthetic zeolites. J. Hazard. Mater. 2009, 162, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Jeon, C.-S.; Baek, K.; Park, J.-K.; Oh, Y.-K.; Lee, S.-D. Adsorption characteristics of As(V) on iron-coated zeolite. J. Hazard. Mater. 2009, 163, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.C.; Fontes, M.P.F. Arsenic (V) removal from water using hydrotalcites as adsorbents: A critical review. Appl. Clay Sci. 2020, 191, 105615. [Google Scholar] [CrossRef]
- Gillman, G.P. A simple technology for arsenic removal from drinking water using hydrotalcite. Sci. Total Environ. 2006, 366, 926–931. [Google Scholar] [CrossRef]
- Zhu, M.; Paul, K.W.; Kubicki, J.D.; Sparks, D.L. Quantum Chemical Study of Arsenic (III, V) Adsorption on Mn-Oxides: Implications for Arsenic(III) Oxidation. Environ. Sci. Technol. 2009, 43, 6655–6661. [Google Scholar] [CrossRef]
- Youngran, J.; FAN, M.; Van Leeuwen, J.; Belczyk, J.F. Effect of competing solutes on arsenic(V) adsorption using iron and aluminum oxides. J. Environ. Sci. 2007, 19, 910–919. [Google Scholar] [CrossRef]
- Siddiqui, S.I.; Chaudhry, S.A. Iron oxide and its modified forms as an adsorbent for arsenic removal: A comprehensive recent advancement. Process Saf. Environ. Prot. 2017, 111, 592–626. [Google Scholar] [CrossRef]
- Gallegos-Garcia, M.; Ramírez-Muñiz, K.; Song, S. Arsenic Removal from Water by Adsorption Using Iron Oxide Minerals as Adsorbents: A Review. Miner. Process. Extr. Metall. Rev. 2012, 33, 301–315. [Google Scholar] [CrossRef]
- Abdallah, E.A.M.; Gagnon, G.A. Arsenic removal from groundwater through iron oxyhydroxide coated waste productsA paper submitted to the Journal of Environmental Engineering and Science. Can. J. Civ. Eng. 2009, 36, 881–888. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U. Arsenic removal from water/wastewater using adsorbents—A critical review. J. Hazard. Mater. 2007, 142, 1–53. [Google Scholar] [CrossRef]
- Kim, L.; Thanh, N.T.; Toan, P.V.; Minh, H.V.T.; Kumar, P. Removal of Arsenic in Groundwater Using Fe(III) Oxyhydroxide Coated Sand: A Case Study in Mekong Delta, Vietnam. Hydrology 2022, 9, 15. [Google Scholar] [CrossRef]
- Habuda-Stanić, M.; Kalajdžić, B.; Kuleš, M.; Velić, N. Arsenite and arsenate sorption by hydrous ferric oxide/polymeric material. Desalination 2008, 229, 1–9. [Google Scholar] [CrossRef]
- Laksmipathiraj, P.; Narasimhan, B.; Prabhakar, S.; Bhaskarraju, G. Adsorption of arsenate on synthetic goethite from aqueous solutions. J. Hazard. Mater. 2006, 136, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Luengo, C.; Brigante, M.; Avena, M. Adsorption kinetics of phosphate and arsenate on goethite: A comparative study. J. Colloid Interface Sci. 2007, 311, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Mamindy-Pajany, Y.; Hurel, C.; Marmier, N.; Roméo, M. Arsenic (V) adsorption from aqueous solution onto goethite, hematite, magnetite and zero-valent iron: Effects of pH, concentration and reversibility. Desalination 2011, 281, 93–99. [Google Scholar] [CrossRef]
- Kunaschk, M.; Schmalz, V.; Dietrich, N.; Dittmar, T.; Worch, E. Novel regeneration method for phosphate loaded granular ferric (hydr)oxide—A contribution to phosphorus recycling. Water Res. 2015, 71, 219–226. [Google Scholar] [CrossRef]
- Nieto, J.M.; Sarmiento, A.M.; Olías, M.; Canovas, C.R.; Riba, I.; Kalman, J.; Delvalls, T.A. Acid mine drainage pollution in the Tinto and Odiel rivers (Iberian Pyrite Belt, SW Spain) and bioavailability of the transported metals to the Huelva Estuary. Environ. Int. 2007, 33, 445–455. [Google Scholar] [CrossRef]
- Fiol, N.; Villaescusa, I. Determination of sorbent point zero charge: Usefulness in sorption studies. Environ. Chem. Lett. 2009, 7, 79–84. [Google Scholar] [CrossRef]
- Aqion Software 2023. Available online: https://www.aqion.de (accessed on 10 February 2023).
- Lu, P.; Zhu, C. Arsenic Eh–pH diagrams at 25 °C and 1 bar. Environ. Earth Sci. 2011, 62, 1673–1683. [Google Scholar] [CrossRef]
- Kalaitzidou, K.; Mitrakas, M.; Raptopoulou, C.; Tolkou, A.; Palasantza, P.-A.; Zouboulis, A. Pilot-Scale Phosphate Recovery from Secondary Wastewater Effluents. Environ. Process. 2016, 3, 5–22. [Google Scholar] [CrossRef]
- Qureshi, A.A.; Javed, S.; Javed, H.M.A.; Jamshaid, M.; Ali, U.; Akram, M.A. Systematic Investigation of Structural, Morphological, Thermal, Optoelectronic, and Magnetic Properties of High-Purity Hematite/Magnetite Nanoparticles for Optoelectronics. Nanomaterials 2022, 12, 1635. [Google Scholar] [CrossRef]
- Lian, J.; Duan, X.; Ma, J.; Peng, P.; Kim, T.; Zheng, W. Hematite (α-Fe2O3) with Various Morphologies: Ionic Liquid-Assisted Synthesis, Formation Mechanism, and Properties. ACS Nano 2009, 3, 3749–3761. [Google Scholar] [CrossRef]
- Lalley, J.; Han, C.; Mohan, G.R.; Dionysiou, D.D.; Speth, T.F.; Garland, J.; Nadagouda, M.N. Phosphate removal using modified Bayoxide® E33 adsorption media. Environ. Sci. Water Res. Technol. 2015, 1, 96–107. [Google Scholar] [CrossRef]
- Domènech, C.; Ayora, C.; de Pablo, J. Sludge weathering and mobility of contaminants in soil affected by the Aznalcollar tailing dam spill (SW Spain). Chem. Geol. 2002, 190, 355–370. [Google Scholar] [CrossRef]
- Di Iorio, E.; Cho, H.G.; Liu, Y.; Cheng, Z.; Angelico, R.; Colombo, C. Arsenate retention mechanisms on hematite with different morphologies evaluated using AFM, TEM measurements and vibrational spectroscopy. Geochim. Cosmochim. Acta 2018, 237, 155–170. [Google Scholar] [CrossRef]
- Lin, T.F.; Liu, C.C.; Hsieh, W.H. Adsorption kinetics and equilibrium of arsenic onto an iron-based adsorbent and an ion exchange resin. Water Supply 2006, 6, 201–207. [Google Scholar] [CrossRef]
- Jagirani, M.S.; Balouch, A.; Abdullah; Mahar, A.M.; Mustafai, F.A.; Rajar, K.; Tunio, A.; Sabir, S.; Samoon, M.K. Review: Arsenic Remediation by Synthetic and Natural Adsorbents. Pak. J. Anal. Environ. Chem. 2017, 18, 18–36. [Google Scholar] [CrossRef]
- Nicomel, N.; Leus, K.; Folens, K.; Van Der Voort, P.; Du Laing, G. Technologies for Arsenic Removal from Water: Current Status and Future Perspectives. Int. J. Environ. Res. Public Health 2015, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Puigdomènech, I.; Colàs, E.; Grivé, M.; Campos, I.; García, D. A tool to draw chemical equilibrium diagrams using SIT: Applications to geochemical systems and radionuclide solubility. MRS Proc. 2014, 1665, 111–116. [Google Scholar] [CrossRef]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: An Introduction Emphasizing Chemical Equilibria in Natural Waters, 2nd ed.; Wiley-VCH Verlag GmbH: New York, NY, USA, 1981; ISBN 0471091731. [Google Scholar]
- Drever, J.I. The Geochemistry of Natural Waters, 3rd ed.; Prentice Hall: Englewood Cliffs, NJ, USA, 1997; ISBN 0132727900. [Google Scholar]
- Pham, T.T.; Ngo, H.H.; Tran, V.S.; Nguyen, M.K. Removal of As (V) from the aqueous solution by a modified granular ferric hydroxide adsorbent. Sci. Total Environ. 2020, 706, 135947. [Google Scholar] [CrossRef]
- Guan, X.-H.; Wang, J.; Chusuei, C.C. Removal of arsenic from water using granular ferric hydroxide: Macroscopic and microscopic studies. J. Hazard. Mater. 2008, 156, 178–185. [Google Scholar] [CrossRef] [PubMed]
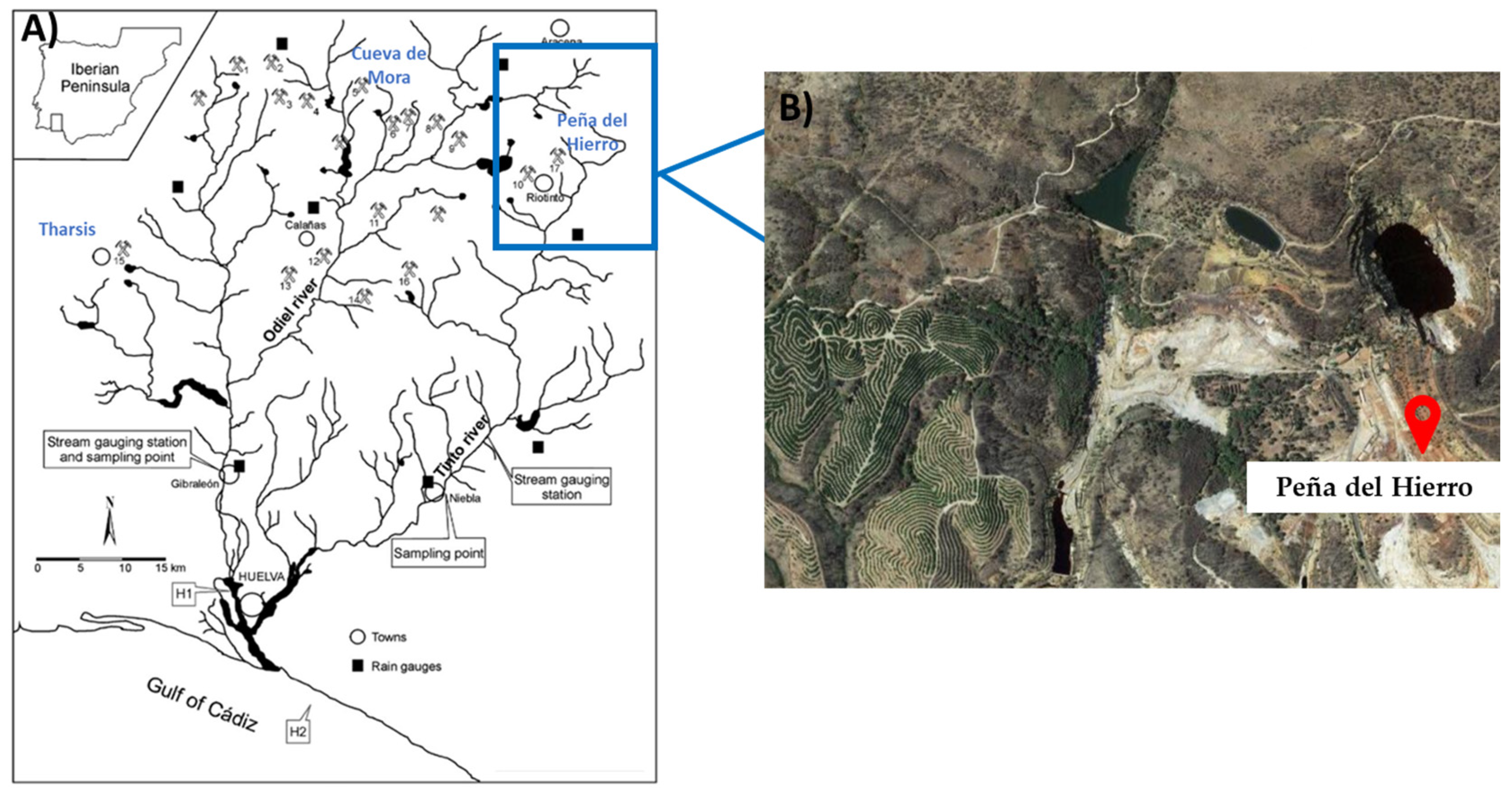
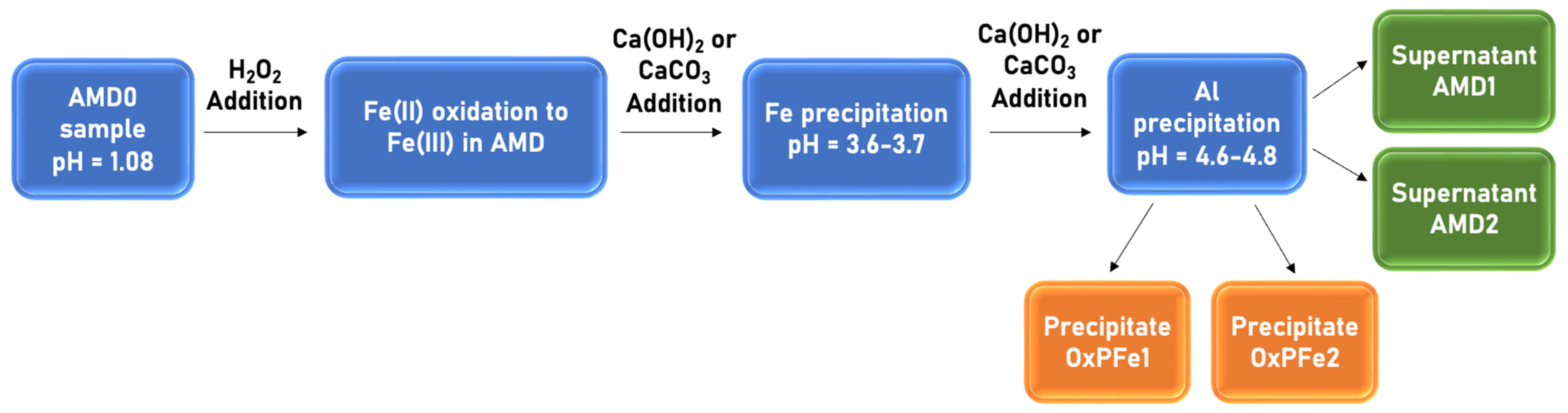




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytical Parameter | PFe-AMD0 C (mg L−1) | PFe-AMD1 C (mg L−1) | PFe-AMD2 C (mg L−1) |
|---|---|---|---|
| Fe | 22,146 | 0.29 | 0.49 |
| Al | 2379.54 | 221.13 | 20.65 |
| S | 519.98 * | 195.98 * | 339.63 * |
| Mg | 357.22 | 32.2 | 36.65 |
| Cu | 96.74 | 36.08 | 31.65 |
| Ca | 78.64 | 390.60 | 628.56 |
| Zn | 71.27 | 39.70 | 36.99 |
| As | 32.36 | 0.01 | 0.01 |
| Ni | 1.13 | 0.34 | 0.32 |
| Cd | 0.58 | 0.11 | 0.11 |
| Pb | 0.57 | 0.01 | 0.004 |
| Na | 38.23 | 4.03 | 6.13 |
| K | 29.18 | 3.18 | 2.25 |
| SO42− | 350.67 | 203.47 | 328.87 |
| pH | 1.08 | 6.66 | 4.09 |
| ORP (mV) | 640 | 280 | 361 |
| Conductivity (mS cm−1) | 49.55 | 4.02 | 5.28 |
| Sample/Element | Ca (%) | Fe (%) | Al (%) | S (%) |
|---|---|---|---|---|
| Nanohematite | <0.5 | 52.2 | <0.5 | NA * |
| Bayoxide® | <0.5 | 63.6 | <0.5 | NA * |
| OxPFe1 | 14.1 | 11.7 | 1.4 | 13.6 |
| OxPFe2 | 12.3 | 11.7 | 1.4 | 14.3 |
| Adsorbent | d10 (µm) | d50 (µm) | d90 (µm) |
|---|---|---|---|
| Nanohematite | 3.66 ± 0.08 | 19.31 ± 0.44 | 47.20 ± 3.14 |
| Bayoxide® | 1.79 ± 0.02 | 15.30 ± 0.28 | 34.50 ± 0.88 |
| OxPFe1 | 2.27 ± 0.05 | 10.40 ± 0.36 | 34.80 ± 1.73 |
| OxPFe2 | 0.883 ± 0.002 | 2.08 ± 0.01 | 27.70 ± 0.54 |
| Adsorbent | pHPZC | Validity Range * |
|---|---|---|
| Nanohematite | 4.2 | 3–10 |
| Bayoxide® | 8.2 | 4–10 |
| OxPFe1 | 5.7 | 3–11 |
| OxPFe2 | 5.8 | 3–10 |
| Phases (Mass %) | Gypsum | Fe(OH)3 | Jurbanite | Total (%) | SSerr |
|---|---|---|---|---|---|
| OxPFe1 | 60.7 | 22.4 | 10.0 | 97.3 | <0.01 |
| OxPFe2 | 54.0 | 22.4 | 17.3 | 97.3 | 0.56 |
| Adsorbent | qmax (mg g−1) | b (L mg−1) | SSerr | qmax·b (L g−1) |
|---|---|---|---|---|
| Nanohematite | 5.06 | 149.62 | 0.67 | 757 |
| Bayoxide® | 17.40 | 24.94 | 9.21 | 434 |
| OxPFe1 | 24.84 | 9.04 | 29.9 | 224 |
| OxPFe2 | 17.95 | 4.81 | 20.9 | 86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Rivero, K.; Bastos-Arrieta, J.; Florido, A.; Martí, V. Potential Use of Precipitates from Acid Mine Drainage (AMD) as Arsenic Adsorbents. Water 2023, 15, 3179. https://doi.org/10.3390/w15183179
Torres-Rivero K, Bastos-Arrieta J, Florido A, Martí V. Potential Use of Precipitates from Acid Mine Drainage (AMD) as Arsenic Adsorbents. Water. 2023; 15(18):3179. https://doi.org/10.3390/w15183179
Chicago/Turabian StyleTorres-Rivero, Karina, Julio Bastos-Arrieta, Antonio Florido, and Vicenç Martí. 2023. "Potential Use of Precipitates from Acid Mine Drainage (AMD) as Arsenic Adsorbents" Water 15, no. 18: 3179. https://doi.org/10.3390/w15183179