Rapid and Sensitive Analysis of Hormones and Other Emerging Contaminants in Groundwater Using Ultrasound-Assisted Emulsification Microextraction with Solidification of Floating Organic Droplet Followed by GC-MS Detection


Abstract
1. Introduction
2. Experimental
2.1. Reagents and Solvents
2.2. Groundwater Samples
2.3. The Procedure of Ultrasound-Assisted Emulsification Microextraction with Solidification of Floating Organic Droplet (USAEME-SFOD) Coupled with In Situ Derivatization
2.4. GC-MS Conditions
3. Results and Discussion
3.1. Optimization of Extraction and Derivatization Procedure
3.1.1. Selection of an Extraction Solvent
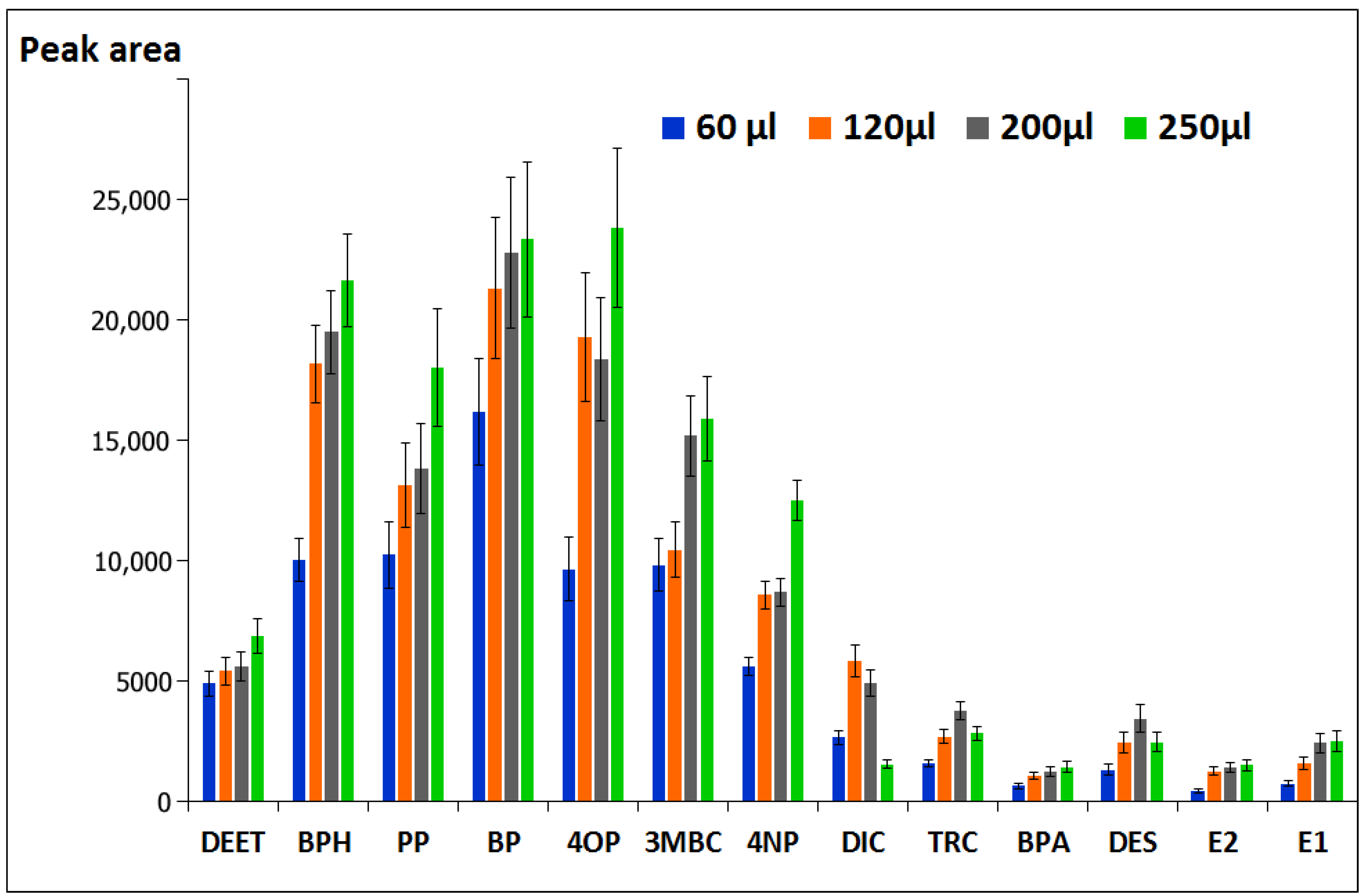
3.1.2. Selection of Solvent Volume
3.1.3. Effect of Derivatization Reagent Volume
3.1.4. Effect of Type and Amount of Buffering Salt
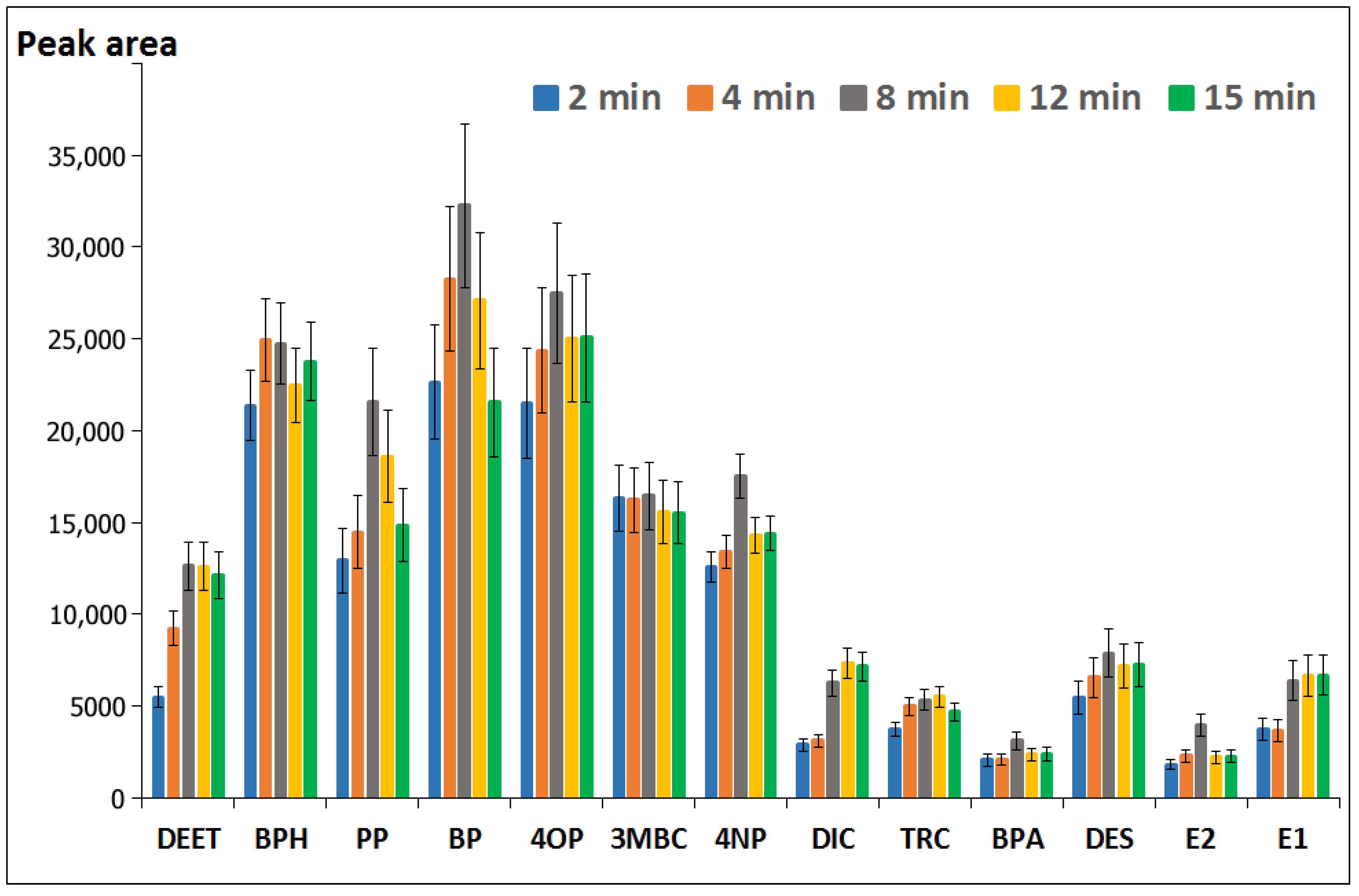
3.1.5. Effect of the Simultaneous Derivatization/Extraction Time
3.2. Method Validation
3.3. Groundwater Analysis
4. Conclusions
- New analytical methodology based on ultrasound-assisted emulsification microextraction with solidification of organic drop followed by GC-MS determination has been proposed for the determination of three hormones and ten other ECs having a high environmental impact. Scrutiny of the available literary sources showed that the present work is the first to describe the combination of the USAEME and SFOD methods for the extraction of target compounds from any matrix.
- High sensitivity of the developed procedure and satisfactory precision and accuracy enabled its use for the determination of ECs in groundwater samples, which are usually characterized by low contamination by anthropogenic compounds.
- Analyses of groundwater have shown that even their deep seams can be contaminated with compounds derived from industrial and everyday human activity. This is a particularly worrying phenomenon since these resources are utilized as sources of high-quality drinking water.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Website United States Geological Survey. Available online: http//:www.toxics.usgs.gov/regional/emc/ (accessed on 4 March 2019).
- Alphenaar, P.A.; van Houten, M. Insight in Emerging Contaminants in Europe; Witteveen Bos: Deventer, The Netherlands, 2016. [Google Scholar]
- Norman Network of Reference Laboratories. Research Centres and Related Organisations for Monitoring of Emerging Environmental Substances. Available online: https://www.norman-network.net (accessed on 4 March 2019).
- Richardson, S.D.; Kimura, S.Y. Water analysis: Emerging contaminants and current issues. Anal. Chem. 2016, 88, 546–582. [Google Scholar] [CrossRef] [PubMed]
- Barber, L.B.; Vajda, A.M.; Douville, C.H.; Norris, D.O.; Writer, J.H. Fish Endocrine disruption responses to a major wastewater treatment facility upgrade. Environ. Sci. Technol. 2012, 46, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Laurenson, J.P.; Bloom, R.A.; Page, S.; Sadrieh, N. Ethinyl estradiol and other human pharmaceutical estrogens in the aquatic environment: A review of recent risk assessment data. AAPS J. 2014, 16, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Xiao, L.; Xia, Y.; Zhou, K.; Wang, H.; Huang, M.; Ge, G.; Wu, Y.; Wu, G.; Yang, L. Diethylstilbestrol can effectively accelerate estradiol-17-O-glucuronidation, while potently inhibiting estradiol-3-O-glucuronidation. Toxicol. Appl. Pharmacol. 2015, 283, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.J.; Cheng, X.J.; Xia, H.F.; Ma, X. The endocrine disruptor diethylstilbestrol induces adipocyte differentiation and promotes obesity in mice. Toxicol. Appl. Pharmacol. 2012, 263, 102–110. [Google Scholar] [CrossRef]
- Routledge, J.E.; Parker, J.; Odum, J.; Ashby, J.; Sumpter, J.P. Some alkyl hydroxybenoate preservatives (parabens) are estrogenic. Toxicol. Appl. Pharmacol. 1998, 153, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Gogoi, A.; Mazumder, P.; Tyagi, V.K.; Tushara Chaminda, G.G.; Kyoungjin Ane, A.; Kumar, M. Occurrence and fate of emerging contaminants in water environment: A review. Groundw. Sustain. Dev. 2018, 6, 169–180. [Google Scholar] [CrossRef]
- Watson, C.; Zoeller, T.; Belcher, S. In vitro molecular mechanisms of bisphenol A action. Reprod. Toxicol. 2007, 24, 178–198. [Google Scholar]
- Fenichel, P.; Chevalier, N.; Brucker-Davis, F. Bisphenol A: An endocrine and metabolic disruptor. Ann. Endocrinol. 2013, 74, 211–220. [Google Scholar] [CrossRef]
- Boas, M.; Feldt-Rasmussen, U.; Main, K.M. Thyroid effects of endocrine disrupting chemicals. Mol. Cell. Endocrinol. 2012, 355, 240–248. [Google Scholar] [CrossRef]
- Veiga-Lopez, A.; Pu, Y.; Gingrich, J.; Padmanabhan, V. Obesogenic endocrine disrupting chemicals: Identifying knowledge gaps. Trends Endocrinol. Metab. 2018, 29, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Walley, S.N.; Roepke, T.A. Perinatal exposure to endocrine disrupting compounds and the control of feeding behavior—An overview. Horm. Behav. 2018, 101, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, B.; Zohrabi, P.; Kim, K.H.; Shamsipur, M.; Deep, A.; Hong, J. Recent advances in liquid-phase microextraction techniques for the analysis of environmental pollutants. Trends Anal. Chem. 2017, 97, 83–95. [Google Scholar] [CrossRef]
- Seidi, S.; Rezazadeh, M.; Yamini, Y. Pharmaceutical applications of liquid-phase microextraction. Trends Anal. Chem. 2018, 108, 296–305. [Google Scholar] [CrossRef]
- Rezaee, M.; Assadi, Y.; Hosseini, M.R.M.; Aghaee, E.; Ahmadi, F.; Berijani, S. Determination of organic compounds in water using dispersive liquid–liquid microextraction. J. Chromatogr. A 2006, 1116, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Regueiro, J.; Llompart, M.; Garcia-Jares, C.; Garcia-Monteagudo, J.C.; Cela, R. Ultrasound-assisted emulsification–microextraction of emergent contaminants and pesticides in environmental waters. J. Chromatogr. A 2008, 1190, 27–38. [Google Scholar] [CrossRef]
- Khalili Zanjani, M.R.; Yamini, Y.; Shariati, S.; Jönsson, J.A. A new liquid-phase microextraction method based on solidification of floating organic drop. Anal. Chim. Acta. 2007, 585, 286–293. [Google Scholar] [CrossRef]
- Yang, D.; Yang, Y.; Li, Y.; Yin, S.; Chen, Y.; Wang, J.; Xiao, J.; Sun, C. Dispersive liquid–liquid microextraction based on solidification of floating organic drop combined with high performance liquid chromatography for analysis of 15 phthalates in water. J. AOAC Int. 2019, 102, 942–951. [Google Scholar] [CrossRef]
- Jafariyan, R.; Shabani, A.M.H.; Dadfarnia, S.; Tafti, E.N.; Shirani, M. Dispersive liquid–liquid microextraction based on solidification of floating organic drop as an efficient preconcentration method for spectrophotometric determination of aluminium. Anal. Bioanal. Chem. Res. 2019, 6, 289–299. [Google Scholar]
- Ezoddin, M.; Adlnasab, L.; Kaveh, A.A.; Karimi, M.A. Ultrasonically formation of supramolecular based ultrasound energy assisted solidification of floating organic drop microextraction for preconcentration of methadone in human plasma and saliva samples prior to gas chromatography–mass spectrometry. Ultrason. Sonochem. 2019, 50, 182–187. [Google Scholar] [CrossRef]
- Guiñez, M.; Bazan, C.; Martinez, L.D.; Cerutti, S. Determination of nitrated and oxygenated polycyclic aromatic hydrocarbons in water samples by a liquid–liquid phase microextraction procedure based on the solidification of a floating organic drop followed by solvent assisted back-extraction and liquid chromatography–tandem mass spectrometry. Microchem. J. 2018, 139, 164–173. [Google Scholar]
- Asadi, M. Determination of ochratoxin A in fruit juice by high-performance liquid chromatography after vortex-assisted emulsification microextraction based on solidification of floating organic drop. Mycotoxin Res. 2018, 34, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Aydin, I.; Chormey, D.S.; Budak, T.; Firat, M.; Turak, F.; Bakirdere, S. Development of an accurate and sensitive analytical method for the determination of cadmium at trace levels using dispersive liquid–liquid microextraction based on the solidification of floating organic drops combined with slotted quartz tube flame atomic absorption spectrometry. J. AOAC Int. 2018, 101, 843–847. [Google Scholar] [PubMed]
- Coutts, R.T.; Hargesheimer, E.E.; Pasutto, F.M. Gas chromatographic analysis of trace phenols by direct acetylation in aqueous solution. J. Chromatogr. A 1979, 179, 291–299. [Google Scholar] [CrossRef]
- Faraji, H.; Tehrani, M.S.; Husain, S.W. Pre-concentration of phenolic compounds in water samples by novel liquid–liquid microextraction and determination by gas chromatography–mass spectrometry. J. Chromatogr. A 2009, 1216, 8569–8574. [Google Scholar] [CrossRef]
- Casado, J.; Nescatelli, R.; Rodríguez, I.; Ramil, M.; Marini, F.; Cela, R. Determination of benzotriazoles in water samples by concurrent derivatization–dispersive liquid–liquid microextraction followedby gas chromatography–mass spectrometry. J. Chromatogr. A 2014, 1336, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.J.; Du, X.; Zhang, J.W.; Li, J.C.; Wang, L.Z. Ultrasound-assisted emulsification-microextraction combined with flame atomic absorption spectrometry for determination of trace cadmium in water samples. Talanta 2009, 80, 980–984. [Google Scholar] [CrossRef]
- Kapelewska, J.; Kotowska, U.; Karpińska, J.; Kowalczuk, D.; Arciszewska, A.; Świrydo, A. Occurrence, removal, mass loading and environmental risk assessment of emerging organic contaminants in leachates, groundwaters and wastewaters. Microchem. J. 2018, 137, 292–301. [Google Scholar] [CrossRef]
- Lapworth, D.J.; Baran, N.; Stuart, M.E.; Ward, R.S. Emerging organic contaminants in groundwater: A review of sources, fate and occurrence. Environ. Pollut. 2012, 163, 287–303. [Google Scholar] [CrossRef]
- Bexfield, L.M.; Toccalino, P.L.; Belitz, K.; Foreman, W.T.; Furlong, E.T. Hormones and pharmaceuticals in groundwater used as a source of drinking water across the United States. Environ. Sci. Technol. 2019, 53, 2950–2960. [Google Scholar] [CrossRef]
- Schulze, S.; Zahn, D.; Montes, R.; Rodil, R.; Quintana, J.B.; Knepper, T.P.; Reemtsma, T.; Berger, U. Occurrence of emerging persistent and mobile organic contaminants in European water samples. Water Res. 2019, 153, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.E.; Manamsa, K.; Talbot, J.C.; Crane, E.J. Emerging Contaminants in Groundwater; British Geological Survey Open Report; British Geological Survey: Nottingham, UK, 2011.
- Sorensen, J.P.R.; Lapworth, D.J.; Nkhuwa, D.C.W.; Stuart, M.E.; Gooddy, D.C.; Bell, R.A.; Chirwa, M.; Kabika, J.; Liemisa, M.; Chibesa, M.; et al. Emerging contaminants in urban groundwater sources in Africa. Water Res. 2015, 72, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Chen, Y.Y.; Chiou, M.R.; Chen, M.Y.; Fan, H.J. Occurrence and treatment efficiency of pharmaceuticals in landfill leachates. Waste Manag. 2016, 55, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Buszka, P.M.; Yeskis, D.J.; Kolpin, D.W.; Furlong, E.T.; Zaugg, S.D.; Meyer, M.T. Waste-indicator and pharmaceutical compounds in landfill-leachate-affected ground water near Elkhart, Indiana, 2000–2002. Bull. Environ. Contam. Toxicol. 2009, 82, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Kapelewska, J.; Kotowska, U.; Wiśniewska, K. Determination of personal care products and hormones in leachate and groundwater from Polish MSW landfills by ultrasound-assisted emulsification microextraction and GC-MS. Environ. Sci. Pollut. Res. 2016, 23, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, Z. Underground Waters of Provincial Cities of Poland; Państwowy Instytut Geologiczny: Warszawa, Poland, 2007. (In Polish) [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Group | Formula | Molar Weight (MW), (g/mol) | Chemical Abstracts Service Number | Retention Time (min) | Quantification and Identification Ion (m/z) |
|---|---|---|---|---|---|---|
| E2 | Natural steroid hormone | C18H24O2 | 272 | 50-28-2 | 25.05 | 43, 146, 272 |
| E1 | Natural steroid hormone | C18H22O2 | 270 | 53-16-7 | 24.81 | 185, 270, 272 |
| DES | Artificial non-steroid hormone | C18H20O2 | 268 | 56-53-1 | 21.87 | 268, 310, 352 |
| DIC | Non-steroidal anti-inflammatory drug | C14H11Cl2NO2 | 296 | 15,307-79-6 | 16.24 | 214, 242, 277 |
| PP | Preservative | C10H12O3 | 180 | 94-13-3 | 6.71 | 121, 138, 180 |
| BP | Preservative | C11H14O3 | 194 | 94-26-8 | 8.90 | 121, 138, 194 |
| BPH | UV filter | C13H10O | 182 | 119-61-9 | 6.41 | 77, 105, 182 |
| 3MBC | UV filter | C18H22O | 254 | 36,861-47-9 | 14.47 | 128, 171, 254 |
| DEET | Repellent | C12H17NO | 191 | 134-62-3 | 5.46 | 91, 119, 190 |
| 4OP | Nonionic surfactant | C14H22O | 206 | 1806-26-4 | 10.96 | 43, 107, 206 |
| 4NP | Nonionic surfactant | C15H24O | 220 | 84,852-15-3 | 12.85 | 43, 107, 220 |
| BPA | Substrate in the production of plastics | C15H16O2 | 228 | 80-05-7 | 19.53 | 213, 228, 270 |
| TRC | Antiseptic | C12H7Cl3O2 | 289.5 | 3380-34-5 | 16.39 | 218, 288, 290 |
| Solvent | CAS | Molar Mass (g/mol) | Melting Point (°C) | Boiling Point (°C) | Density (g/mL) |
|---|---|---|---|---|---|
| 1-Undecanol | 112-42-5 | 172.31 | 13 | 243 | 0.830 |
| 2-Dodecanol | 10,203-28-8 | 186.34 | 18 | 250 | 0.829 |
| 1-Dodecanol | 112-53-8 | 186.34 | 24 | 259 | 0.831 |
| n-Hexadecane | 544-76-3 | 226.41 | 18 | 286.8 | 0.773 |
| Analyte | Equation of the Calibration Curve * | Coefficient of Determination (r2) * | ||
|---|---|---|---|---|
| Range I (0.001–0.05 μg/L) | Range II (0.05–10 μg/L) | Range I (0.001–0.05 μg/L) | Range II (0.05–10 μg/L) | |
| E2 | - | y = 1940.2x − 144.7 | - | 0.9982 |
| E1 | y = 2835.0x + 254.5 | y = 5214.1x − 570.5 | 0.9970 | 0.9965 |
| DES | y = 7036.5x + 234.7 | y = 5105.1x − 861.9 | 0.9949 | 0.9974 |
| DIC | y = 3102.3 + 292.2 | y = 1490.7x + 500.0 | 0.9894 | 0.9929 |
| PP | y = 6120.7x + 309.9 | y = 5807.8x + 799.4 | 0.9891 | 0.9993 |
| BP | y = 30,653.4x + 1269.4 | y = 9395.7x + 2330.7 | 0.9872 | 0.9960 |
| BPH | y = 191,925.7x + 7247.8 | y = 7793.9x + 16284.3 | 0.9944 | 0.9993 |
| 3MBC | y = 4671.2x + 128.3 | y = 2580.6x + 113.7 | 0.9901 | 0.9978 |
| DEET | y = 8894.3x + 1231.8 | y = 3955.3x + 1761.6 | 0.9853 | 0.9988 |
| 4OP | y = 31,697.4x + 658.1 | y = 18,968.0x − 298.7 | 0.9935 | 0.9962 |
| 4NP | y = 10,575.9x + 236.2 | y = 8932.6x + 293.0 | 0.9890 | 0.9904 |
| BPA | y = 119,133.7x + 3285.2 | y = 10,428.7x + 6435.3 | 0.9916 | 0.9974 |
| TRC | y = 6070.4x + 215.5 | y = 4535.3x + 674.8 | 0.9869 | 0.9994 |
| Analyte | USAEME-SFOD/GC-MS (This Work) * | USAEME/GC-MS (From Literature [31]) * | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | LoD (ng/L) | LoQ (ng/L) | Recovery (%) | RSD (%) | LoD (ng/L) | LoQ (ng/L) | ||
| 0.03 µg/L | 4.00 µg/L | 1 µg/L | |||||||
| E2 | 121.3 | 90.2 | 15.5 | 5.9 | 19.5 | 103 | 14.3 | 130.73 | 435.77 |
| E1 | - | 111.5 | 16.7 | 1.54 | 5.10 | 103 | 10.3 | 8.92 | 29.73 |
| DES | 96.7 | 90.1 | 16.8 | 0.04 | 0.12 | 101 | 13.7 | 88.36 | 294.54 |
| DIC | 93.3 | 99.1 | 11.4 | 0.17 | 0.56 | 110 | 12.1 | 149.55 | 498.48 |
| PP | 116.7 | 95.5 | 13.5 | 0.05 | 0.16 | 97 | 12.4 | 23.62 | 47.24 |
| BP | 120.0 | 92.2 | 13.8 | 0.04 | 0.15 | 126 | 14.0 | 10.21 | 34.03 |
| BPH | 110.0 | 103.7 | 8.9 | 0.03 | 0.11 | 105 | 16.2 | 2.97 | 9.90 |
| 3MBC | 120.7 | 93.7 | 11.0 | 0.04 | 0.15 | 116 | 11.2 | 3.01 | 10.00 |
| DEET | 113.8 | 99.5 | 10.4 | 0.02 | 0.05 | 117 | 17.3 | 1.50 | 4.99 |
| 4OP | 113.3 | 93.1 | 13.9 | 0.02 | 0.07 | 105 | 10.3 | 1.50 | 5.00 |
| 4NP | 112.9 | 97.7 | 6.7 | 0.01 | 0.05 | 101 | 14.7 | 2.94 | 9.80 |
| BPA | 123.1 | 94.7 | 15.8 | 0.04 | 0.12 | 107 | 15.6 | 1.49 | 4.98 |
| TRC | 123.3 | 96.1 | 10.2 | 0.04 | 0.14 | 134 | 15.8 | 2.48 | 8.26 |
| Analyte | Groundwater Samples from Drilling Wells (NS = 6) | Groundwater Samples from Shallow Wells (NS = 5) | Groundwater Samples from MSW Monitoring Wells (NS = 12) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Range (ng/L) | Median (ng/L) | d.f. | Range (ng/L) | Median (ng/L) | d.f. | Range (ng/L) | Median (ng/L) | d.f. | |
| E2 | n.d. | - | 0 | n.d. | - | 0 | n.d. | - | 0 |
| E1 | n.d. | - | 0 | n.d. | - | 0 | n.d.–309 | 107 | 2 |
| DES | n.d. | - | 0 | n.d. | - | 0 | n.d. | - | 0 |
| DIC | n.d. | - | 0 | n.d. | - | 0 | n.d.–312 | 280 | 3 |
| PP | n.d. | - | 0 | n.d. | - | 0 | n.d.–0.5 | - | 1 |
| BP | n.d. | - | 0 | n.d. | - | 0 | n.d.–0.2 | 0.2 | 2 |
| BPH | n.d. | - | 0 | n.d.–124 | - | 1 | 0.5–3300 | 33 | 12 |
| 3MBC | n.d. | - | 0 | n.d. | - | 0 | n.d. | - | 0 |
| DEET | n.d.–2 | 2 | 2 | n.d.–21 | 6 | 2 | n.d.–3 | 2 | 10 |
| 4OP | n.d. | - | 0 | n.d.–17 | 11 | 2 | n.d.–25 | 10 | 3 |
| 4NP | n.d. | - | 0 | n.d.–8 | - | 1 | n.d.–9 | 7 | 2 |
| BPA | n.d.–98 | 53 | 2 | n.d.–689 | 124 | 4 | 0.2–1050 | 79 | 12 |
| TRC | n.d. | - | 0 | n.d. | - | 0 | n.d.–38 | 28 | 3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotowska, U.; Kapelewska, J.; Kotowski, A.; Pietuszewska, E. Rapid and Sensitive Analysis of Hormones and Other Emerging Contaminants in Groundwater Using Ultrasound-Assisted Emulsification Microextraction with Solidification of Floating Organic Droplet Followed by GC-MS Detection. Water 2019, 11, 1638. https://doi.org/10.3390/w11081638
Kotowska U, Kapelewska J, Kotowski A, Pietuszewska E. Rapid and Sensitive Analysis of Hormones and Other Emerging Contaminants in Groundwater Using Ultrasound-Assisted Emulsification Microextraction with Solidification of Floating Organic Droplet Followed by GC-MS Detection. Water. 2019; 11(8):1638. https://doi.org/10.3390/w11081638
Chicago/Turabian StyleKotowska, Urszula, Justyna Kapelewska, Adam Kotowski, and Ewelina Pietuszewska. 2019. "Rapid and Sensitive Analysis of Hormones and Other Emerging Contaminants in Groundwater Using Ultrasound-Assisted Emulsification Microextraction with Solidification of Floating Organic Droplet Followed by GC-MS Detection" Water 11, no. 8: 1638. https://doi.org/10.3390/w11081638
APA StyleKotowska, U., Kapelewska, J., Kotowski, A., & Pietuszewska, E. (2019). Rapid and Sensitive Analysis of Hormones and Other Emerging Contaminants in Groundwater Using Ultrasound-Assisted Emulsification Microextraction with Solidification of Floating Organic Droplet Followed by GC-MS Detection. Water, 11(8), 1638. https://doi.org/10.3390/w11081638