
Composition and Sources of Particulate Matter Measured near Houston, TX: Anthropogenic-Biogenic Interactions

 ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
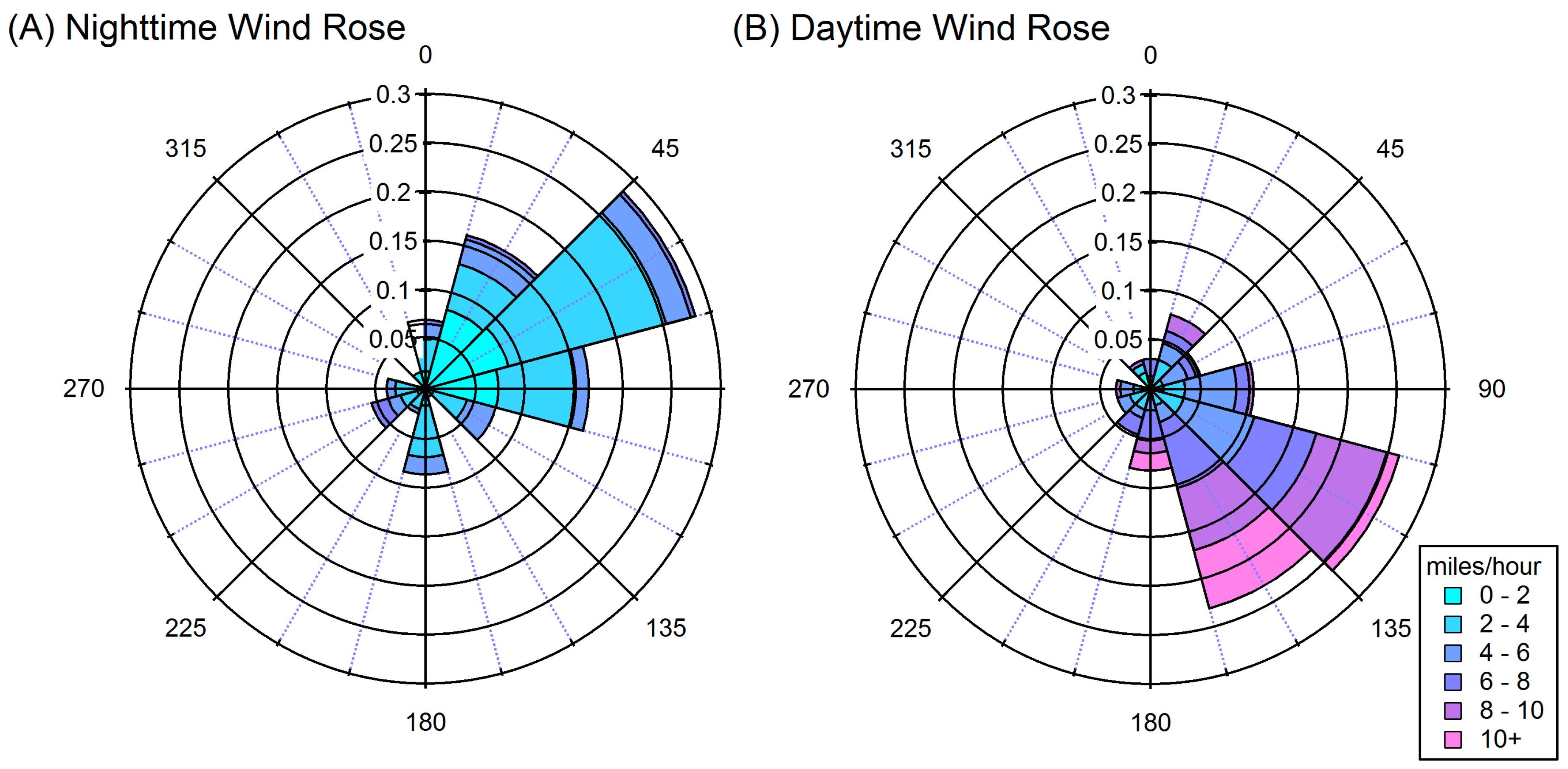
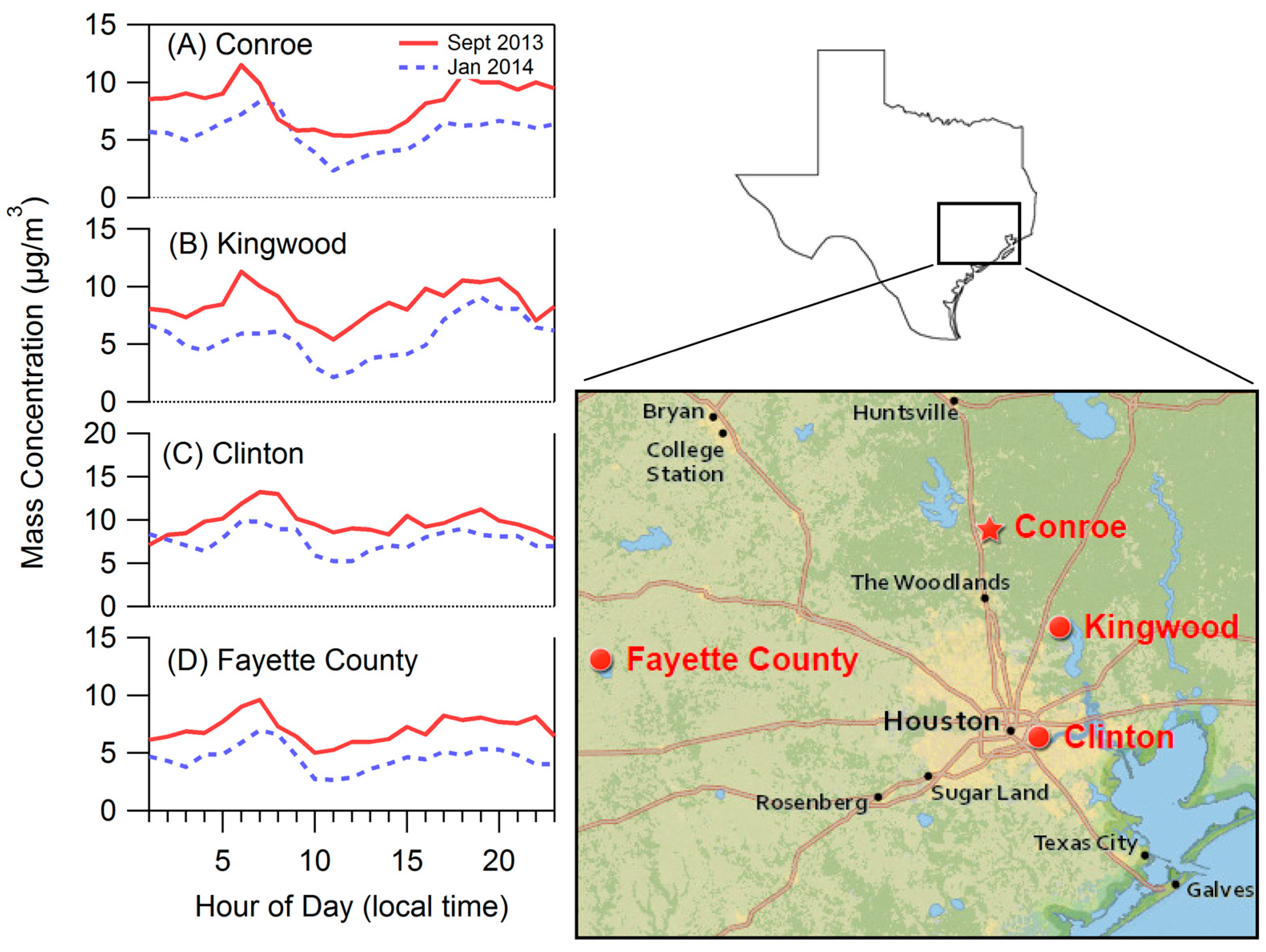
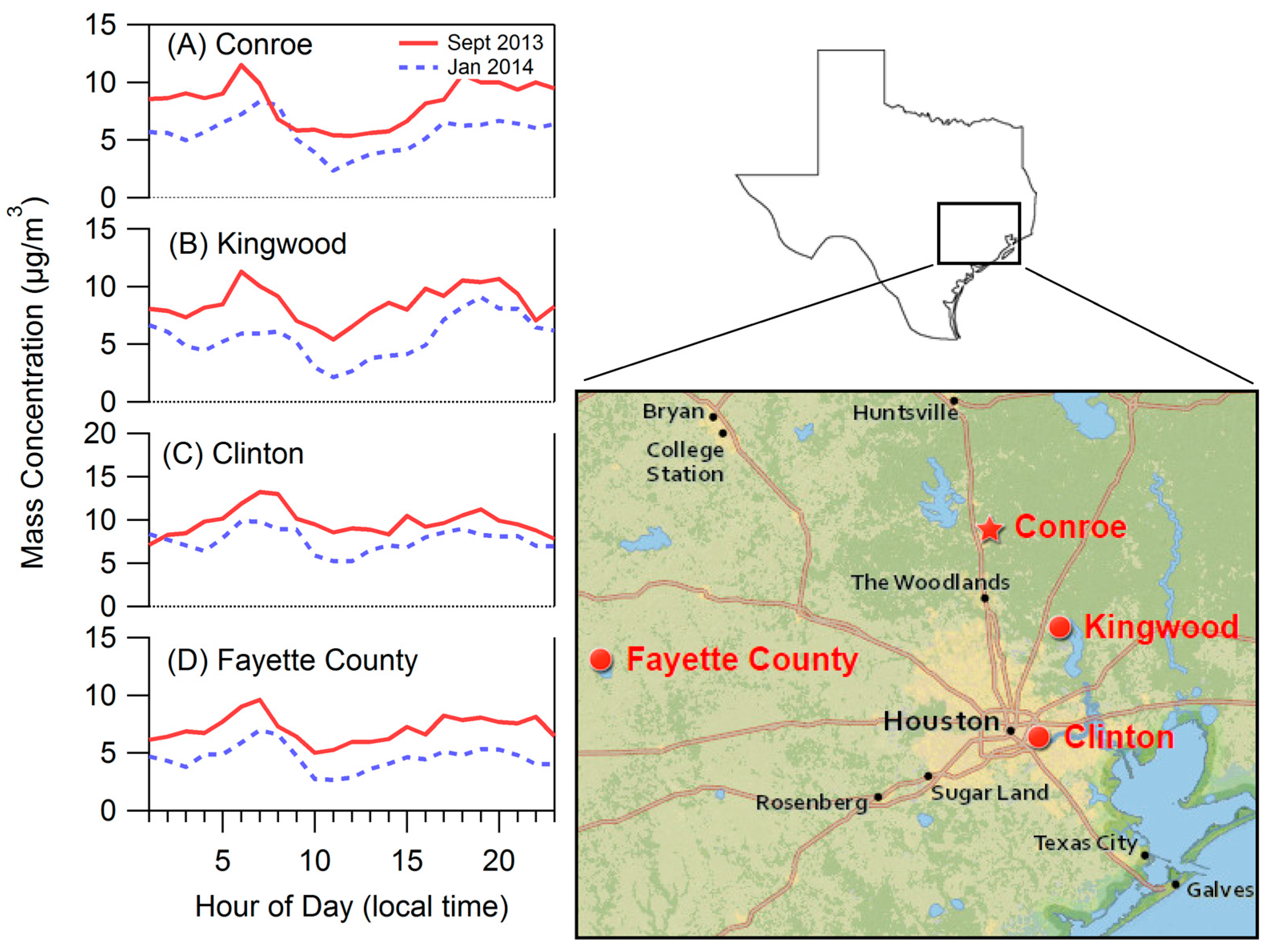
2.1. Site Description
2.2. Instrumentation and Data Analysis
2.2.1. Aerosol Chemical Speciation Monitor
2.2.2. Positive Matrix Factorization
2.2.3. High Resolution Time of Flight Chemical Ionization Mass Spectrometer
2.2.4. Filter Measurements
2.2.5. Diurnal Patterns: Analysis of Statistical Significance and Patterns
3. Results
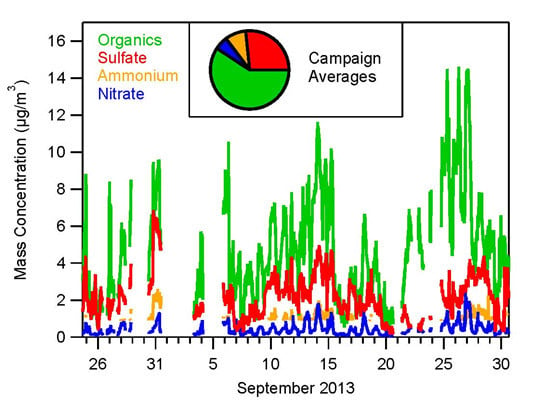
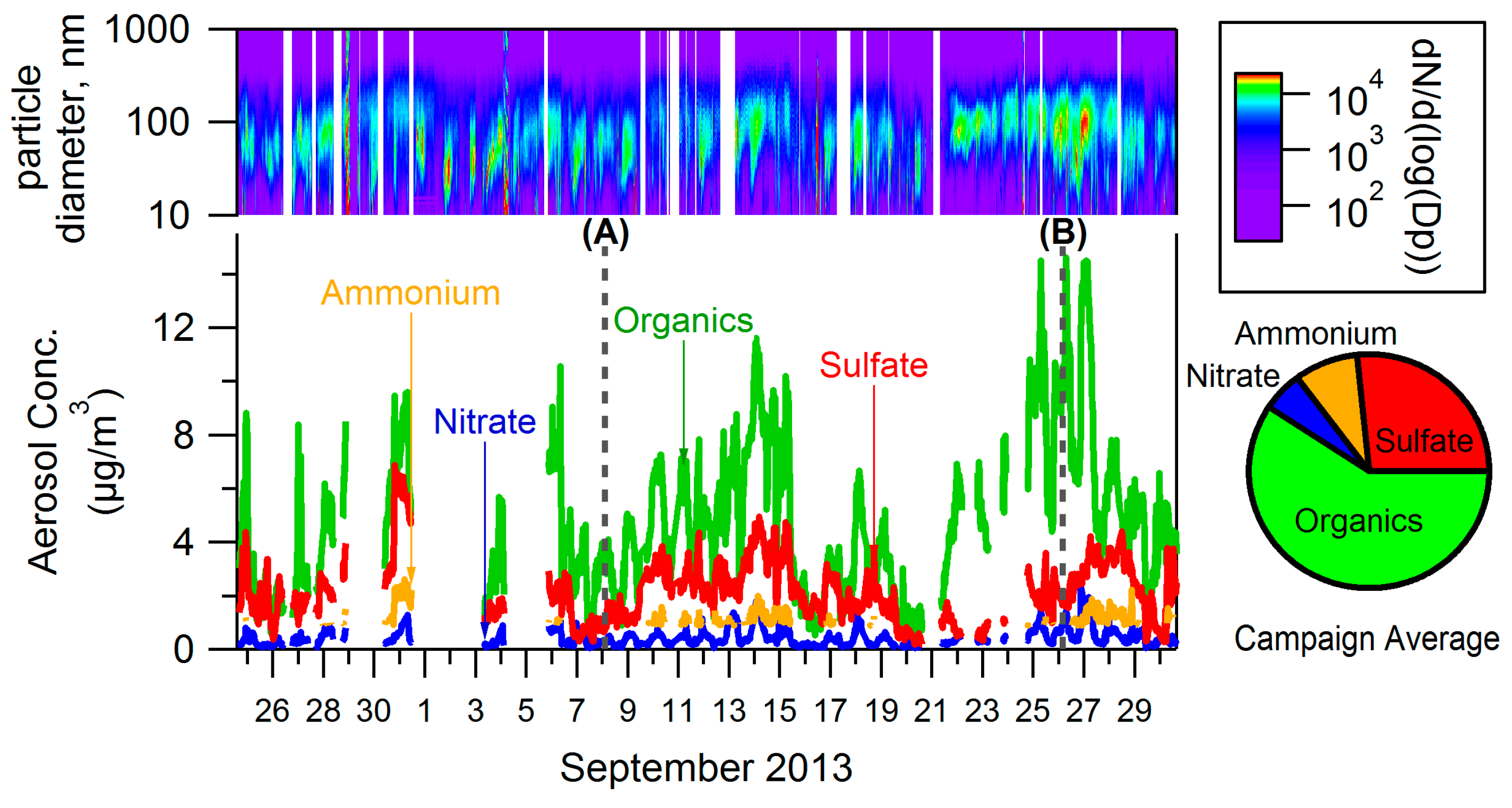
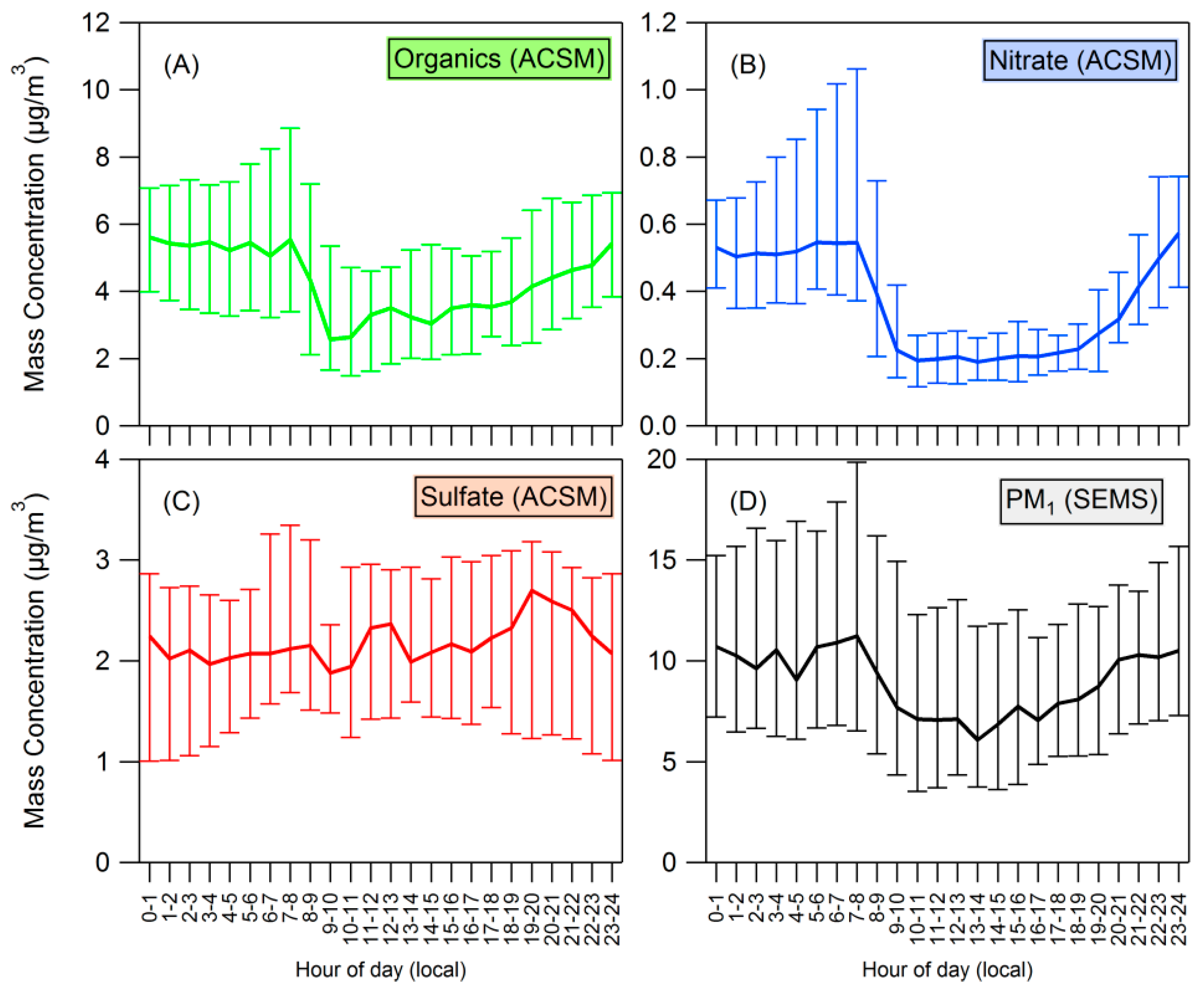
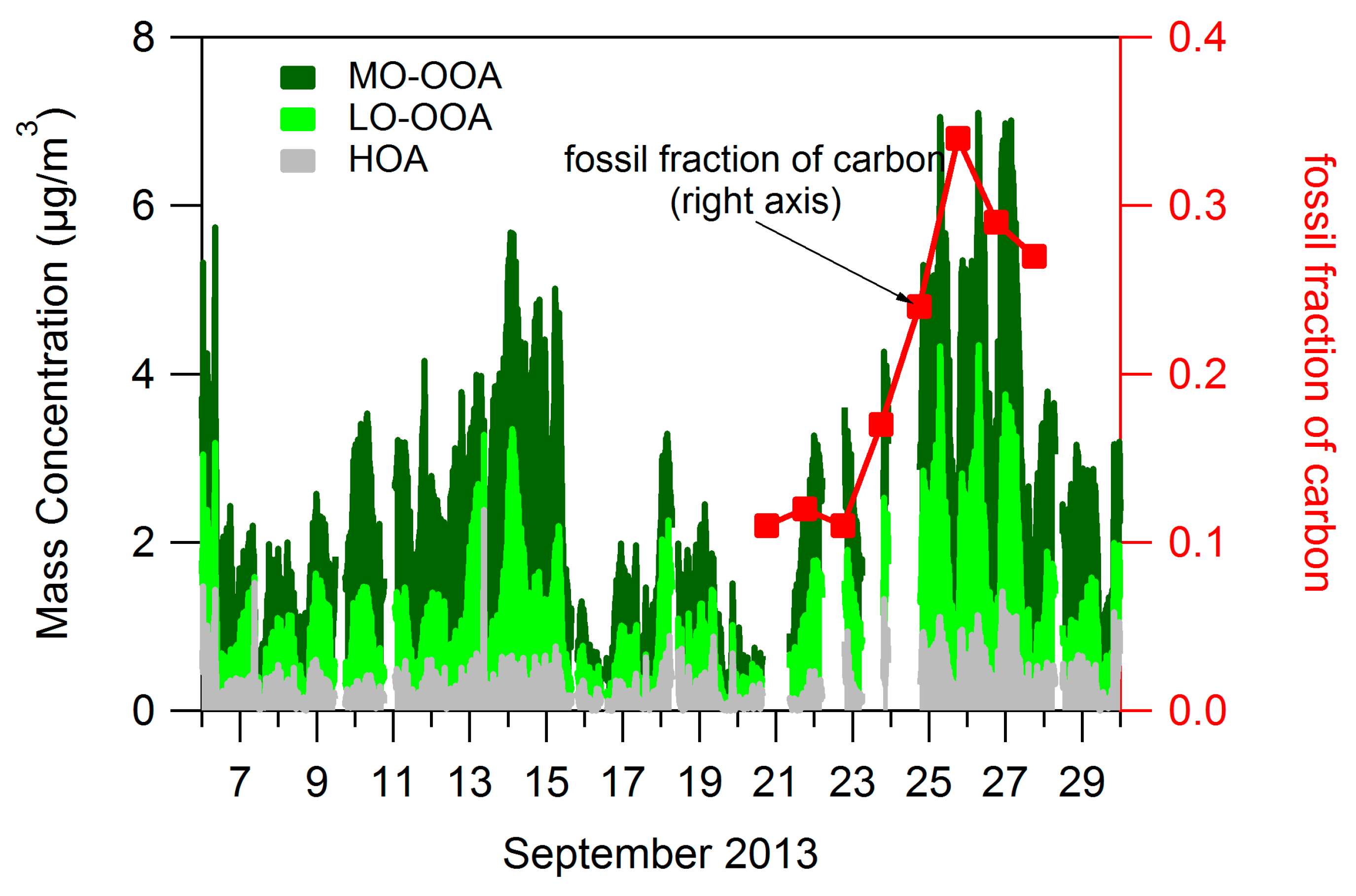
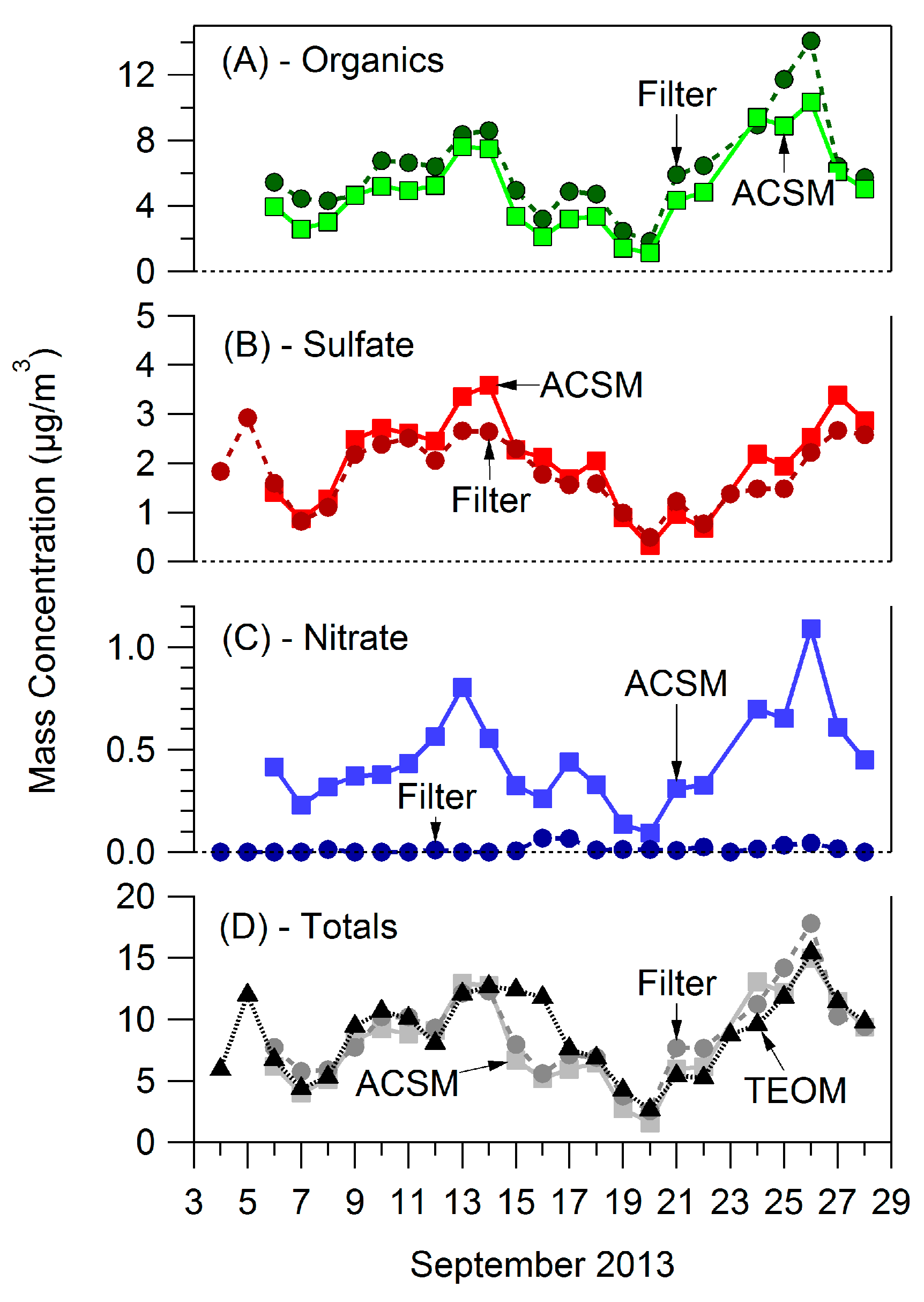
3.1. Bulk Concentrations and Diurnal Cycle
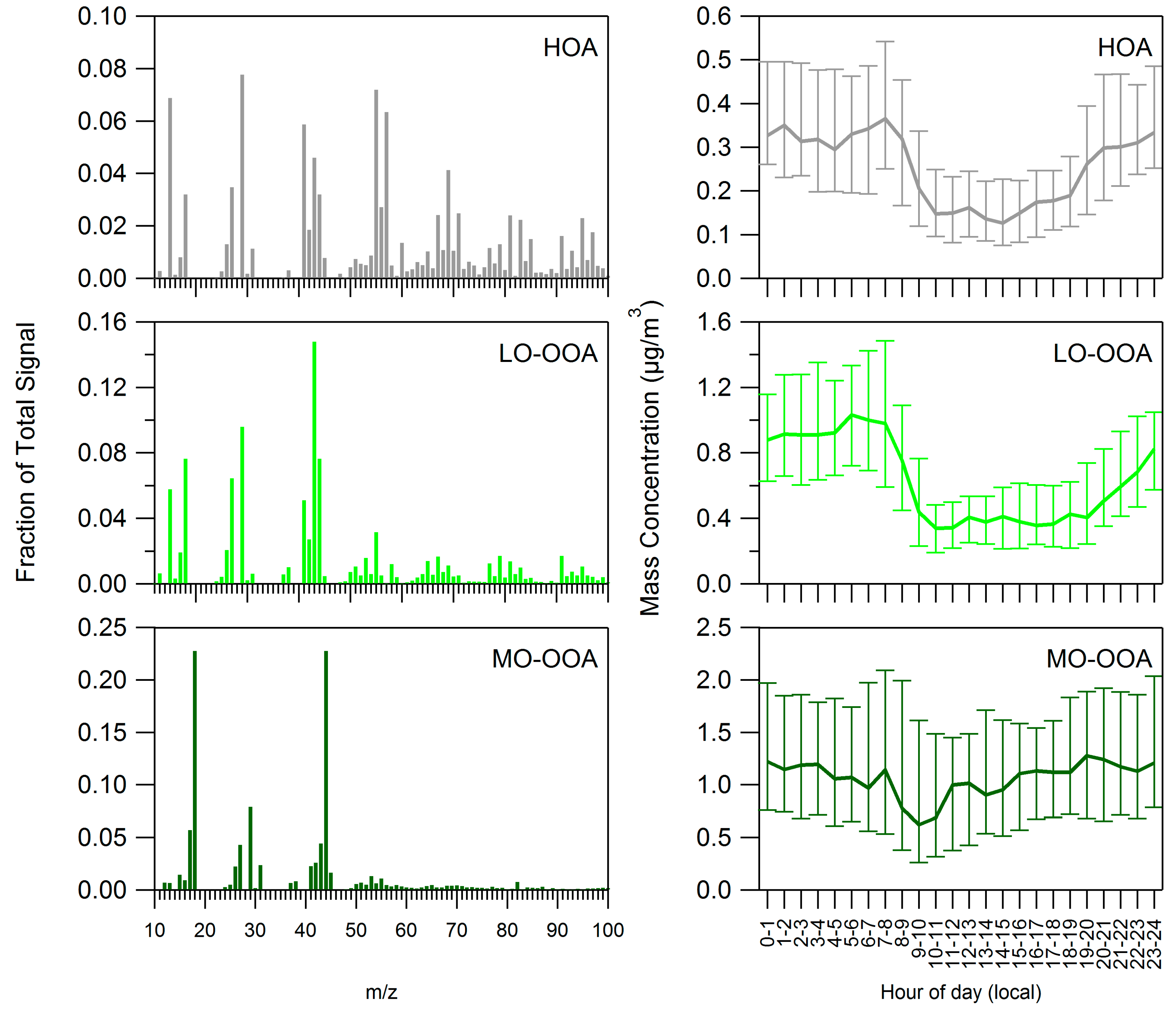
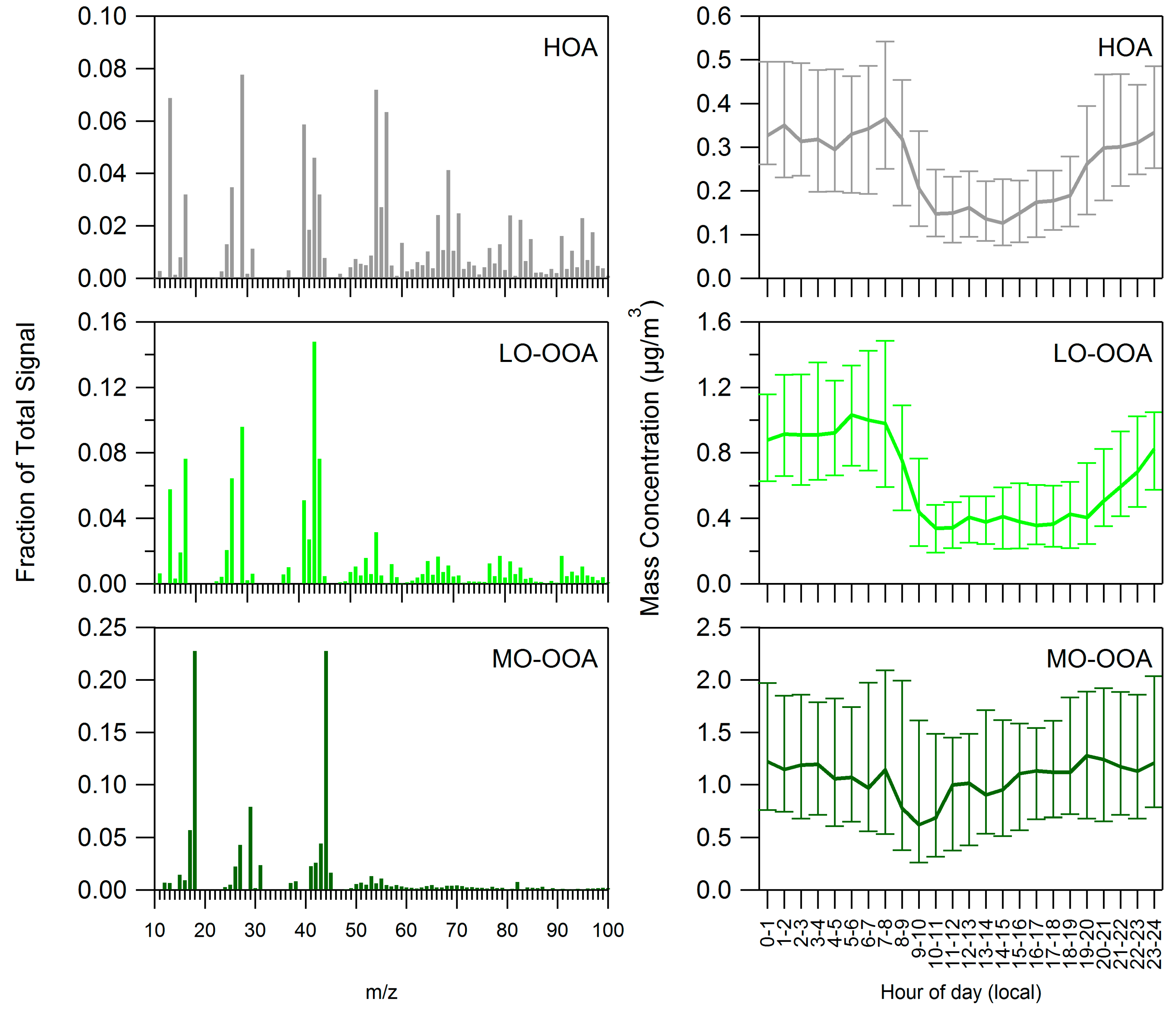
3.2. Positive Matrix Factorization
4. Discussion
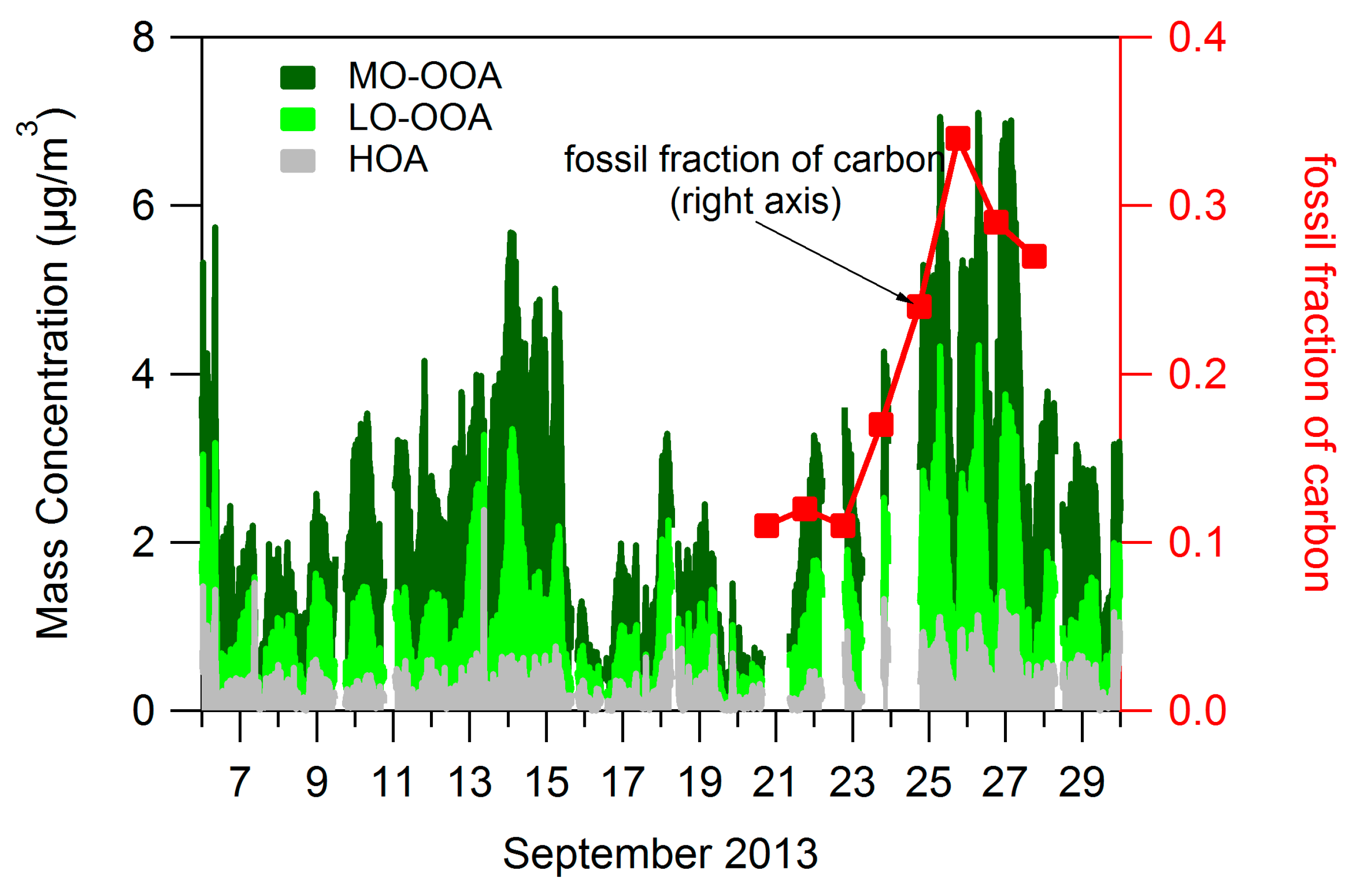
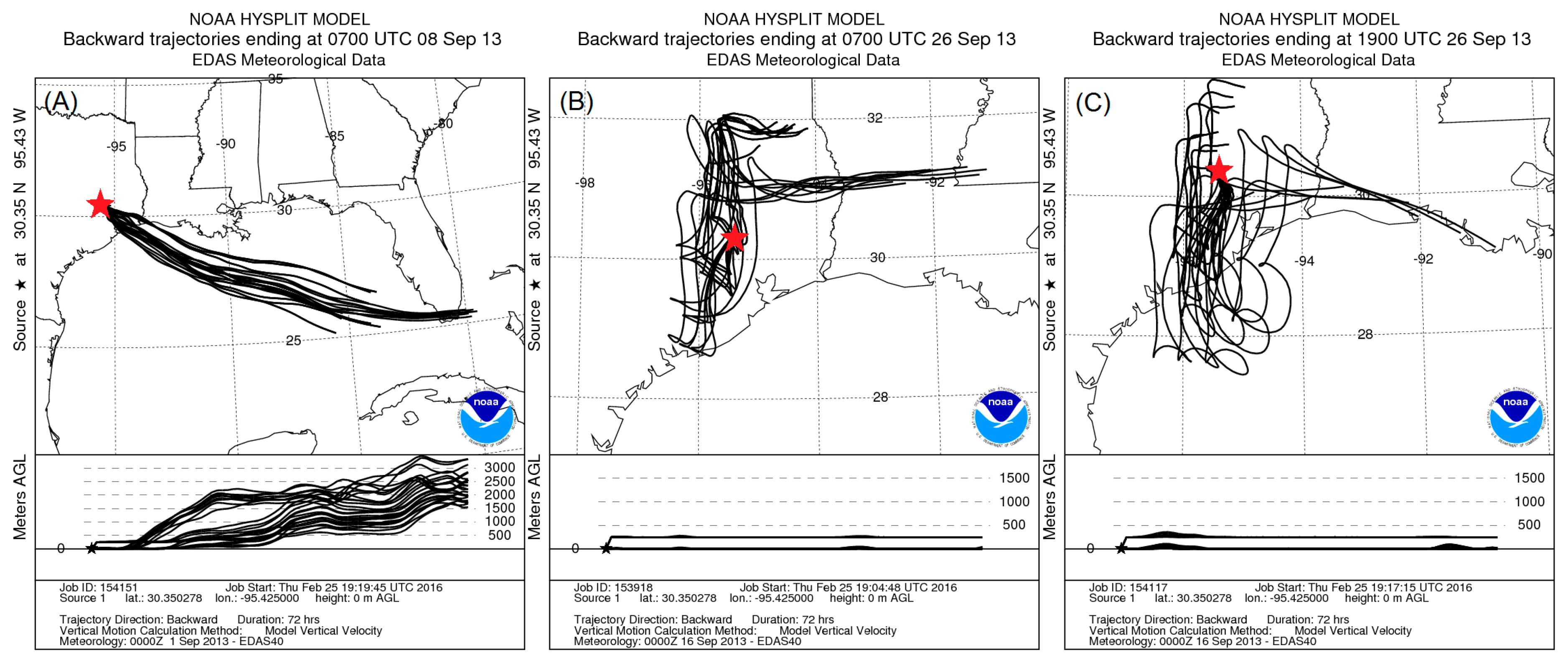
4.1. Composition of PM and Source Regions
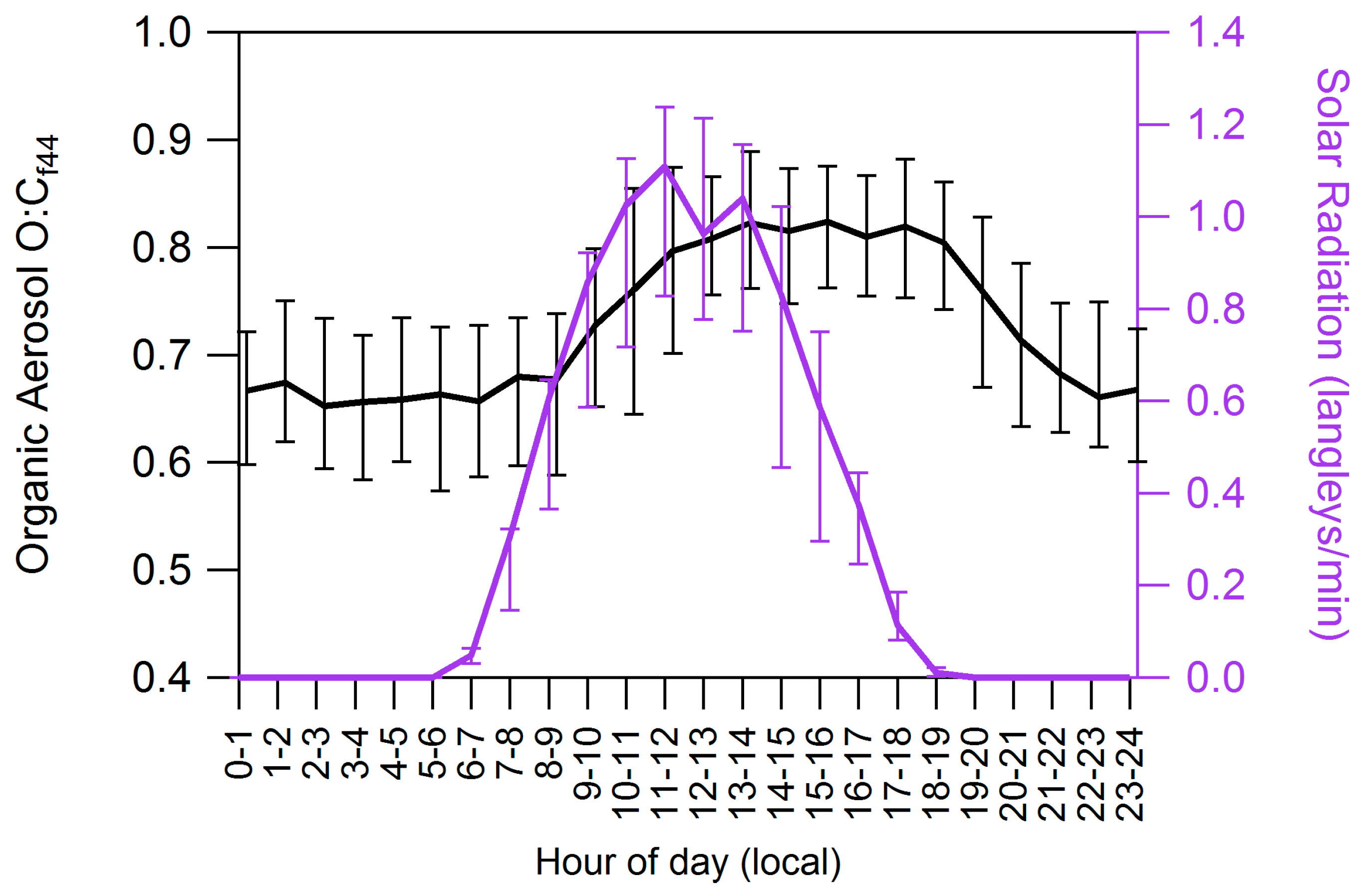
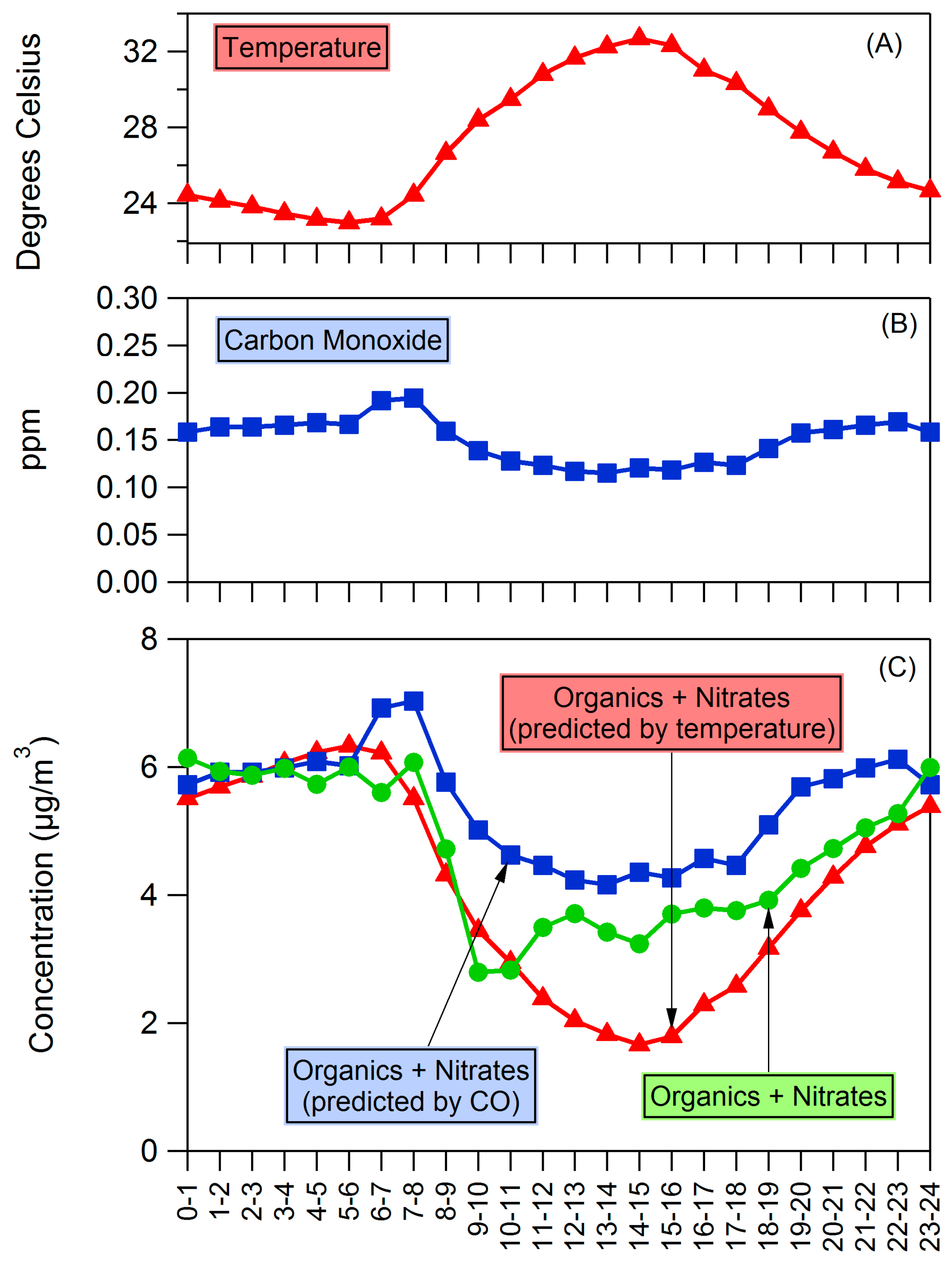
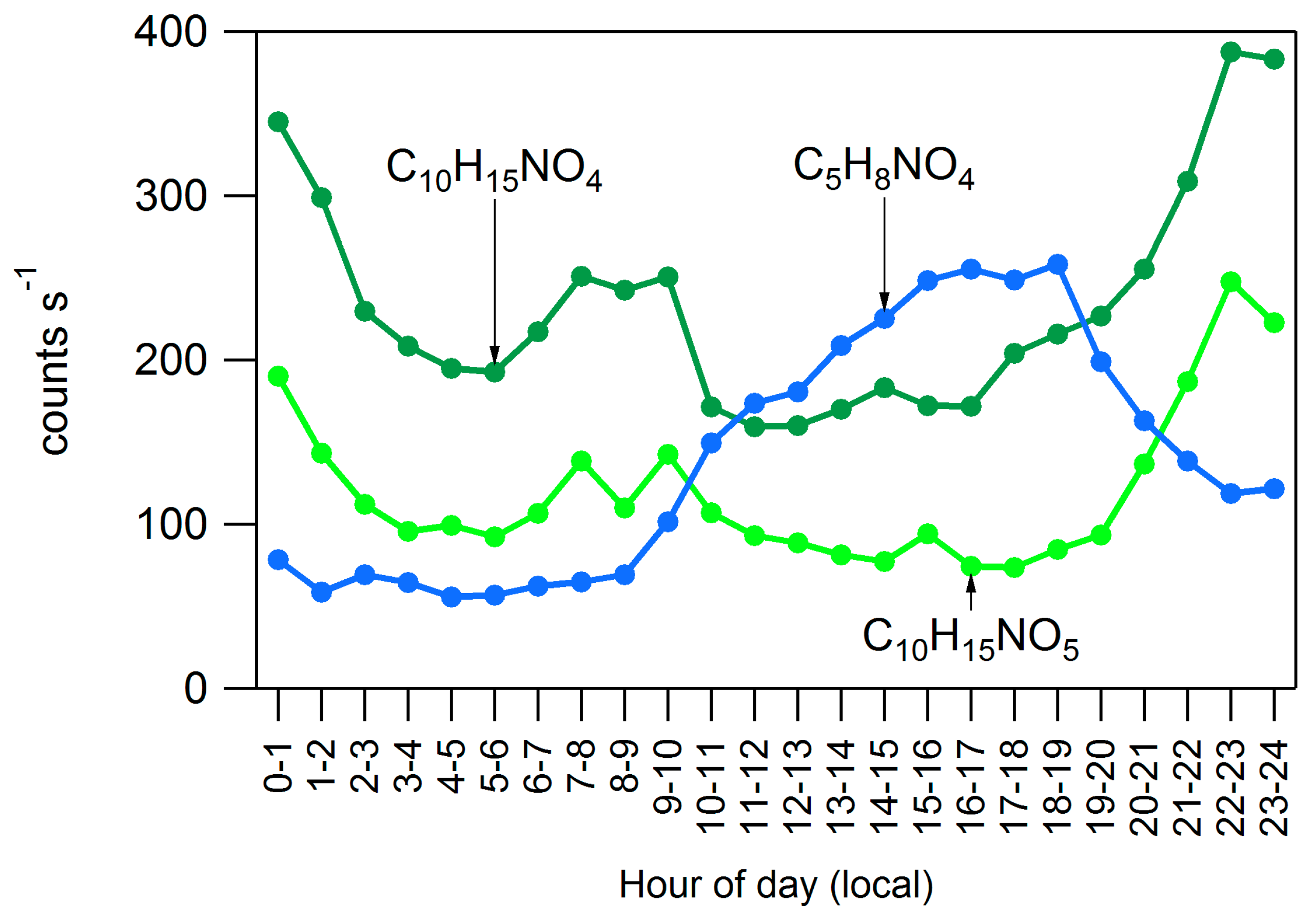
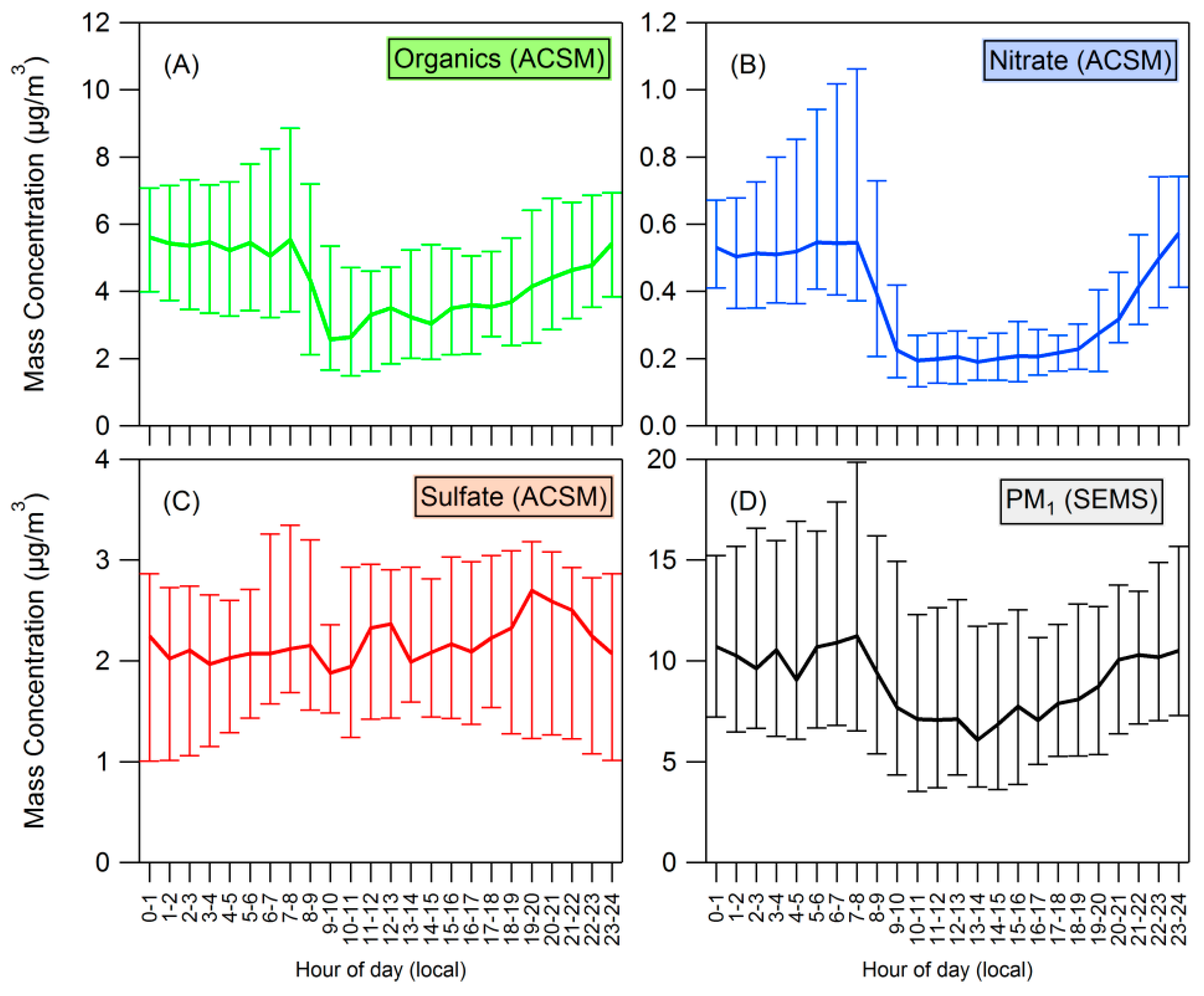
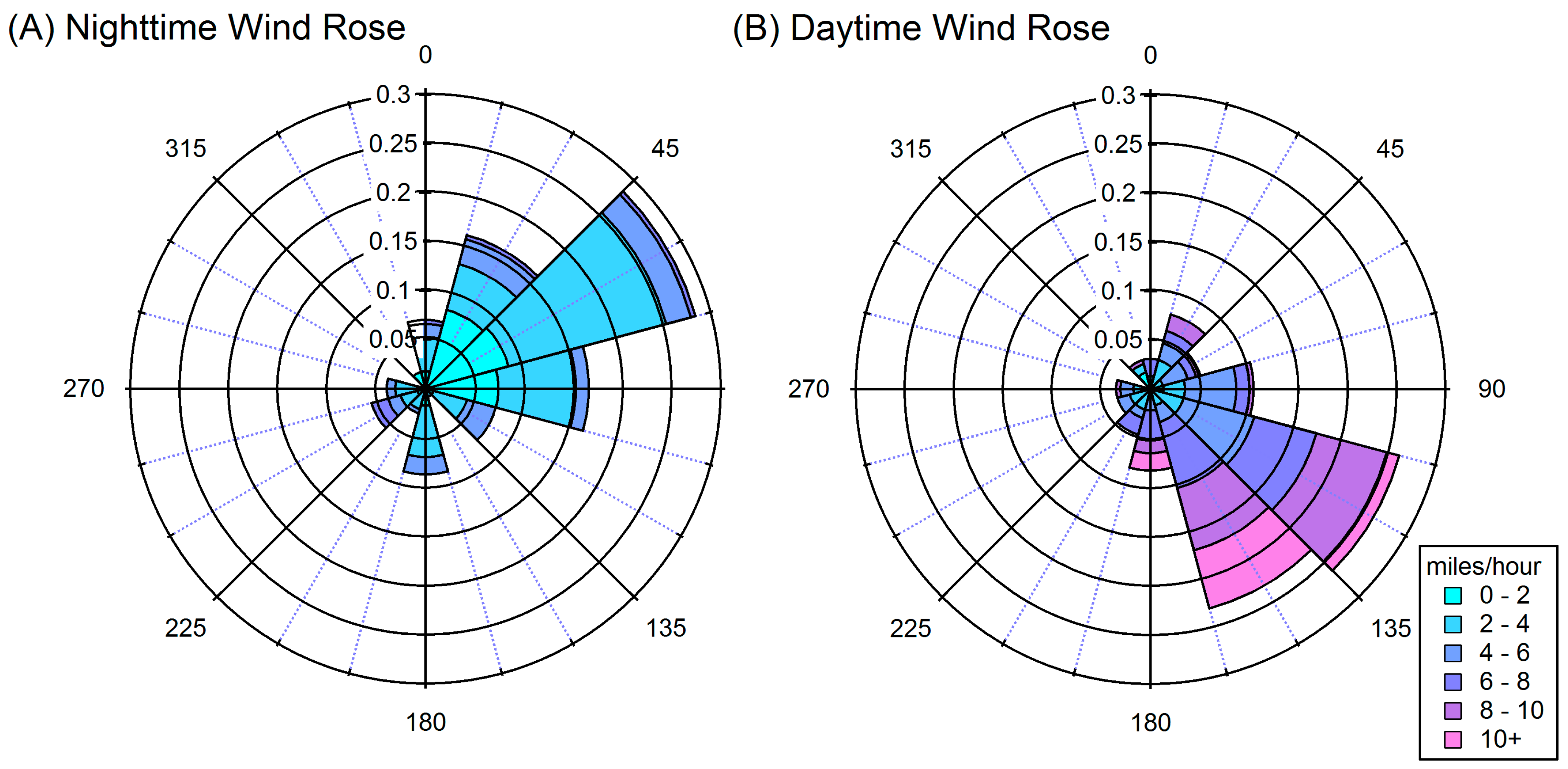
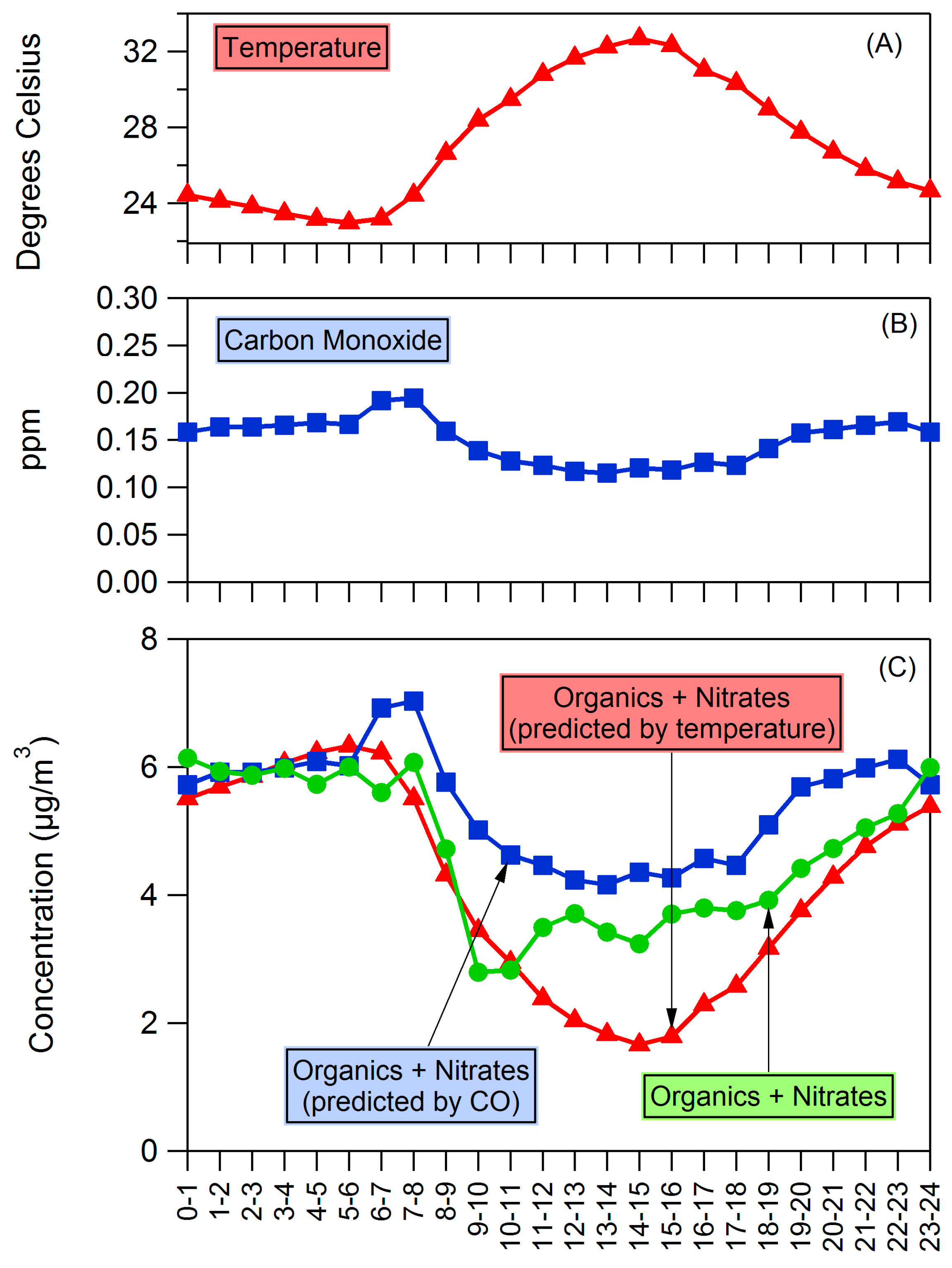
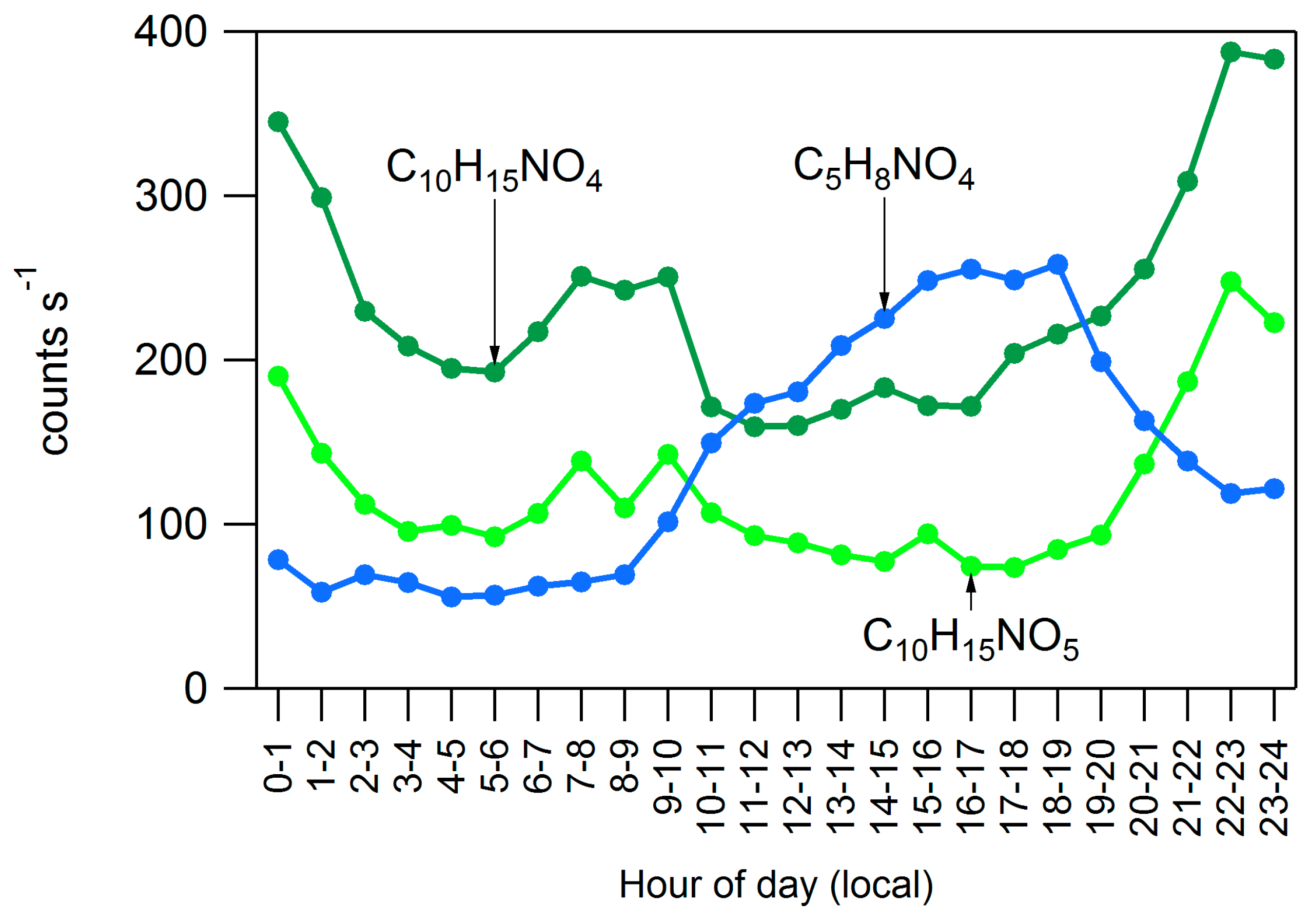
4.2. Influences on Diurnal Cycle
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
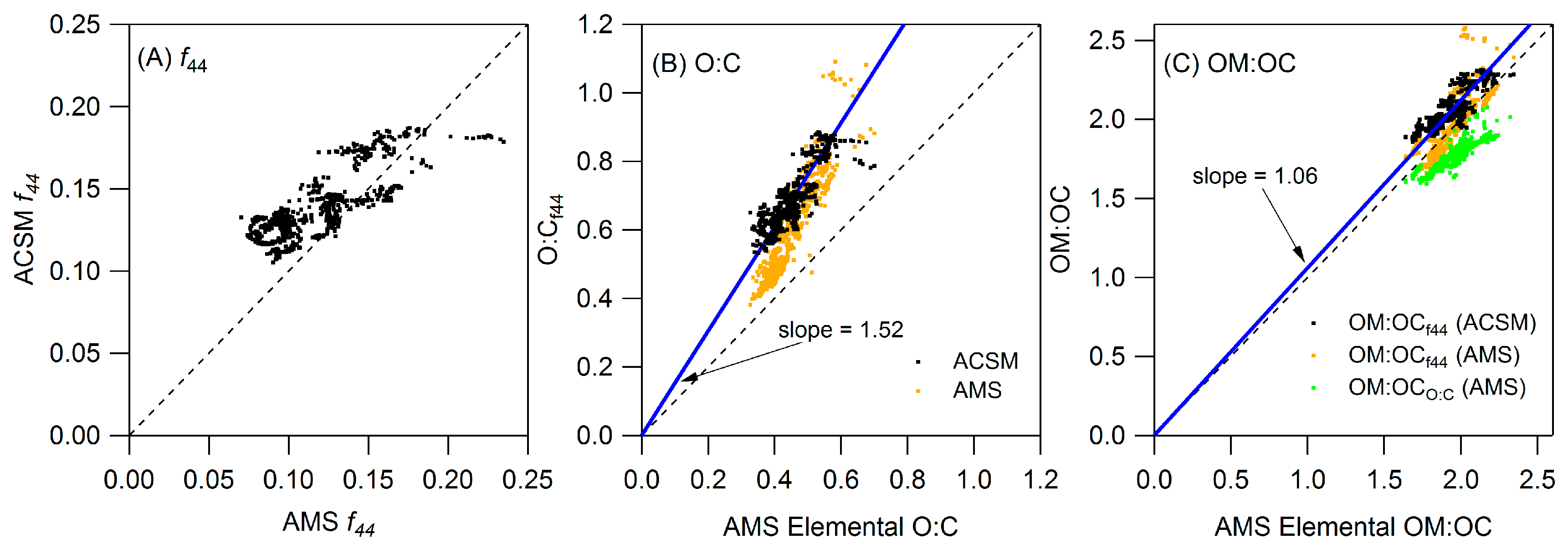
Appendix A: ACSM Calibration and Data Preparation
A1. ACSM Calibration
A2. Adjustments to the Standard Fragmentation Table
A3. Data Averaging
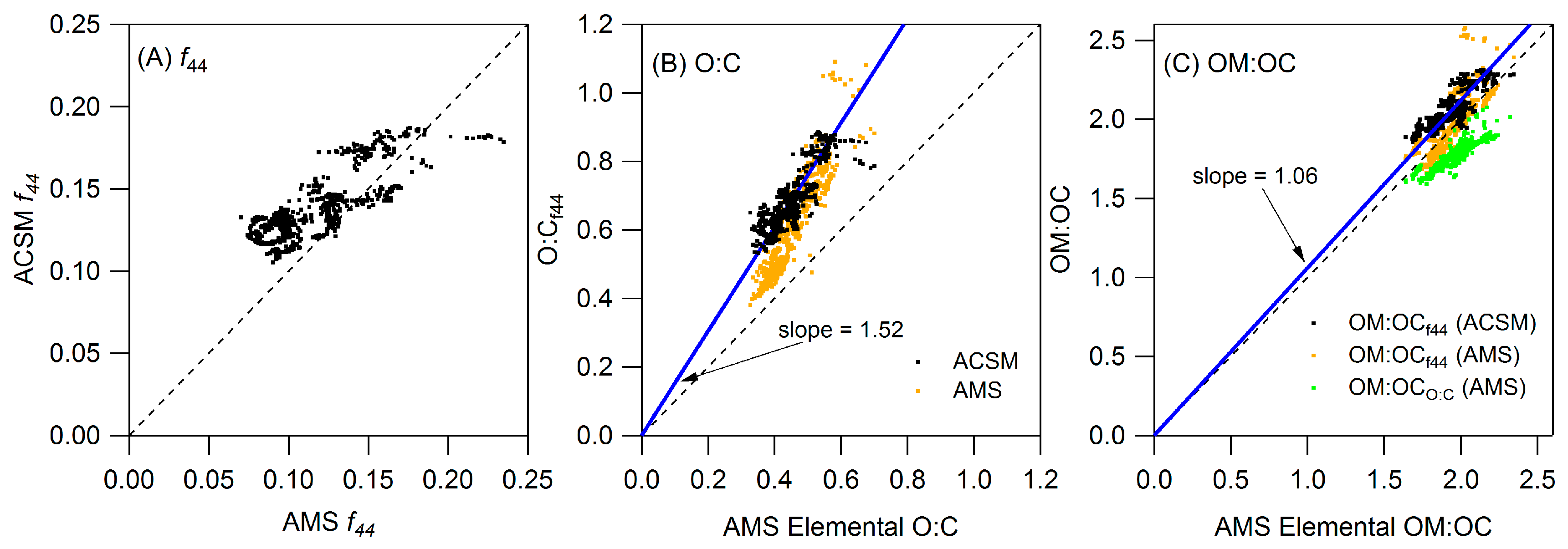
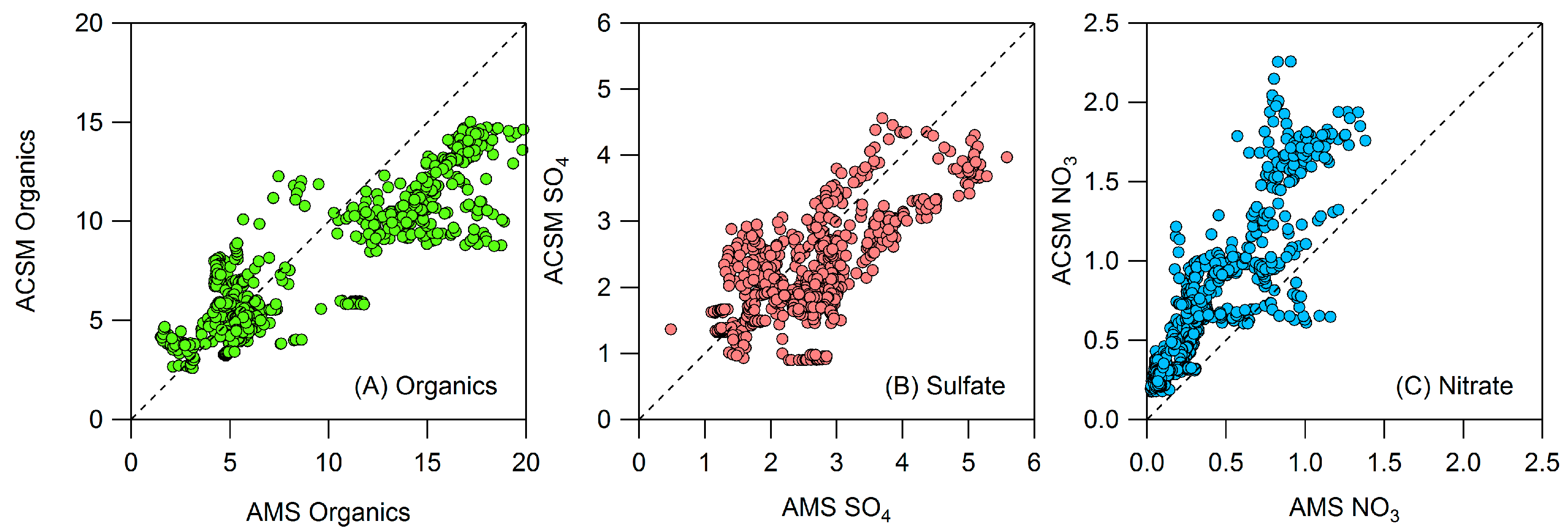
Appendix B: Comparison of Co-Located Instruments


References
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Dockery, D.; Pope, C.A.; Xiping, X.; Spengler, J.D.; Ware, J.H.; Fay, M.E.; Ferris, B.G.; Speizer, F.E. An association between air pollution and mortality in six USA cities. N. Engl. J. Med. 1993, 329, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.Q.; Yu, S.; Kan, H. Ozone exposure and mortality. N. Engl. J. Med. 2009, 360, 2788–2789. [Google Scholar] [PubMed]
- NAAQS Table. Available online: https://www.epa.gov/criteria-air-pollutants/naaqs-table (accessed on 26 February 2016).
- United States Environmental Protection Agency. National Ambient Air Quality Standards for Particulate Matter. Fed. Reg. 2013, 78, 3085–3287. [Google Scholar]
- United States Environmental Protection Agency. National Ambient Air Quality Standards for Ozone. Fed. Reg. 2015, 80, 75233–75411. [Google Scholar]
- Allen, D.T.; McDonald-Buller, E.C.; McGaughey, G.R. State of the Science of Air Quality in Texas: Scientific Findings from the Air Quality Research Program (AQRP) and Recommendations for Future Research; Air Quality Research Program: Austin, TX, USA, 2016. [Google Scholar]
- Allen, D.; Estes, M.; Smith, J.; Jeffries, H. Accelerated Science Evaluation of Ozone Formation in the Houston-Galveston Area; University of Texas: Austin, TX, USA, 2001. [Google Scholar]
- Allen, D.T.; Fraser, M. An overview of the gulf coast aerosol research and characterization study: The Houston fine particulate matter supersite. J. Air Waste Manag. Assoc. 2006, 56, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Olaguer, E.; Kolb, C.; Lefer, B.; Rappenglueck, B.; Zhang, R.; Pinto, J. Overview of the SHARP campaign: Motivation, design, and major outcomes. J. Geophys. Res. Atmos. 2014, 119, 2597–2610. [Google Scholar] [CrossRef]
- Allen, D.T.; Torres, V.M.; Thomas, J.; Sullivan, D.W.; Harrison, M.; Hendler, A.; Herndon, S.C.; Kolb, C.E.; Fraser, M.P.; Hill, A.D.; et al. Measurements of methane emissions at natural gas production sites in the United States. Proc. Natl. Acad. Sci. USA 2013, 110, 17768–17773. [Google Scholar] [CrossRef] [PubMed]
- Pasci, A.; Kimura, Y.; McGaughey, G.; McDonald-Buller, E.; Allen, D.T. Regional ozone impacts of increased natural gas use in the Texas power sector and development in the Eagle Ford shale. Environ. Sci. Technol. 2015, 49, 3966–3973. [Google Scholar]
- Zavala-Araiza, D.; Sullivan, D.W.; Allen, D.T. Atmospheric hydrocarbon emissions and concentrations in the Barnett Shale natural gas production region. Environ. Sci. Technol. 2014, 48, 5314–5321. [Google Scholar] [CrossRef] [PubMed]
- Bahreini, R.; Ervens, B.; Middlebrook, A.M.; Warneke, C.; de Gouw, J.A.; DeCarlo, P.F.; Jimenez, J.L.; Brock, C.A.; Neuman, J.A.; Ryerson, T.B.; et al. Organic aerosol formation in urban and industrial plumes near Houston and Dallas, Texas. J. Geophys. Res. Atmos. 2009, 114, 1–17. [Google Scholar] [CrossRef]
- Murphy, B.N.; Donahue, N.M.; Robinson, A.L.; Pandis, S.N. A naming convention for atmospheric organic aerosol. Atmos. Chem. Phys. 2014, 14, 5825–5839. [Google Scholar] [CrossRef]
- Parrish, D.D.; Allen, D.T.; Bates, T.S.; Estes, M.; Fehsenfeld, F.C.; Feingold, G.; Ferrare, R.; Hardesty, R.M.; Meagher, J.F.; Nielsen-Gammon, J.W.; et al. Overview of the second texas air quality study (TexAQS II) and the Gulf of Mexico atmospheric composition and climate study (GoMACCS). J. Geophys. Res. Atmos. 2009, 114, 1–28. [Google Scholar] [CrossRef]
- Weber, R.; Sullivan, A.; Peltier, R.; Russell, A.; Yan, B.; Zheng, M.; de Gouw, J.; Warneke, C.; Brock, C.; Holloway, J.; et al. A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States. J. Geophys. Res. Atmos. 2007, 112. [Google Scholar] [CrossRef]
- Xu, L.; Guo, H.; Boyd, C.M.; Bougiatioti, A.; Cerully, K.M.; Hite, J.R.; Isaacman-Vanwertz, G.; Kreisberg, N.M.; Olson, K.; Koss, A.; et al. Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States. Proc. Natl. Acad. Sci. USA 2015, 112, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Suresh, S.; Guo, H.; Weber, R.J.; Ng, N.L. Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: Spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates. Atmos. Chem. Phys. 2015, 15, 7307–7336. [Google Scholar] [CrossRef]
- Boyd, C.M.; Sanchez, J.; Xu, L.; Eugene, A.J.; Nah, T.; Tuet, W.Y.; Guzman, M.I.; Ng, N.L. Secondary organic aerosol formation from the β-pinene+NO3 system: Effect of humidity and peroxy radical fate. Atmos. Chem. Phys. 2015, 15, 7497–7522. [Google Scholar] [CrossRef]
- DISCOVER-AQ Home. Available online: http://discover-aq.larc.nasa.gov/ (accessed on 26 February 2016).
- Texas Air Monitoring Information System (TAMIS) Web Interface. Available online: http://www17.tceq.texas.gov/tamis/ (accessed on 26 February 2016).
- Kebabian, P.L.; Wood, E.C.; Herndon, S.C.; Freedman, A. A Practical Alternative to Detection of Nitrogen Dioxide: Cavity Attenuated Phase Shift Spectroscopy. Environ. Sci. Technol. 2008, 42, 6040–6045. [Google Scholar] [CrossRef] [PubMed]
- Ng, N.L.; Herndon, S.C.; Trimborn, A.; Canagaratna, M.R.; Croteau, P.L.; Onasch, T.B.; Sueper, D.; Worsnop, D.R.; Zhang, Q.; Sun, Y.L.; et al. An Aerosol Chemical Speciation Monitor (ACSM) for routine monitoring of the composition and mass concentrations of ambient aerosol. Aerosol Sci. Technol. 2011, 45, 780–794. [Google Scholar] [CrossRef]
- Bertram, T.H.; Kimmel, J.R.; Crisp, T.A.; Ryder, O.S.; Yatavelli, R.L.N.; Thornton, J.A.; Cubison, M.J.; Gonin, M.; Worsnop, D.R. A field-deployable, chemical ionization time-of-flight mass spectrometer. Atmos. Meas. Tech. 2011, 4, 1471–1479. [Google Scholar] [CrossRef]
- Yatavelli, R.L.N.; Lopez-Hilfiker, F.; Wargo, J.D.; Kimmel, J.R.; Cubison, M.J.; Bertram, T.H.; Jimenez, J.L.; Gonin, M.; Worsnop, D.R.; Thornton, J.A. A chemical ionization high-resolution time-of-flight mass spectrometer coupled to a Micro Orifice Volatilization Impactor (MOVI-HRToF-CIMS) for analysis of gas and particle-phase organic species. Aerosol Sci. Technol. 2012, 46, 1313–1327. [Google Scholar] [CrossRef]
- Lee, B.H.; Lopez-Hilfiker, F.D.; Mohr, C.; Kurtén, T.; Worsnop, D.R.; Thornton, J.A. An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: Application to atmospheric inorganic and organic compounds. Environ. Sci. Technol. 2014, 48, 6309–6317. [Google Scholar] [CrossRef] [PubMed]
- Aljawhary, D.; Lee, A.K.Y.; Abbatt, J.P.D. High-resolution chemical ionization mass spectrometry (ToF-CIMS): Application to study SOA composition and processing. Atmos. Meas. Tech. 2013, 6, 3211–3224. [Google Scholar] [CrossRef]
- Allan, J.D.; Delia, A.E.; Coe, H.; Bower, K.N.; Alfarra, M.R.; Jimenez, J.L.; Middlebrook, A.M.; Drewnick, F.; Onasch, T.B.; Canagaratna, M.R.; et al. A generalised method for the extraction of chemically resolved mass spectra from Aerodyne aerosol mass spectrometer data. J. Aerosol Sci. 2004, 35, 909–922. [Google Scholar] [CrossRef]
- Leong, Y.J.; Sanchez, N.P.; Wallace, H.W.; Karakurt Cevik, B.; Hernandez, C.S.; Han, Y.; Choi, Y.; Flynn, J.H.; Massoli, P.; Floerchinger, C.; et al. Overview of Surface Measurements and Spatial Characterization of Submicron Particulate Matter during the DISCOVER-AQ 2013 Campaign in Houston, TX, USA, 2016. in preparation.
- Aiken, A.C.; Decarlo, P.F.; Kroll, J.H.; Worsnop, D.R.; Huffman, J.A.; Docherty, K.S.; Ulbrich, I.M.; Mohr, C.; Kimmel, J.R.; Sueper, D.; et al. O/C and OM/OC Ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry. Environ. Sci. Technol. 2008, 42, 4478–4485. [Google Scholar] [CrossRef] [PubMed]
- Canagaratna, M.R.; Jimenez, J.L.; Kroll, J.H.; Chen, Q.; Kessler, S.H.; Massoli, P.; Hildebrandt Ruiz, L.; Fortner, E.; Williams, L.R.; Wilson, K.R.; et al. Elemental ratio measurements of organic compounds using aerosol mass spectrometry: Characterization, improved calibration, and implications. Atmos. Chem. Phys. 2015, 15, 253–272. [Google Scholar] [CrossRef]
- Paatero, P.; Tapper, U. Positive matrix factorization: A nonnegative factor model with optimal utilization of error estimates of data values. Environmetrics 1994, 5, 111–126. [Google Scholar] [CrossRef]
- Hildebrandt, L.; Engelhart, G.J.; Mohr, C.; Kostenidou, E.; Lanz, V.A.; Bougiatioti, A.; DeCarlo, P.F.; Prevot, A.S.H.; Baltensperger, U.; Mihalopoulos, N.; et al. Aged organic aerosol in the eastern Mediterranean: The finokalia aerosol measurement experiment–2008. Atmos. Chem. Phys. 2010, 10, 4167–4186. [Google Scholar] [CrossRef]
- Hildebrandt, L.; Kostenidou, E.; Lanz, V.A.; Prevot, A.S.H.; Baltensperger, U.; Mihalopoulos, N.; Donahue, N.M.; Pandis, S.N. Sources and atmospheric processing of organic aerosol in the Mediterranean: Insights from aerosol mass spectrometer factor analysis. Atmos. Chem. Phys. 2011, 11, 12499–12515. [Google Scholar] [CrossRef]
- Lanz, V.A.; Alfarra, M.R.; Baltensperger, U.; Buchmann, B.; Hueglin, C.; Prevot, A.S.H. Source apportionment of submicron organic aerosols at an urban site by factor analytical modelling of aerosol mass spectra. Atmos. Chem. Phys. 2007, 7, 1503–1522. [Google Scholar] [CrossRef]
- Lanz, V.A.; Prévôt, A.S.H.; Alfarra, M.R.; Weimer, S.; Mohr, C.; DeCarlo, P.F.; Gianini, M.F.D.; Hueglin, C.; Schneider, J.; Favez, O.; et al. Characterization of aerosol chemical composition with aerosol mass spectrometry in Central Europe: An overview. Atmos. Chem. Phys. 2010, 10, 10453–10471. [Google Scholar] [CrossRef]
- Ulbrich, I.M.; Canagaratna, M.R.; Zhang, Q.; Worsnop, D.R.; Jimenez, J.L. Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data. Atmos. Chem. Phys. 2009, 9, 2891–2918. [Google Scholar] [CrossRef]
- Faxon, C.; Bean, J.; Hildebrandt Ruiz, L. Inland concentrations of ClNO2 in Southeast Texas suggest chlorine chemistry significantly contributes to atmospheric reactivity. Atmosphere 2015, 6, 1487–1506. [Google Scholar] [CrossRef]
- Birch, M.; Cary, R. Elemental carbon-based method for monitoring occupational exposures to particulate diesel exhaust. Aerosol Sci. Technol. 1996, 25, 221–241. [Google Scholar] [CrossRef]
- Zaveri, R.A.; Shaw, W.J.; Cziczo, D.J.; Schmid, B.; Ferrare, R.A.; Alexander, M.L.; Alexandrov, M.; Alvarez, R.J.; Arnott, W.P.; Atkinson, D.B.; et al. Overview of the 2010 Carbonaceous Aerosols and Radiative Effects Study (CARES). Atmos. Chem. Phys. 2012, 12, 7647–7687. [Google Scholar] [CrossRef]
- Zotter, P.; El-Haddad, I.; Zhang, Y.; Hayes, P.L.; Zhang, X.; Lin, Y.; Wacker, L.; Schnelle-Kreis, J.; Abbaszade, G.; Zimmermann, R.; et al. Diurnal cycle of fossil and nonfossil carbon using radiocarbon analyses during CalNex. J. Geophys. Res. Atmos. 2014, 119, 6818–6835. [Google Scholar] [CrossRef]
- Barrett, T.E.; Robinson, E.M.; Usenko, S.; Sheesley, R.J. Source contributions to wintertime elemental and organic carbon in the western arctic based on radiocarbon and tracer apportionment. Environ. Sci. Technol. 2015, 49, 11631–11639. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, Ö.; Kruså, M.; Zencak, Z.; Sheesley, R.J.; Granat, L.; Engström, E.; Praveen, P.S.; Rao, P.S.P.; Leck, C.; Rodhe, H. Brown clouds over South Asia: Biomass or fossil fuel combustion? Science 2009, 323, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Atkinson-Palombo, C.M.; Miller, J.A.; Balling, R.C., Jr. Quantifying the ozone “weekend effect” at various locations in Phoenix, Arizona. Atmos. Environ. 2006, 40, 7644–7658. [Google Scholar] [CrossRef]
- Wilks, D.S. Statistical Methods in the Atmospheric Science; Academic Press: San Diego, CA, USA, 1995. [Google Scholar]
- Ng, N.L.; Canagaratna, M.R.; Jimenez, J.L.; Zhang, Q.; Ulbrich, I.M.; Worsnop, D.R. Real-time methods for estimating organic component mass concentrations from aerosol mass spectrometer data. Environ. Sci. Technol. 2011, 45, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Rollins, A.W.; Browne, E.C.; Min, K.-E.; Pusede, S.E.; Wooldridge, P.J.; Gentner, D.R.; Goldstein, A.H.; Liu, S.; Day, D.A.; Russell, L.M.; et al. Evidence for NOx control over nighttime SOA formation. Science 2012, 337, 1210–1212. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, D.T.; Allen, D.T.; Ehrmanf, S.H.; Pratsins, S.E. The sources and size distributions of organonitrates in Los Angeles aerosol. Atmos. Environ. 1991, 25A, 2855–2861. [Google Scholar] [CrossRef]
- Garnes, L.A.; Allen, D.T. Size distributions of organonitrates in ambient aerosol collected in Houston, Texas. Aerosol Sci. Technol. 2002, 36, 983–992. [Google Scholar] [CrossRef]
- Laurent, J.-P.; Allen, D.T. Size distributions of organic functional groups in ambient aerosol collected in Houston, Texas. Aerosol Sci. Technol. 2004, 38, 60–67. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Holmes, J.R.; Bockian, A.H. Formation of photochemical aerosol from hydrocarbons. chemical reactivity and products. Environ. Sci. Technol. 1975, 9, 568–576. [Google Scholar] [CrossRef]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. NOAA’S HYSPLIT atmospheric transport and dispersion modeling system. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Pankow, J.F. An absorption model of gas/particle partitioning of organic compounds in the atmosphere. Atmos. Environ. 1994, 28, 185–188. [Google Scholar] [CrossRef]
- Donahue, N.M.; Robinson, A.L.; Stanier, C.O.; Pandis, S.N. Coupled partitioning, dilution, and chemical aging of semivolatile organics. Environ. Sci. Technol. 2006, 40, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.N.; Pandis, S.N. Exploring summertime organic aerosol formation in the eastern United States using a regional-scale budget approach and ambient measurements. J. Geophys. Res. 2010, 115, D24216. [Google Scholar] [CrossRef]
- Tucker, S.C.; Banta, R.M.; Langford, A.O.; Senff, C.J.; Brewer, W.A.; Williams, E.J.; Lerner, B.M.; Osthoff, H.D.; Hardesty, R.M. Relationships of coastal nocturnal boundary layer winds and turbulence to Houston ozone concentrations during TexAQS 2006. J. Geophys. Res. 2010, 115, 1–17. [Google Scholar] [CrossRef]
- Vizuete, W.; Junquera, V.; McDonald-Buller, E.; McGaughey, G.; Yarwood, G.; Allen, D. Effects of temperature and land use on predictions of biogenic emissions in Eastern Texas, USA. Atmos. Environ. 2002, 36, 3321–3337. [Google Scholar] [CrossRef]
- Lee, B.H.; Mohr, C.; Lopez-Hilfiker, F.D.; Lutz, A.; Hallquist, M.; Lee, L.; Romer, P.; Cohen, R.C.; Iyer, S.; Kurtén, T.; et al. Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets. Proc. Natl. Acad. Sci. USA 2016, 113, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Pankow, J.F.; Asher, W.E. SIMPOL.1: A simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds. Atmos. Chem. Phys. 2008, 8, 2773–2796. [Google Scholar] [CrossRef]
- Ng, N.L.; Chhabra, P.S.; Chan, A.W.H.; Surratt, J.D.; Kroll, J.H.; Kwan, A.J.; McCabe, D.C.; Wennberg, P.O.; Sorooshian, A.; Murphy, S.M.; et al. Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes. Atmos. Chem. Phys. Discuss. 2007, 7, 10131–10177. [Google Scholar] [CrossRef]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bean, J.K.; Faxon, C.B.; Leong, Y.J.; Wallace, H.W.; Cevik, B.K.; Ortiz, S.; Canagaratna, M.R.; Usenko, S.; Sheesley, R.J.; Griffin, R.J.; et al. Composition and Sources of Particulate Matter Measured near Houston, TX: Anthropogenic-Biogenic Interactions. Atmosphere 2016, 7, 73. https://doi.org/10.3390/atmos7050073
Bean JK, Faxon CB, Leong YJ, Wallace HW, Cevik BK, Ortiz S, Canagaratna MR, Usenko S, Sheesley RJ, Griffin RJ, et al. Composition and Sources of Particulate Matter Measured near Houston, TX: Anthropogenic-Biogenic Interactions. Atmosphere. 2016; 7(5):73. https://doi.org/10.3390/atmos7050073
Chicago/Turabian StyleBean, Jeffrey K., Cameron B. Faxon, Yu Jun Leong, Henry William Wallace, Basak Karakurt Cevik, Stephanie Ortiz, Manjula R. Canagaratna, Sascha Usenko, Rebecca J. Sheesley, Robert J. Griffin, and et al. 2016. "Composition and Sources of Particulate Matter Measured near Houston, TX: Anthropogenic-Biogenic Interactions" Atmosphere 7, no. 5: 73. https://doi.org/10.3390/atmos7050073
APA StyleBean, J. K., Faxon, C. B., Leong, Y. J., Wallace, H. W., Cevik, B. K., Ortiz, S., Canagaratna, M. R., Usenko, S., Sheesley, R. J., Griffin, R. J., & Hildebrandt Ruiz, L. (2016). Composition and Sources of Particulate Matter Measured near Houston, TX: Anthropogenic-Biogenic Interactions. Atmosphere, 7(5), 73. https://doi.org/10.3390/atmos7050073