
Wintertime Residential Biomass Burning in Las Vegas, Nevada; Marker Components and Apportionment Methods



Abstract
:1. Introduction
1.1. Residential Wintertime Biomass Burning and Its Fine Particle Tracers
1.2. Study Area: Las Vegas
2. Methodology
2.1. Monitoring Location
2.2. Measurement Methods
2.3. Source Apportionment Methods
3. Results
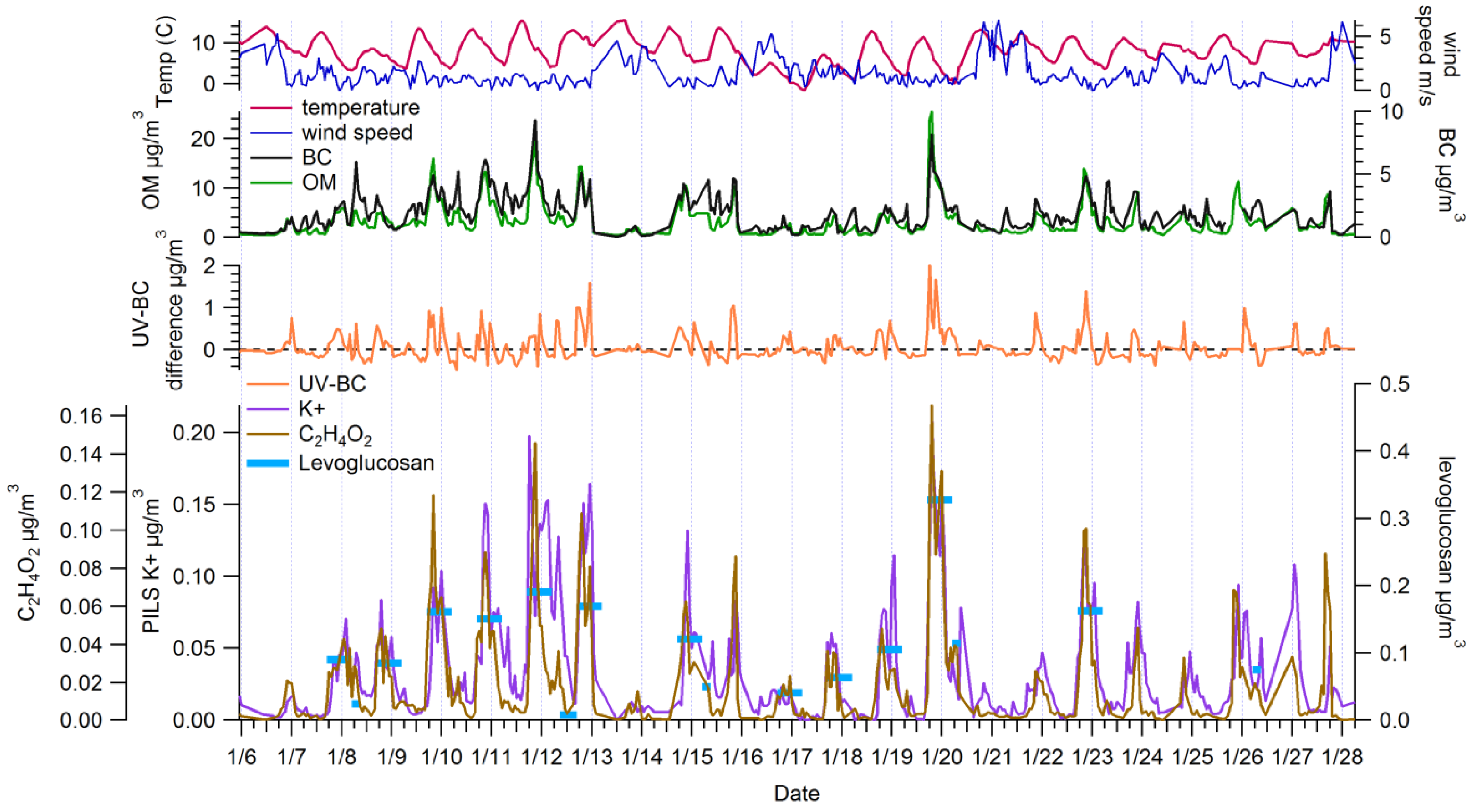
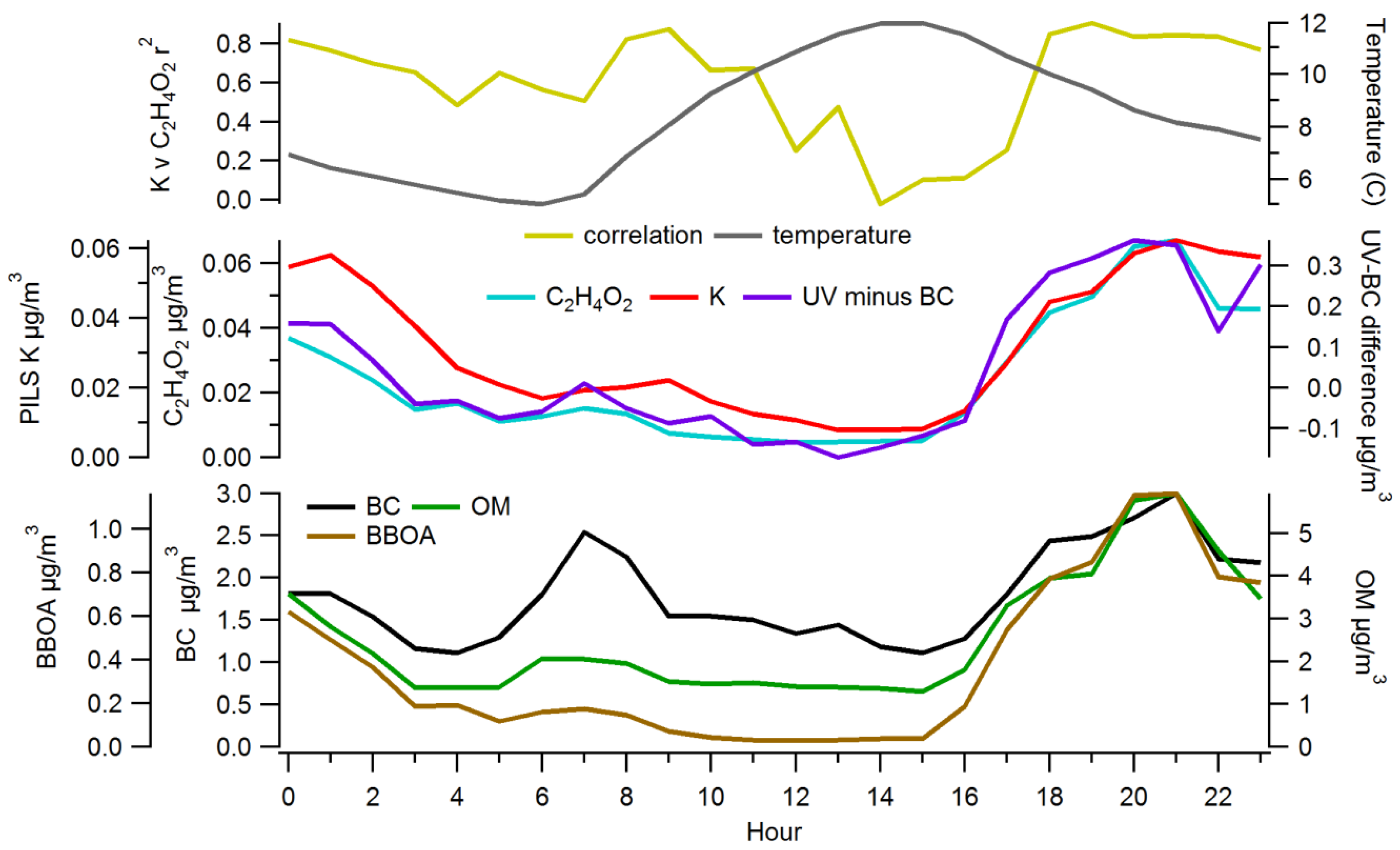
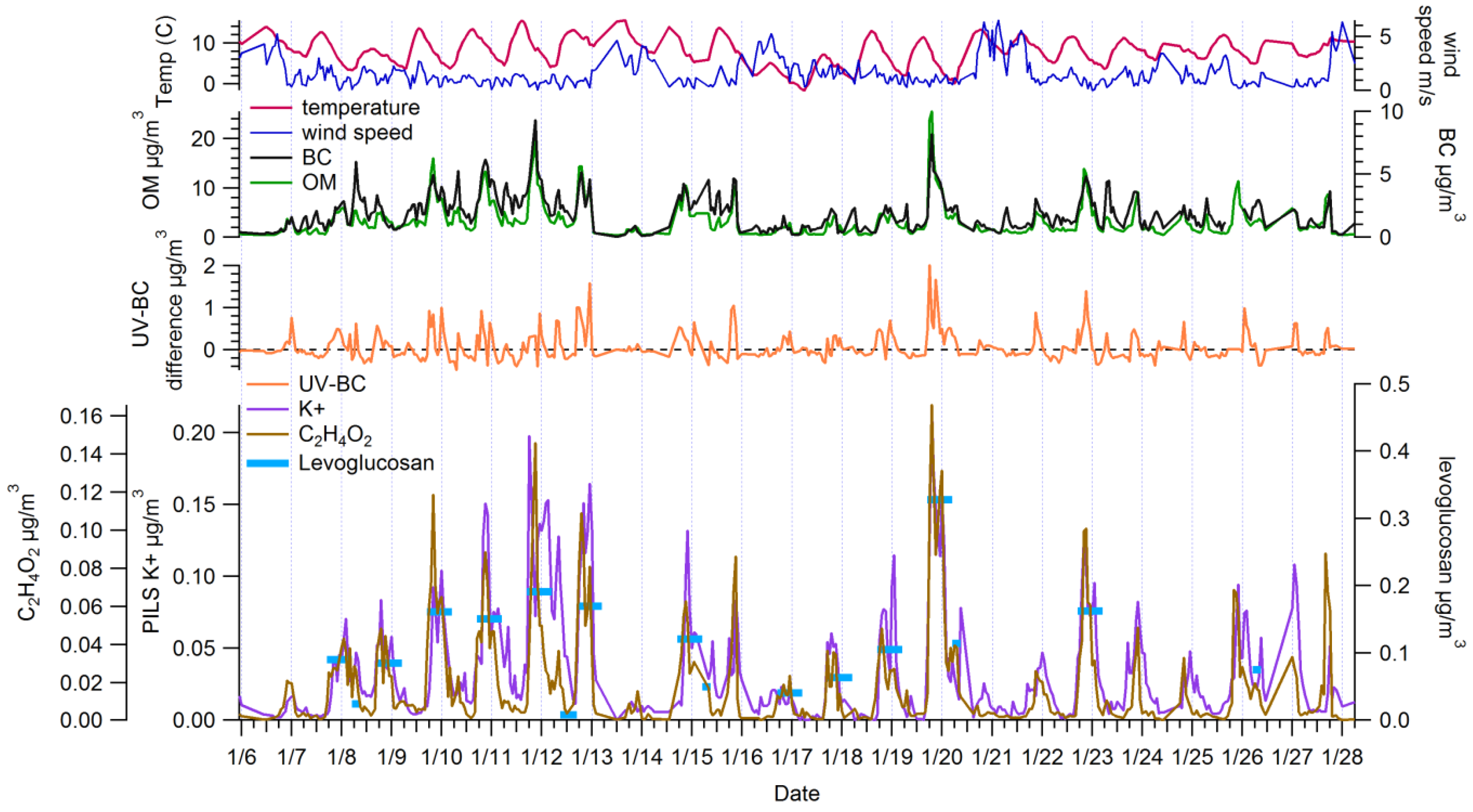
3.1. Ambient Concentrations and Temporal Variability of Biomass Burning Markers
3.2. Comparison among Biomass Burning Markers
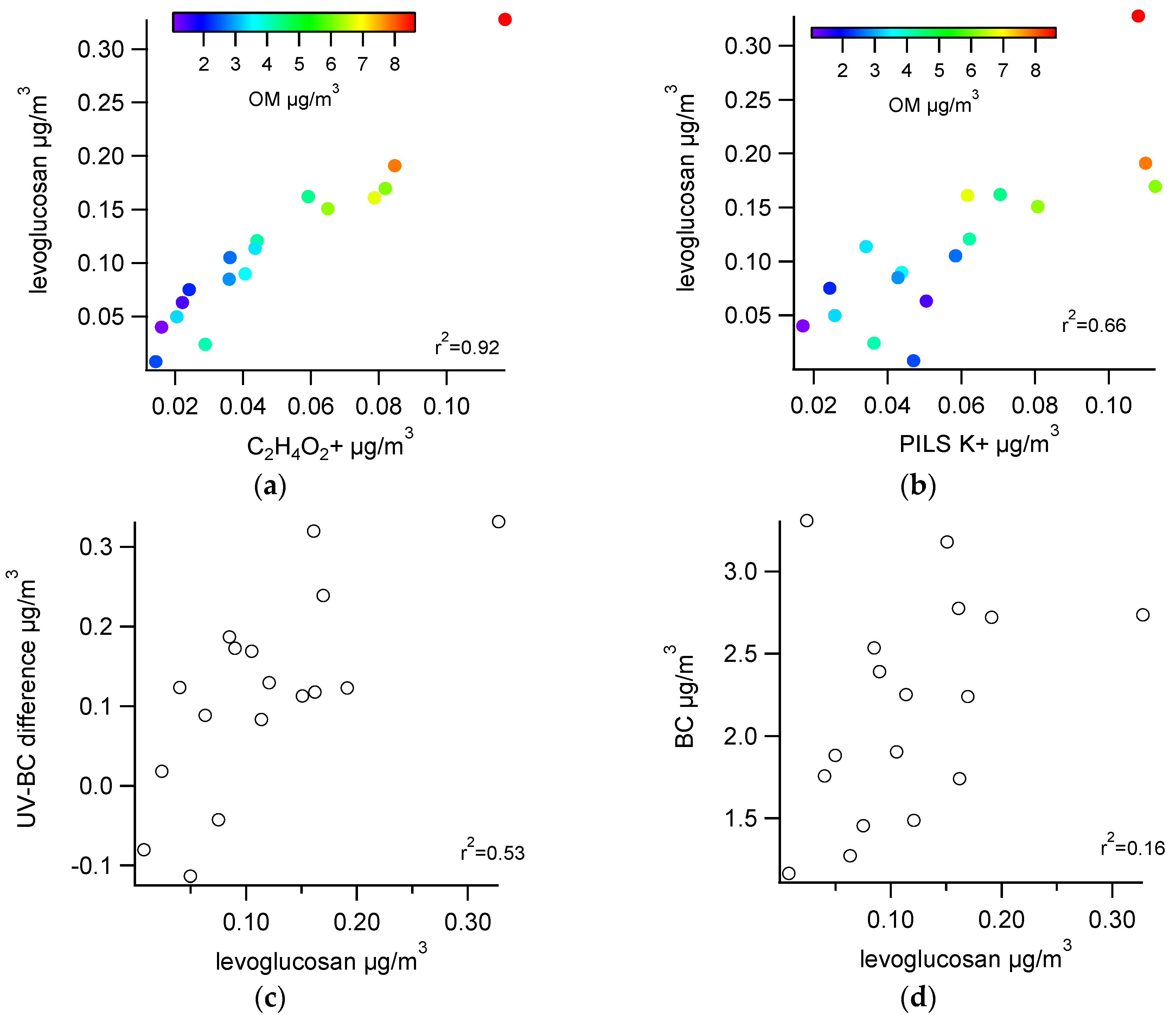
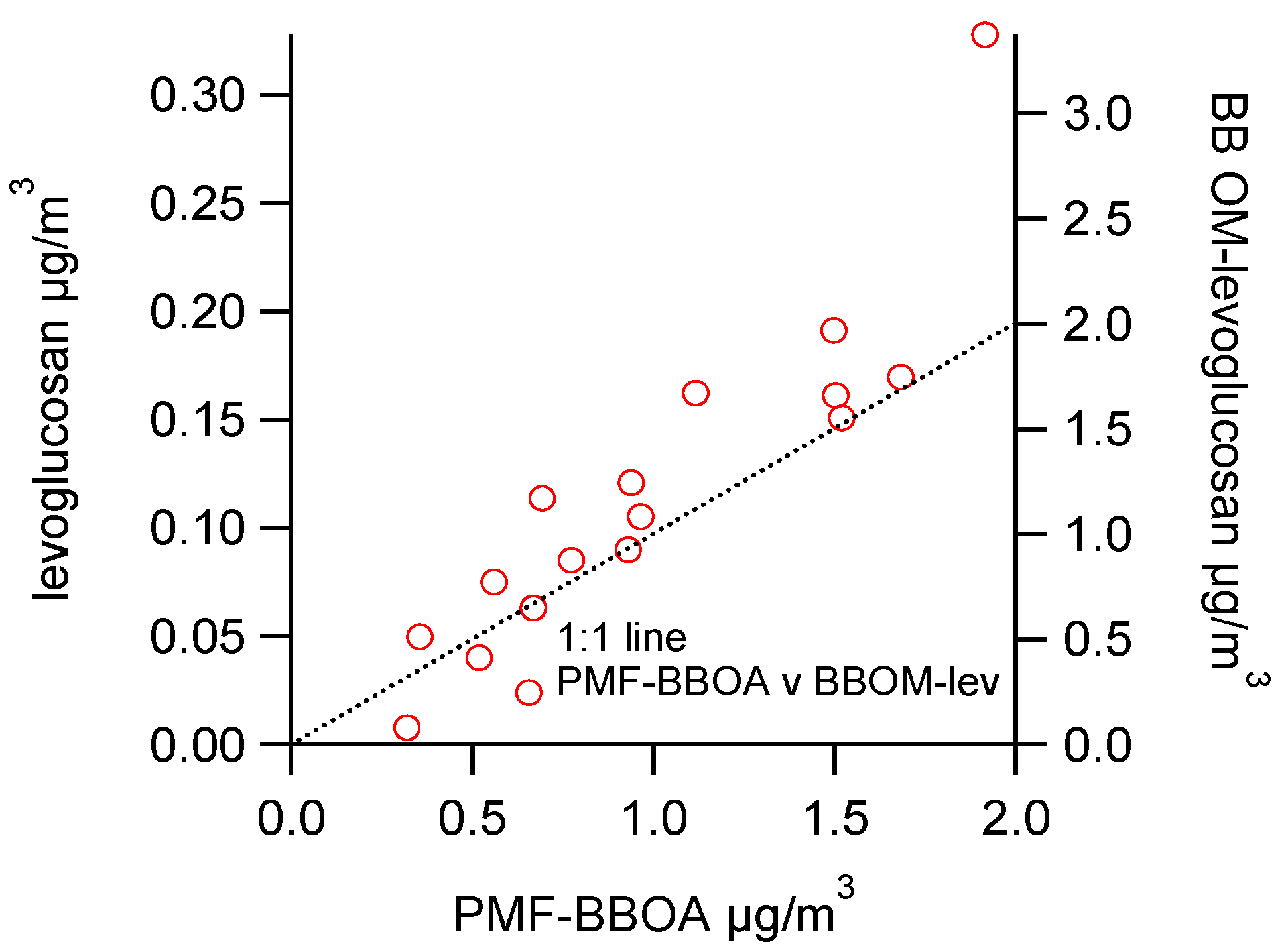
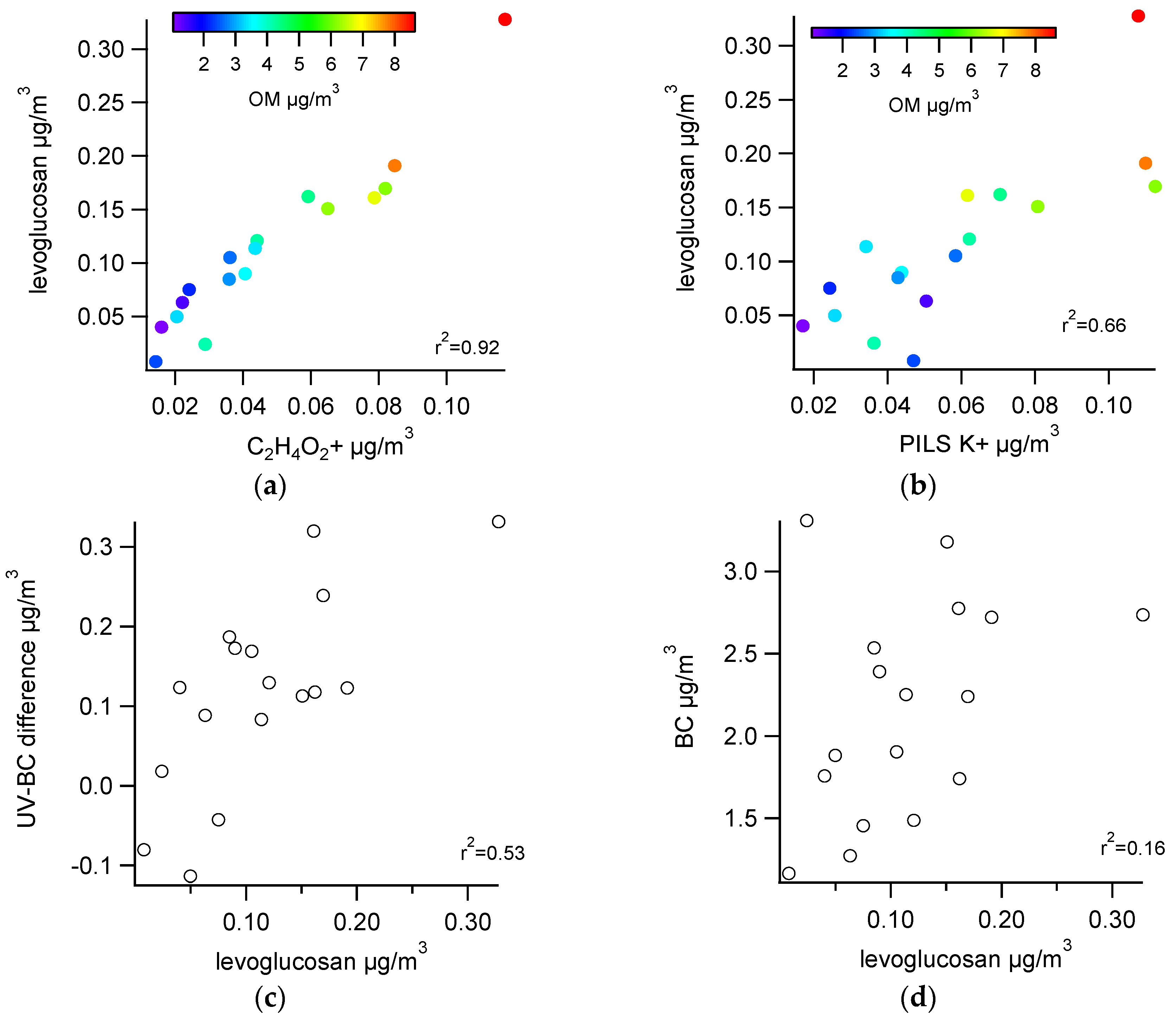
3.2.1. Comparisons with Levoglucosan
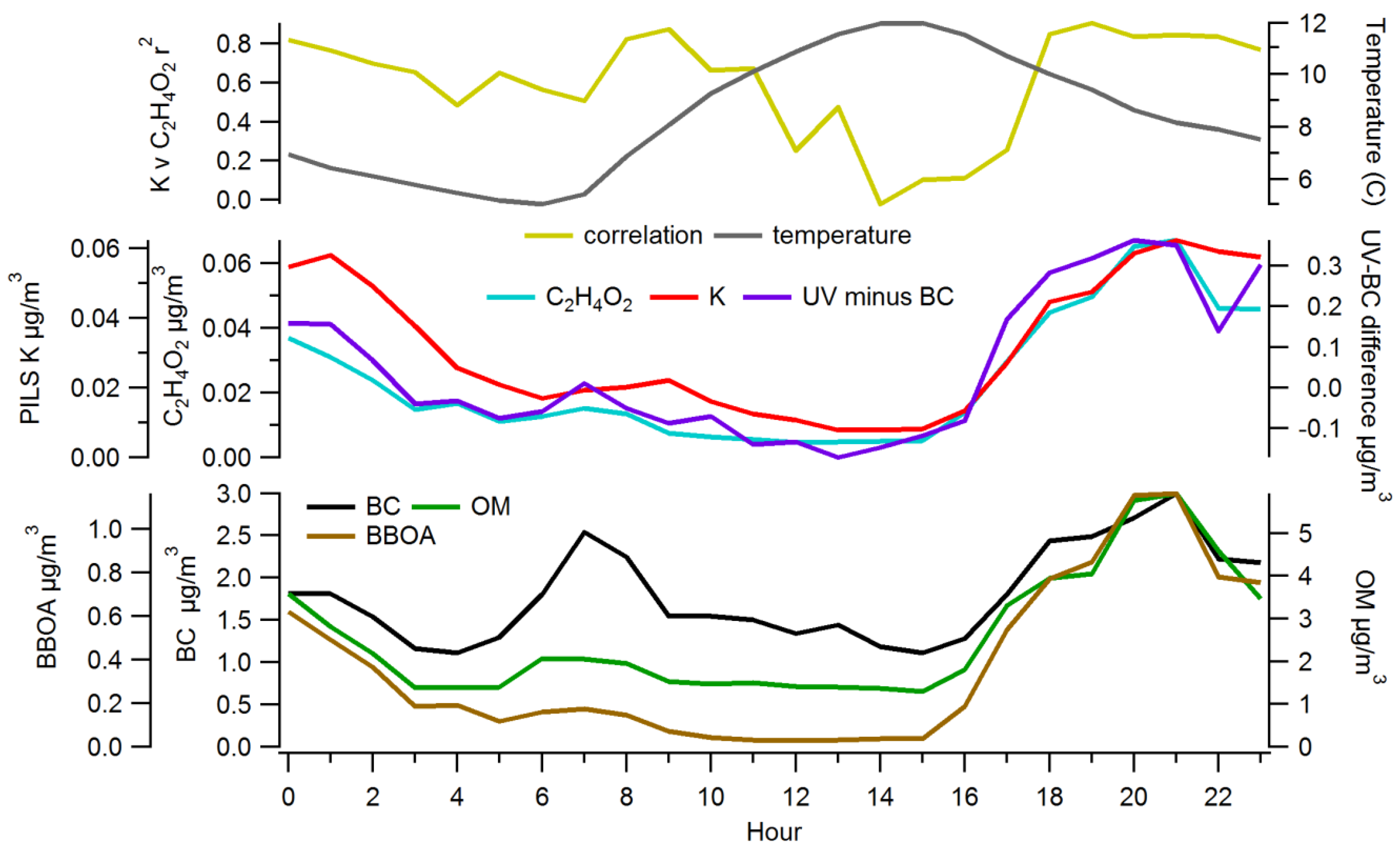
3.2.2. Comparisons among Semi-Continuous Biomass Burning Markers
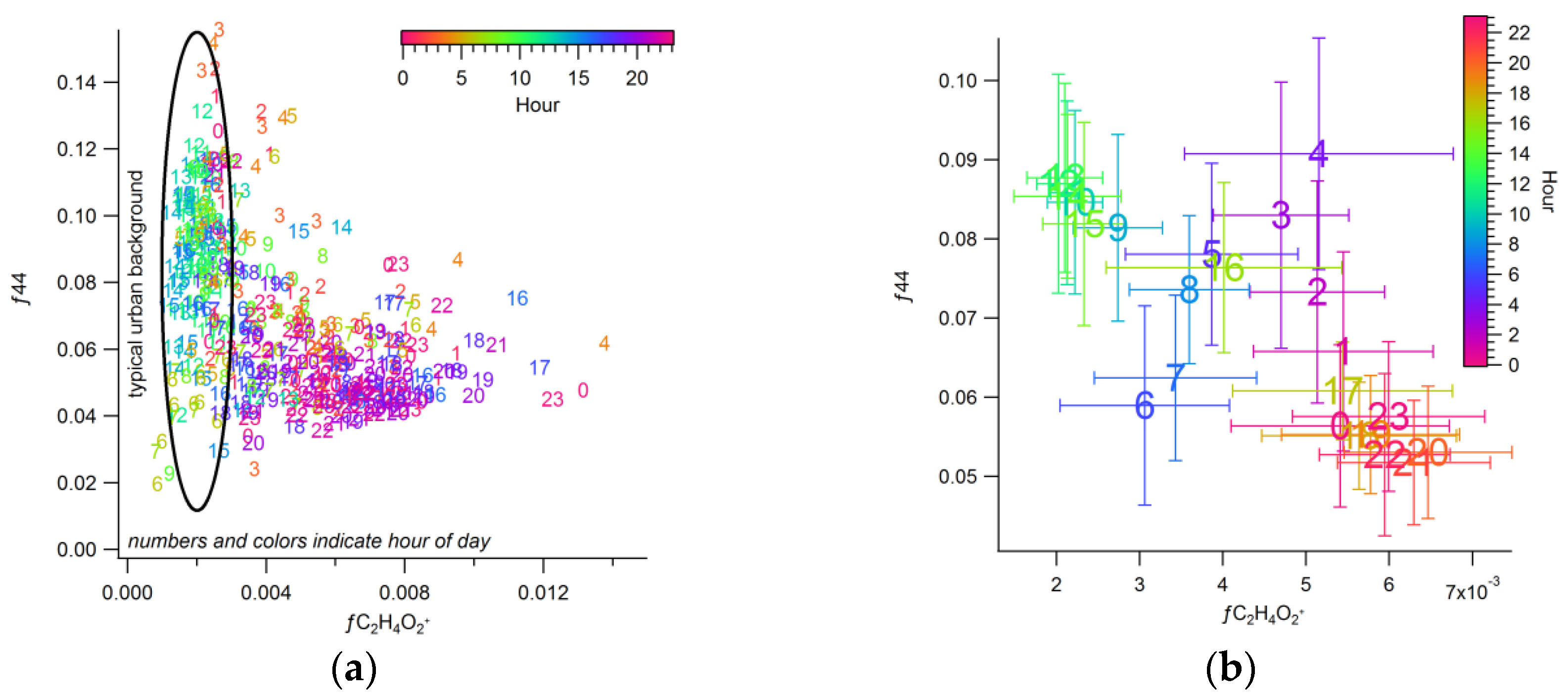
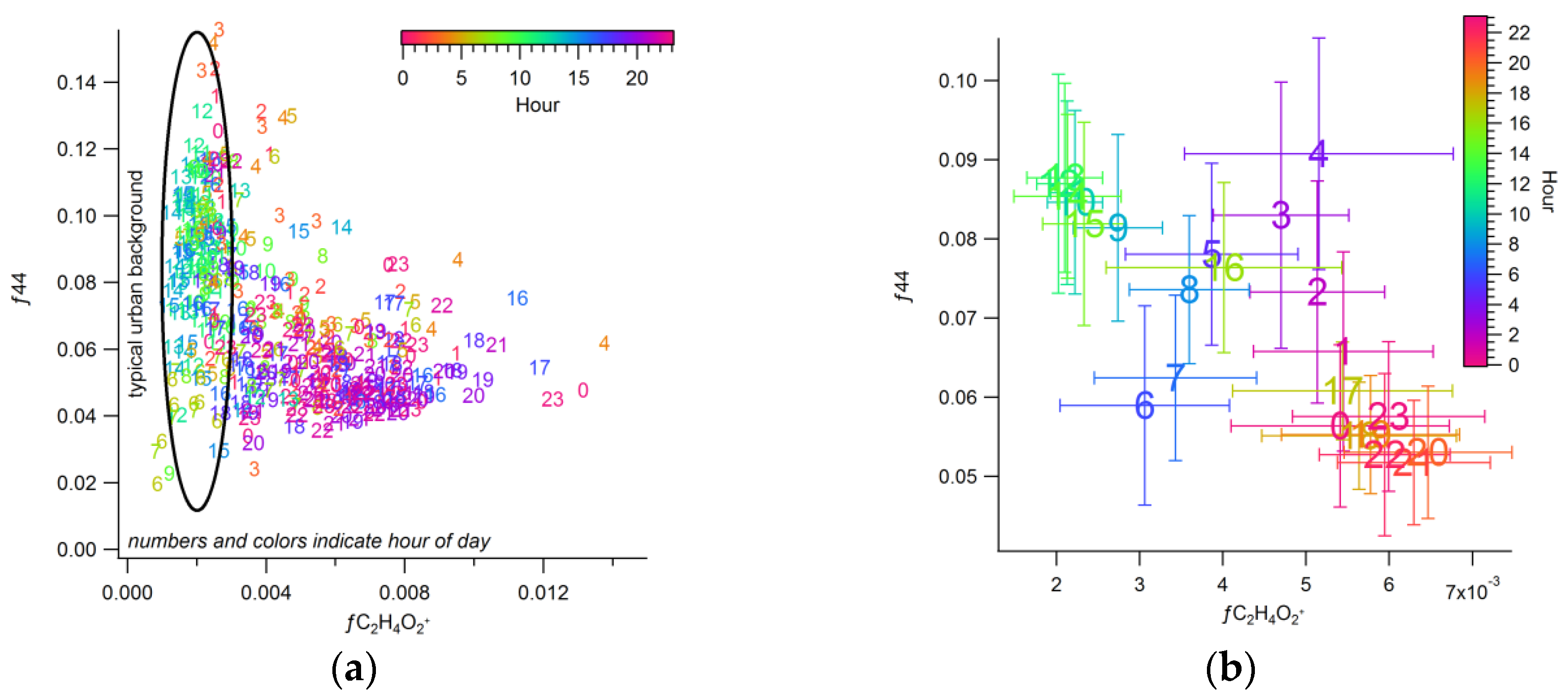
3.2.3. Urban Background Levels of C2H4O2+
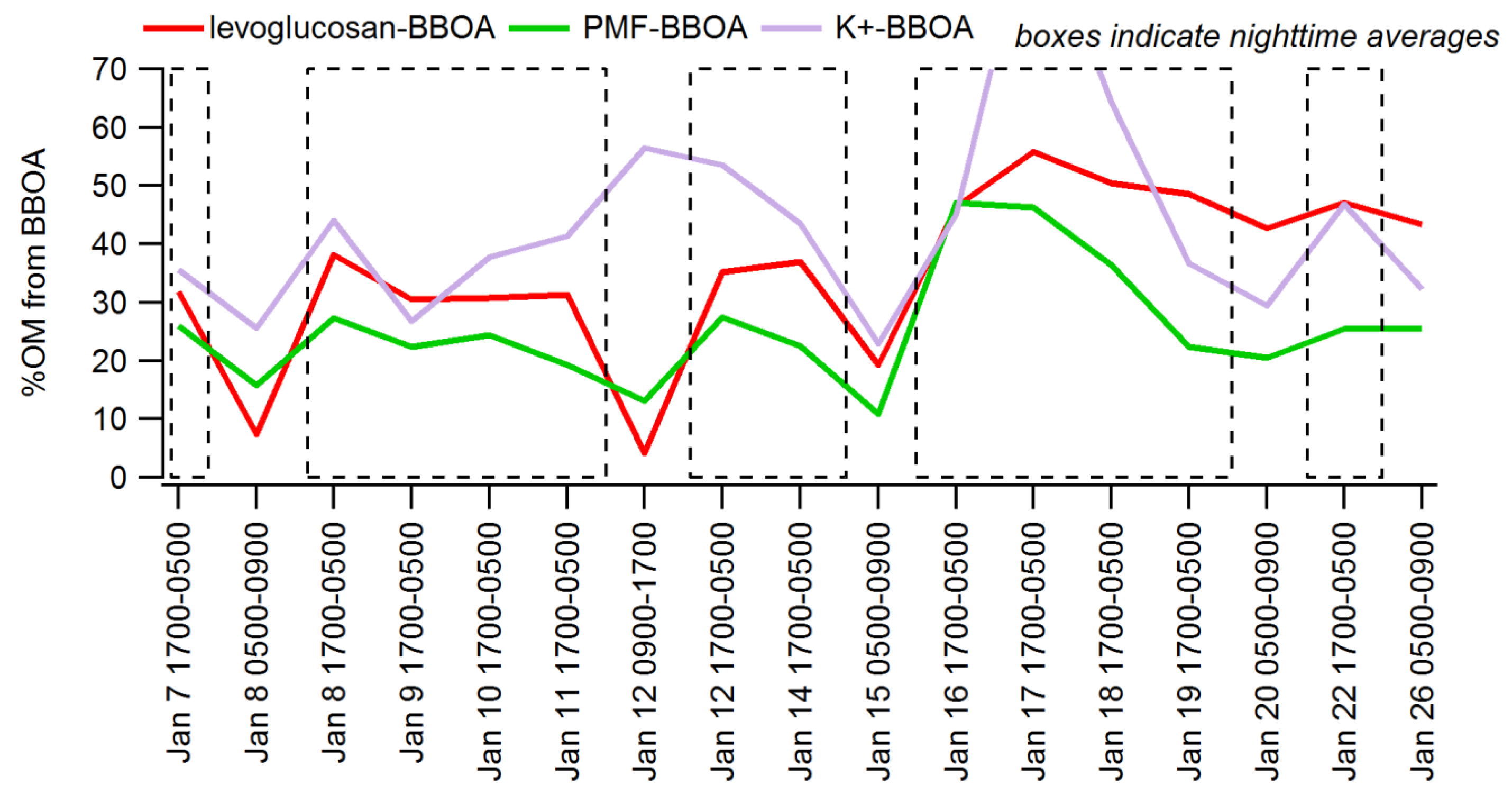
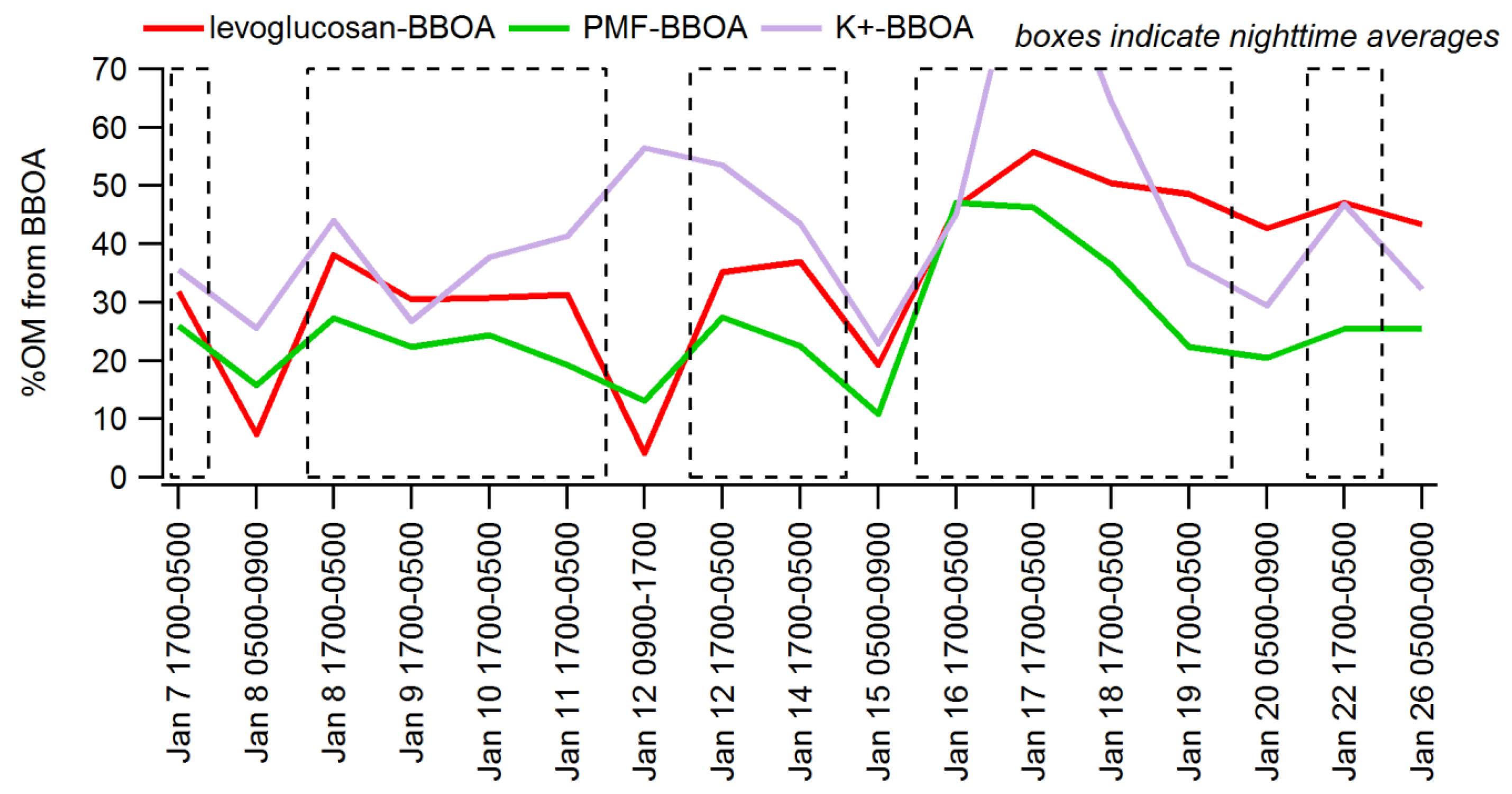
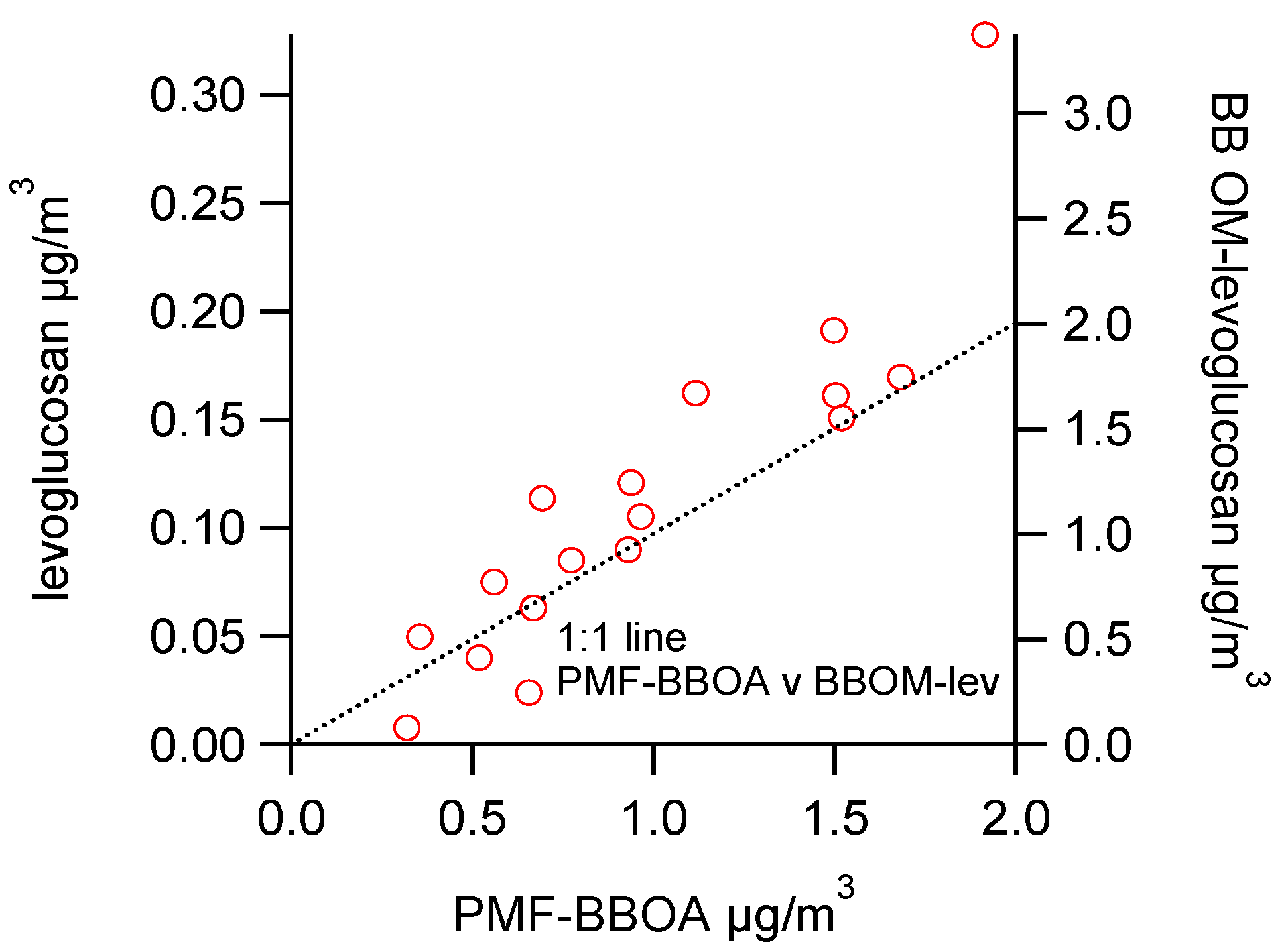
3.3. Apportioning Biomass Burning via Multiple Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bergauff, M.A.; Ward, T.J.; Noonan, C.W.; Palmer, C.P. The effect of a woodstove changeout on ambient levels of PM2.5 and chemical tracers for woodsmoke in Libby, Montana. Atmos Environ. 2009, 43, 2938–2943. [Google Scholar] [CrossRef]
- Ward, T.; Noonan, C. Results of a residential indoor PM2.5 sampling program before and after a woodstove changeout. Indoor Air 2008, 18, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.A.; Miller, P.J.; Rector, L.J.; Brauer, M.; Su, J.G. Characterization of valley winter woodsmoke concentrations in Northern NY using highly time-resolved measurements. Aerosol Air Qual. Res. 2011, 11, 519–530. [Google Scholar] [CrossRef]
- Wang, Y.; Hopke, P.K.; Utell, M.J. Urban-scale spatial-temporal variability of black carbon and winter residential wood combustion particles. Aerosol Air Qual. Res. 2011, 11, 473–481. [Google Scholar] [CrossRef]
- Lighty, J.S.; Veranth, J.M.; Sarofim, A.F. Combustion aerosols: Factors governing their size and composition and implications to human health. J. Air Waste Manag. Assoc. 2000, 50, 1565–1618. [Google Scholar] [CrossRef] [PubMed]
- Barregard, L.; Sallsten, G.; Andersson, L.; Almstrand, A.-C.; Gustafson, P.; Andersson, M.; Olin, A.-C. Experimental exposure to wood smoke: Effects on airway inflammation and oxidative stress. Occup. Environ. Med. 2007, 65, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.; Stiefer, P.S.; Weir, B.R. Carcinogenic Risk and Residential Wood Smoke; Systems Applications International: San Rafael, CA, USA, 1992. [Google Scholar]
- Seagrave, J.; McDonald, J.D.; Bedrick, E.; Edgerton, E.S.; Gigliotti, A.P.; Jansen, J.J.; Ke, L.; Naeher, L.P.; Seilkop, S.K.; Zheng, M.; et al. Lung toxicity of ambient particulate matter from southeastern U.S. sites with different contributing sources: Relationships between composition and effects. Environ. Health Perspect. 2006, 114, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Travis, C.C.; Etnier, E.L.; Meyer, H.R. Health risks of residential wood heat. Environ. Manag. 1985, 9, 209–215. [Google Scholar] [CrossRef]
- Naeher, L.P.; Brauer, M.; Lipsett, M.; Zelikoff, J.T.; Simpson, C.D.; Koenig, J.Q.; Smith, K.R. Woodsmoke health effects: A review. Inhal. Toxicol. 2007, 19, 67–106. [Google Scholar] [CrossRef] [PubMed]
- Fine, P.M.; Cass, G.R.; Simoneit, B.R.T. Chemical characterization of fine particle emissions from the fireplace combustion of wood types grown in the Midwestern and Western United States. Environ. Eng. Sci. 2004, 21, 387–409. [Google Scholar] [CrossRef]
- Simoneit, B.R.T.; Schauer, J.J.; Nolte, C.G.; Oros, D.R.; Elias, V.O.; Fraser, M.P.; Rogge, W.F.; Cass, G.R. Levoglucosan, a tracer for cellulose in biomass burning and atmospheric particles. Atmos. Environ. 1999, 33, 173–182. [Google Scholar] [CrossRef]
- Simoneit, B.R.T. Biomass burning—A review of organic tracers for smoke from incomplete combustion. Appl. Geochem. 2002, 17, 129–162. [Google Scholar] [CrossRef]
- Engling, G.; Herckes, P.; Kreidenweis, S.M.; Malm, W.C.; Collett, J.L. Composition of the fine organic aerosol in Yosemite National Park during the 2002 Yosemite Aerosol Characterization Study. Atmos. Environ. 2006, 40, 2959–2972. [Google Scholar] [CrossRef]
- Schauer, J.J.; Kleeman, M.J.; Cass, G.R.; Simoneit, B.R.T. Measurement of emissions from air pollution sources. 3. C1 through C29 organic compounds from fireplace combustion of wood. Environ. Sci. Technol. 2001, 35, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.P.; Holden, A.S.; Patterson, L.A.; McMeeking, G.R.; Kreidenweis, S.M.; Malm, W.C.; Hao, W.M.; Wold, C.E.; Collett, J.L., Jr. A method for smoke marker measurements and its potential application for determining the contribution of biomass burning from wildfires and prescribed fires to ambient PM2.5 organic carbon. J. Geophys. Res. 2008. [Google Scholar] [CrossRef]
- Hennigan, C.J.; Miracolo, M.A.; Engelhart, G.J.; May, A.A.; Presto, A.A.; Lee, T.; Sullivan, A.P.; McMeeking, G.R.; Coe, H.; Wold, C.E.; et al. Chemical and physical transformations of organic aerosol from the photo-oxidation of open biomass burning emissions in an environmental chamber. Atmos. Chem. Phys. 2011, 11, 7669–7686. [Google Scholar] [CrossRef]
- Hoffmann, D.; Tilgner, A.; Iinuma, Y.; Herrmann, H. Atmospheric stability of levoglucosan: A detailed laboratory and modeling study. Environ. Sci. Technol. 2010, 44, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Slade, J.H.; Knopf, D.A. Multiphase OH oxidation kinetics of organic aerosol: The role of particle phase state and relative humidity. Geophys. Res. Lett. 2014, 41, 5297–5306. [Google Scholar] [CrossRef]
- Kessler, S.H.; Smith, J.D.; Che, D.L.; Worsnop, D.R.; Wilson, K.R.; Kroll, J.H. Chemical sinks of organic aerosol: Kinetics and products of the heterogeneous oxidation of erythritol and levoglucosan. Environ. Sci. Technol. 2010, 44, 7005–7010. [Google Scholar] [CrossRef] [PubMed]
- Slade, J.H.; Knopf, D.A. Heterogeneous OH oxidation of biomass burning organic aerosol surrogate compounds: Assessment of volatilisation products and the role of OH concentration on the reactive uptake kinetics. Phys. Chem. Chem. Phys. 2013, 15, 5898–5915. [Google Scholar] [CrossRef] [PubMed]
- Alfarra, M.R.; Prévôt, A.S.H.; Szidat, S.; Sandradewi, J.; Weimer, S.; Lanz, V.A.; Schreiber, D.; Mohr, M.; Baltensperger, U. Identification of the mass spectral signature of organic aerosols from wood burning emissions. Environ. Sci. Technol. 2007, 41, 5770–5777. [Google Scholar] [CrossRef] [PubMed]
- Weimer, S.; Alfarra, M.R.; Schreiber, D.; Mohr, M.; Prévôt, A.S.H.; Baltensperger, U. Organic aerosol mass spectral signatures from wood-burning emissions: Influence of burning conditions and wood type. J. Geophys. Res. Atmos. 2008. [Google Scholar] [CrossRef]
- Schneider, J.; Weimer, S.; Drewnick, F.; Borrmann, S.; Helas, G.; Gwaze, P.; Schmid, O.; Andreae, M.O.; Kirchner, U. Mass spectrometric analysis and aerodynamic properties of various types of combustion-related aerosol particles. Int. J. Mass Spec. 2006, 258, 37–49. [Google Scholar] [CrossRef]
- Lee, T.; Sullivan, A.P.; Mack, L.; Jimenez, J.L.; Kreidenweis, S.M.; Onasch, T.B.; Worsnop, D.R.; Malm, W.; Wold, C.E.; Hao, W.M.; et al. Chemical smoke marker emissions during flaming and smoldering phases of laboratory open burning of wildland fuels. Aerosol Sci. Technol. 2010. [Google Scholar] [CrossRef]
- Mohr, C.; Huffman, J.A.; Cubison, M.; Aiken, A.C.; Docherty, K.S.; Kimmel, J.R.; Ulbrich, I.M.; Hannigan, M.; Jimenez, J.L. Characterization of primary organic aerosol emissions from meat cooking, trash burning, and motor vehicles with high-resolution aerosol mass spectrometry and comparison with ambient and chamber observations. Environ. Sci. Technol. 2009, 43, 2443–2449. [Google Scholar] [CrossRef] [PubMed]
- Takegawa, N.; Miyakawa, T.; Kawamura, K.; Kondo, Y. Contribution of selected dicarboxylic and omega-oxocarboxylic acids in ambient aerosol to the m/z 44 signal of an aerodyne aerosol mass spectrometer. Aerosol Sci. Technol. 2007, 41, 418–437. [Google Scholar] [CrossRef]
- Cubison, M.J.; Ortega, A.M.; Hayes, P.L.; Farmer, D.K.; Day, D.; Lechner, M.J.; Brune, W.H.; Apel, E.; Diskin, G.S.; Fisher, J.A.; et al. Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies. Atmos. Chem. Phys. 2011, 11, 12049–12064. [Google Scholar] [CrossRef]
- Minguillon, M.C.; Perron, N.; Querol, X.; Szidat, S.; Fahrni, S.M.; Alastuey, A.; Jimenez, J.L.; Mohr, C.; Ortega, A.M.; Day, D.A.; et al. Fossil versus contemporary sources of fine elemental and organic carbonaceous particulate matter during the DAURE campaign in Northeast Spain. Atmos. Chem. Phys. 2011, 11, 12067–12084. [Google Scholar] [CrossRef]
- Kim, E.; Larson, T.V.; Hopke, P.K.; Slaughter, C.; Sheppard, L.E.; Claiborn, C. Source identification of PM2.5 in an arid northwest U.S. city by positive matrix factorization. Atmos. Res. 2003, 66, 291–305. [Google Scholar] [CrossRef]
- Poirot, R. Tracers of Opportunity: Potassium. Available online: http://capita.wustl.edu/PMFine/Workgroup/SourceAttribution/Reports/In-progress/potass/Kcover.htm (accessed on 12 April 2016).
- Liu, W.; Wang, Y.; Russell, A.; Edgerton, E.S. Atmospheric aerosol over two urban-rural pairs in the southeastern United States: Chemical composition and possible sources. Atmos. Environ. 2005, 39, 4453–4470. [Google Scholar] [CrossRef]
- Brown, S.G.; Frankel, A.; Raffuse, S.M.; Roberts, P.T.; Hafner, H.R.; Anderson, D.J. Source apportionment of fine particulate matter in Phoenix, Arizona, using positive matrix factorization. J. Air Waste Manag. Assoc. 2007, 57, 741–752. [Google Scholar] [PubMed]
- Aiken, A.C.; de Foy, B.; Wiedinmyer, C.; DeCarlo, P.F.; Ulbrich, I.M.; Wehrli, M.N.; Szidat, S.; Prévôt, A.S.H.; Noda, J.; Wacker, L.; et al. Mexico City aerosol analysis during MILAGRO using high resolution aerosol mass spectrometry at the urban supersite (T0). Part 2: Analysis of the biomass burning contribution and the non-fossil carbon fraction. Atmos. Chem. Phys. 2010, 10, 5315–5341. [Google Scholar] [CrossRef]
- Zhang, X.; Hecobian, A.; Zheng, M.; Frank, N.H.; Weber, R.J. Biomass burning impact on PM2.5 over the southeastern US during 2007: Integrating chemically speciated FRM filter measurements, MODIS fire counts and PMF analysis. Atmos. Chem. Phys. 2010, 10, 6839–6853. [Google Scholar] [CrossRef]
- Schauer, J.J.; Kleeman, M.J.; Cass, G.R.; Simoneit, B.R.T. Measurement of emissions from air pollution sources. 1. C1 through C29 organic compounds from meat charbroiling. Environ. Sci. Technol. 1999, 33, 1566–1577. [Google Scholar] [CrossRef]
- Fine, P.M.; Cass, G.R.; Simoneit, B.R.T. Chemical characterization of fine particle emissions from the fireplace combustion of woods grown in the southern United States. Environ. Sci. Technol. 2002, 36, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Fine, P.M.; Cass, G.R.; Simoneit, B.R.T. Organic compounds in biomass smoke from residential wood combustion: Emissions characterization at a continental scale. J. Geophys. Res. Atmos. 2002. [Google Scholar] [CrossRef]
- Fine, P.M.; Cass, G.R.; Simoneit, B.R.T. Chemical characterization of fine particle emissions from fireplace combustion of woods grown in the northeastern United States. Environ. Sci. Technol. 2001, 35, 2665–2675. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.A.; Lawrence, J.; Koutrakis, P. Field validation of a semi-continuous method for aerosol black carbon (Aethalometer) and termporal patterns of summertime hourly black carbon measurements in Southwestern Pennsylvania. Atmos. Environ. 1999, 33, 817–823. [Google Scholar] [CrossRef]
- Sandradewi, J.; Prévôt, A.S.H.; Weingartner, E.; Schmidhauser, R.; Gysel, M.; Baltensperger, U. A study of wood burning and traffic aerosols in an Alpine valley using a multi-wavelength Aethalometer. Atmos. Environ. 2008, 42, 101–112. [Google Scholar] [CrossRef]
- Harrison, R.M.; Beddowsa, D.C.S.; Jones, A.M.; Calvo, A.; Alves, C.; Pio, C. An evaluation of some issues regarding the use of aethalometers to measure woodsmoke concentrations. Atmos. Environ. 2013, 80, 540–548. [Google Scholar] [CrossRef]
- Sandradewi, J.; Prévôt, A.S.H.; Alfarra, M.R.; Szidat, S.; Wehrli, M.N.; Ruff, M.; Weimer, S.; Lanz, V.A.; Weingartner, E.; Perron, N.; et al. Comparison of several wood smoke markers and source apportionment methods for wood burning particulate mass. Atmos. Chem. Phys. Discuss. 2008, 8, 8091–8118. [Google Scholar] [CrossRef]
- Favez, O.; El Haddad, I.; Piot, C.; Boréave, A.; Abidi, E.; Marchand, N.; Jaffrezo, J.-L.; Besombes, J.-L.; Personnaz, M.-B.; Sciare, J.; et al. Inter-comparison of source apportionment models for the estimation of wood burning aerosols during wintertime in an Alpine city (Grenoble, France). Atmos. Chem. Phys. 2010, 10, 5295–5314. [Google Scholar] [CrossRef]
- Green, M.C.; Chow, J.C.; Hecobian, A.; Etyemezian, V.; Kuhns, H.; Watson, J.G. Las Vegas Valley Visibility and PM2.5 Study; Final Report Prepared for the Clark County Department of Air Quality Management, Las Vegas, NV; Desert Research Institute: Las Vegas, NV, USA, 2002. [Google Scholar]
- Watson, J.G.; Barber, P.W.; Chang, M.C.O.; Chow, J.C.; Etyemezian, V.R.; Green, M.C.; Keislar, R.E.; Kuhns, H.D.; Mazzoleni, C.; Moosmüller, H.; et al. Southern Nevada Air Quality Study; Final Report Prepared for the U.S. Department of Transportation, Washington, DC; Desert Research Institute: Reno, NV, USA, 2007. [Google Scholar]
- Brown, S.G.; Lee, T.; Norris, G.A.; Roberts, P.T.; Collett, J.L., Jr.; Paatero, P.; Worsnop, D.R. Receptor modeling of near-roadway aerosol mass spectrometer data in Las Vegas, Nevada, with EPA PMF. Atmos. Chem. Phys. 2012, 12, 309–325. [Google Scholar] [CrossRef]
- Brown, S.G.; McCarthy, M.C.; DeWinter, J.L.; Vaughn, D.L.; Roberts, P.T. Changes in air quality at near-roadway schools after a major freeway expansion in Las Vegas, Nevada. J. Air Waste Manag. Assoc. 2014, 64, 1002–1012. [Google Scholar] [CrossRef]
- DeCarlo, P.; Kimmel, J.R.; Trimborn, A.; Northway, M.; Jayne, J.T.; Aiken, A.C.; Gonin, M.; Fuhrer, K.; Horvath, T.; Docherty, K.S.; et al. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 2006, 78, 8281–8289. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, J.L.; Jayne, J.T.; Shi, Q.; Kolb, C.E.; Worsnop, D.R.; Yourshaw, I.; Seinfeld, J.H.; Flagan, R.C.; Zhang, X.F.; Smith, K.A.; et al. Ambient aerosol sampling using the Aerodyne Aerosol Mass Spectrometer. J. Geophys. Res. Atmos. 2003. [Google Scholar] [CrossRef]
- Zhang, Q.; Jimenez, J.L.; Canagaratna, M.R.; Ulbrich, I.M.; Ng, N.L.; Worsnop, D.R.; Sun, Y. Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: A review. Anal. Bioanal. Chem. 2011, 401, 3045–3067. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, Q.; MacDonald, A.M.; Hayden, K.; Li, S.M.; Liggio, J.; Liu, P.S.K.; Anlauf, K.G.; Leaitch, W.R.; Steffen, A.; et al. Size-resolved aerosol chemistry on Whistler Mountain, Canada with a high-resolution aerosol mass spectrometer during INTEX-B. Atmos. Chem. Phys. 2009, 9, 3095–3111. [Google Scholar] [CrossRef]
- Canagaratna, M.R.; Jayne, J.T.; Jimenez, J.L.; Allan, J.D.; Alfarra, M.R.; Zhang, Q.; Onasch, T.B.; Drewnick, F.; Coe, H.; Middlebrook, A.; et al. Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer. Mass Spectrom. Rev. 2007, 26, 185–222. [Google Scholar] [CrossRef] [PubMed]
- Drewnick, F.; Hings, S.S.; Alfarra, M.R.; Prevot, A.S.H.; Borrmann, S. Aerosol quantification with the Aerodyne Aerosol Mass Spectrometer: Detection limits and ionizer background effects. Atmos. Measure. Tech. 2009, 2, 33–46. [Google Scholar] [CrossRef]
- Olson, D.A.; Vedantham, R.; Norris, G.A.; Brown, S.G.; Roberts, P. Determining source impacts near roadways using wind regression and organic source markers. Atmos. Environ. 2012, 47, 261–268. [Google Scholar] [CrossRef]
- Orsini, D.A.; Ma, Y.L.; Sullivan, A.; Sierau, B.; Baumann, K.; Weber, R.J. Refinements to the particle-into-liquid sampler (PILS) for ground and airborne measurements of water soluble aerosol composition. Atmos. Environ. 2003, 37, 1243–1259. [Google Scholar] [CrossRef]
- Weber, R.J.; Orsini, D.; Daun, Y.; Lee, Y.N.; Klotz, P.J.; Brechtel, F. A particle-into-liquid collector for rapid measurement of aerosol bulk chemical composition. Aerosol Sci. Technol. 2001, 35, 718–727. [Google Scholar] [CrossRef]
- Weber, R.; Orsini, D.; Duan, Y.; Baumann, K.; Kiang, C.S.; Chameides, W.; Lee, Y.N.; Brechtel, F.; Klotz, P.; Jongejan, P.; et al. Intercomparison of near real time monitors of PM2.5 nitrate and sulfate at the U.S. Environmental Protection Agency Atlanta Supersite. J. Geophys. Res. Atmos. 2003. [Google Scholar] [CrossRef]
- Sorooshian, A.; Brechtel, F.J.; Ma, Y.L.; Weber, R.J.; Corless, A.; Flagan, R.C.; Seinfeld, J.H. Modeling and characterization of a particle-into-liquid sampler (PILS). Aerosol Sci. Technol. 2006, 40, 396–409. [Google Scholar] [CrossRef]
- Lee, T.; Yu, X.-Y.; Kreidenweis, S.M.; Malm, W.C.; Collett, J.L. Semi-continuous measurement of PM2.5 ionic composition at several rural locations in the United States. Atmos. Environ. 2008, 42, 6655–6669. [Google Scholar] [CrossRef]
- Norris, G.; Duvall, R.; Brown, S.; Bai, S. EPA Positive Matrix Factorization (PMF) 5.0 Fundamentals and User Guide; Prepared for the U.S. Environmental Protection Agency Office of Research and Development: Washington, DC, USA, 2014. [Google Scholar]
- Brown, S.G.; Eberly, S.; Paatero, P.; Norris, G.A. Methods for estimating uncertainty in PMF solutions: Examples with ambient air and water quality data and guidance on reporting PMF results. Sci. Total Environ. 2015, 518, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, I.M.; Canagaratna, M.R.; Zhang, Q.; Worsnop, D.R.; Jimenez, J.L. Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data. Atmos. Chem. Phys. 2009, 9, 2891–2918. [Google Scholar] [CrossRef]
- Lanz, V.A.; Alfarra, M.R.; Baltensperger, U.; Buchmann, B.; Hueglin, C.; Prévôt, A.S.H. Source apportionment of submicron organic aerosols at an urban site by factor analytical modelling of aerosol mass spectra. Atmos. Chem. Phys. 2007, 7, 1503–1522. [Google Scholar] [CrossRef]
- Puxbaum, H.; Caseiro, A.; Sanchez-Ochoa, A.; Kasper-Giebl, A.; Claeys, M.; Gelencser, A.; Legrand, M.; Preunkert, S.; Pio, C. Levoglucosan levels at background sites in Europe for assessing the impact of biomass combustion on the European aerosol background. J. Geophys. Res. 2007. [Google Scholar] [CrossRef]
- Schmidl, C.; Marr, I.L.; Caseiro, A.; Kotianova, P.; Berner, A.; Bauer, H.; Kasper-Giebl, A.; Puxbaum, H. Chemical characterisation of fine particle emissions from wood stove combustion of common woods growing in mid-European Alpine regions. Atmos. Environ. 2008, 42, 126–141. [Google Scholar] [CrossRef]
- Brown, S.G.; Lee, T.; Roberts, P.T.; Collett, J.L., Jr. Variations in the OM/OC ratio of urban organic aerosol next to a major roadway. J. Air Waste Manag. Assoc. 2013, 63, 1422–1433. [Google Scholar] [CrossRef] [PubMed]
- Oja, V.; Suuberg, E.M. Vapor pressures and enthalpies of sublimation of d-glucose, d-xylose, cellobiose, and levoglucosan. J. Chem. Eng. Data 1999, 44, 26–29. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Range | % OM from BB via Levoglucosan | % OM from BB via PMF-AMS (BBOA) | % OM via K+ |
|---|---|---|---|
| 12 12-h overnight periods | 33% +/− 7% | 26% +/− 9% | 44% +/− 18% |
| All evenings (1800–2300 LST) | n/a | 15% +/− 9% | 26% +/− 24% |
| All hours | n/a | 9% +/− 8% | 25% +/− 25% |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, S.G.; Lee, T.; Roberts, P.T.; Collett, J.L. Wintertime Residential Biomass Burning in Las Vegas, Nevada; Marker Components and Apportionment Methods. Atmosphere 2016, 7, 58. https://doi.org/10.3390/atmos7040058
Brown SG, Lee T, Roberts PT, Collett JL. Wintertime Residential Biomass Burning in Las Vegas, Nevada; Marker Components and Apportionment Methods. Atmosphere. 2016; 7(4):58. https://doi.org/10.3390/atmos7040058
Chicago/Turabian StyleBrown, Steven G., Taehyoung Lee, Paul T. Roberts, and Jeffrey L. Collett. 2016. "Wintertime Residential Biomass Burning in Las Vegas, Nevada; Marker Components and Apportionment Methods" Atmosphere 7, no. 4: 58. https://doi.org/10.3390/atmos7040058
APA StyleBrown, S. G., Lee, T., Roberts, P. T., & Collett, J. L. (2016). Wintertime Residential Biomass Burning in Las Vegas, Nevada; Marker Components and Apportionment Methods. Atmosphere, 7(4), 58. https://doi.org/10.3390/atmos7040058