
Spatial Distribution of Methanesulphonic Acid in the Arctic Aerosol Collected during the Chinese Arctic Research Expedition

Abstract
:1. Introduction
2. Experimental Methods
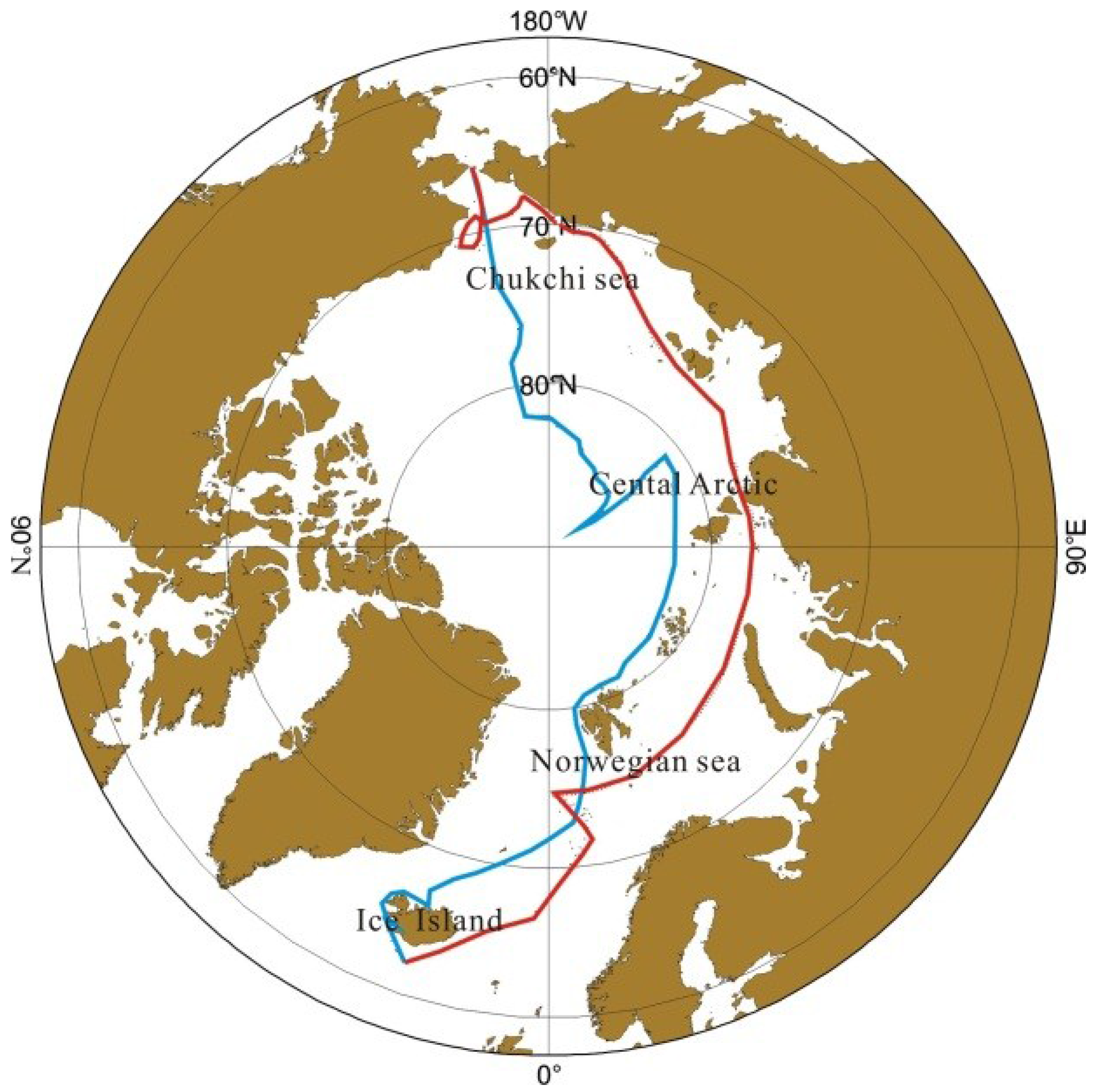
2.1. The Cruise Route
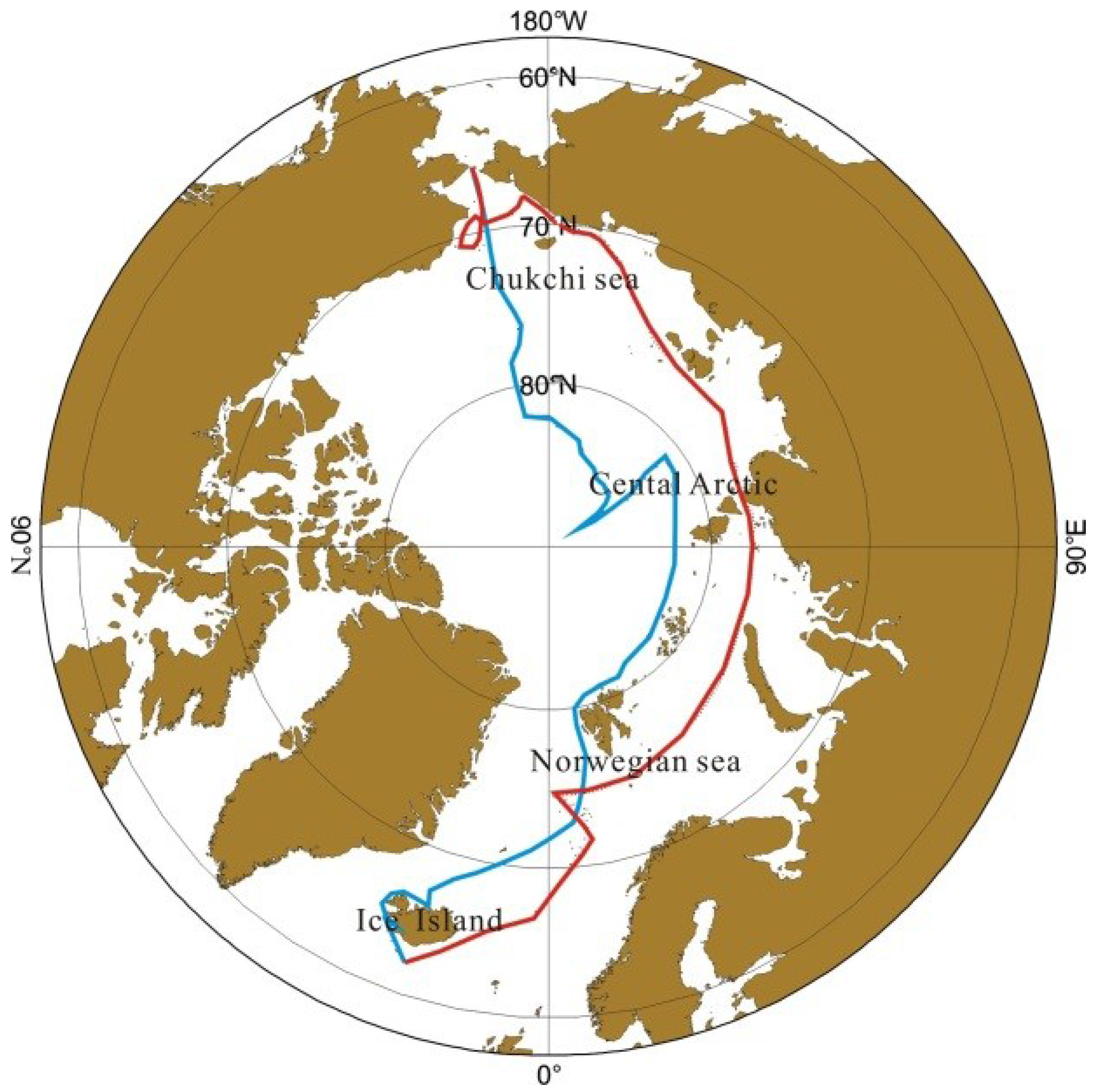
2.2. Sampling and Experimental Methods
3. Results and Discussion
3.1. General Observations
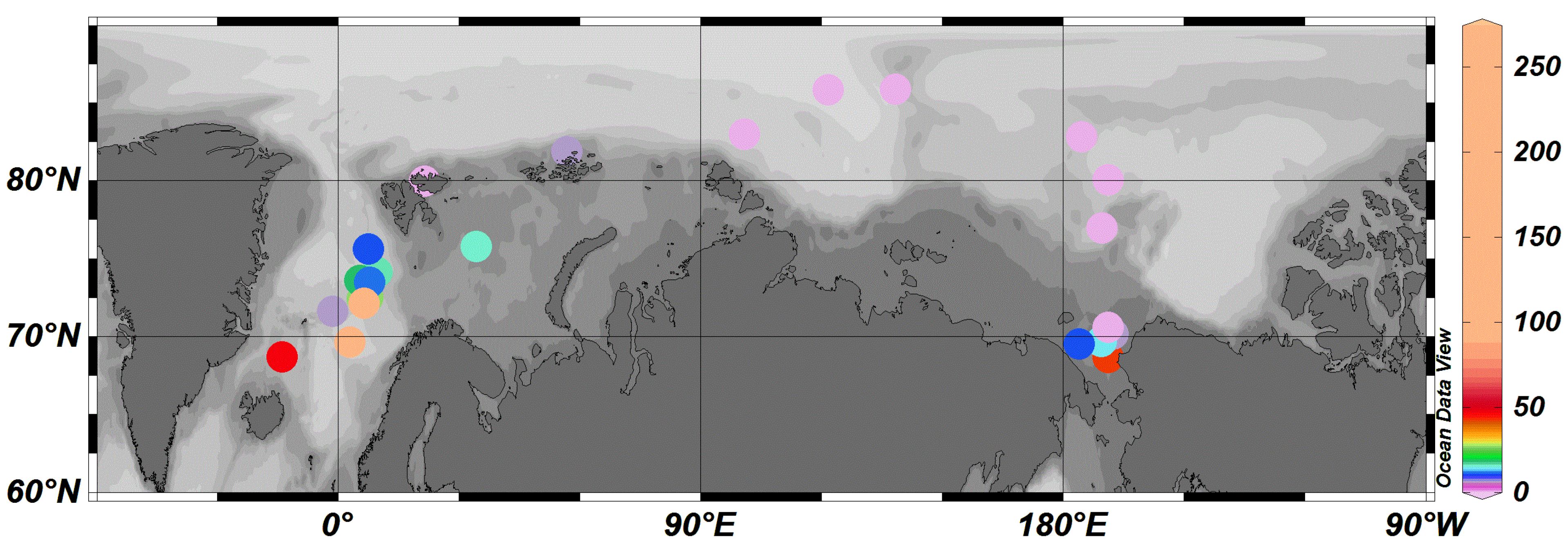
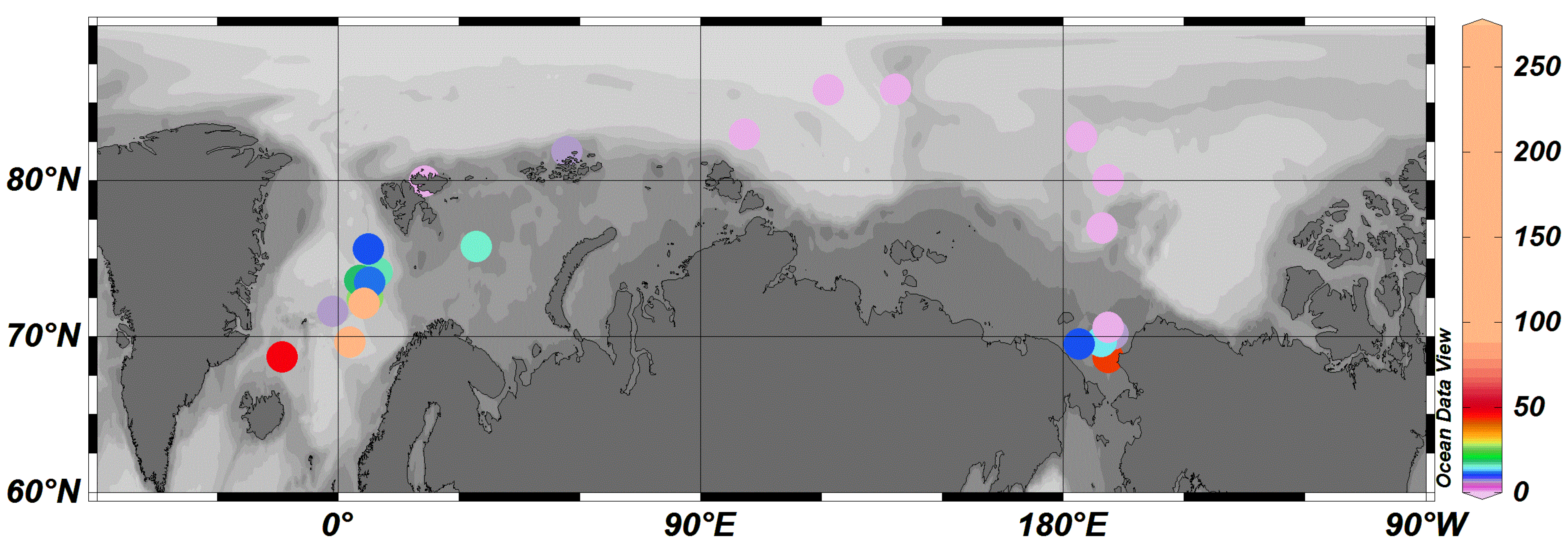
3.2. Regional Characteristics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Sampling Time | Average (ng/m3) | Range (ng/m3) | Reference |
|---|---|---|---|---|
| Chukchi Sea | July, September 2012 | 12 | ND-42 | This study |
| Norwegian Sea | August 2012 | 60 | 10–229 | This study |
| Central Arctic | August 2012 | 1.2 | ND-5 | This study |
| Chukchi Sea | July, August 2010 | 8 | ND-43 | This study |
| Central Arctic | August 2010 | 3 | ND-12 | This study |
| Alert | July–August 1980–1991 | - | 6–18 | [24] |
| Alert | July–August 1998–2009 | 10 | 3–17 | [12] |
| Barrow | July–August 1998–2009 | 12 | ND-40 | [12] |
| Ny-Ålesund | July–August 1998–2004 | 42 | ND-86 | [25] |
| Island | June–August 2011, 2012 | - | 1–10 | [26] |
| Norwegian coast | Fall 2007, 2008 | - | 120 (maximum) | [23] |
| Shemya (52°N, 174°E) | May–September 1981 | 17 | - | [27] |
| Shemya (52°N, 174°E) | October 1981–March 1982 | 14 | - | [27] |
| Bermuda (32.27°N, 64.87°W) | July 1988, 1990 | 43 | - | [7] |
| MaceHead, Ireland (53.32°N, 9.85°W) | August 1989–August 1990 | 69 | - | [7] |
| Equator | Summer | * | 11 | [15] |
| 63°S, 23°W | Summer | * | 260 (maximum) | [15] |
3.3. The Role of Biotic Factors
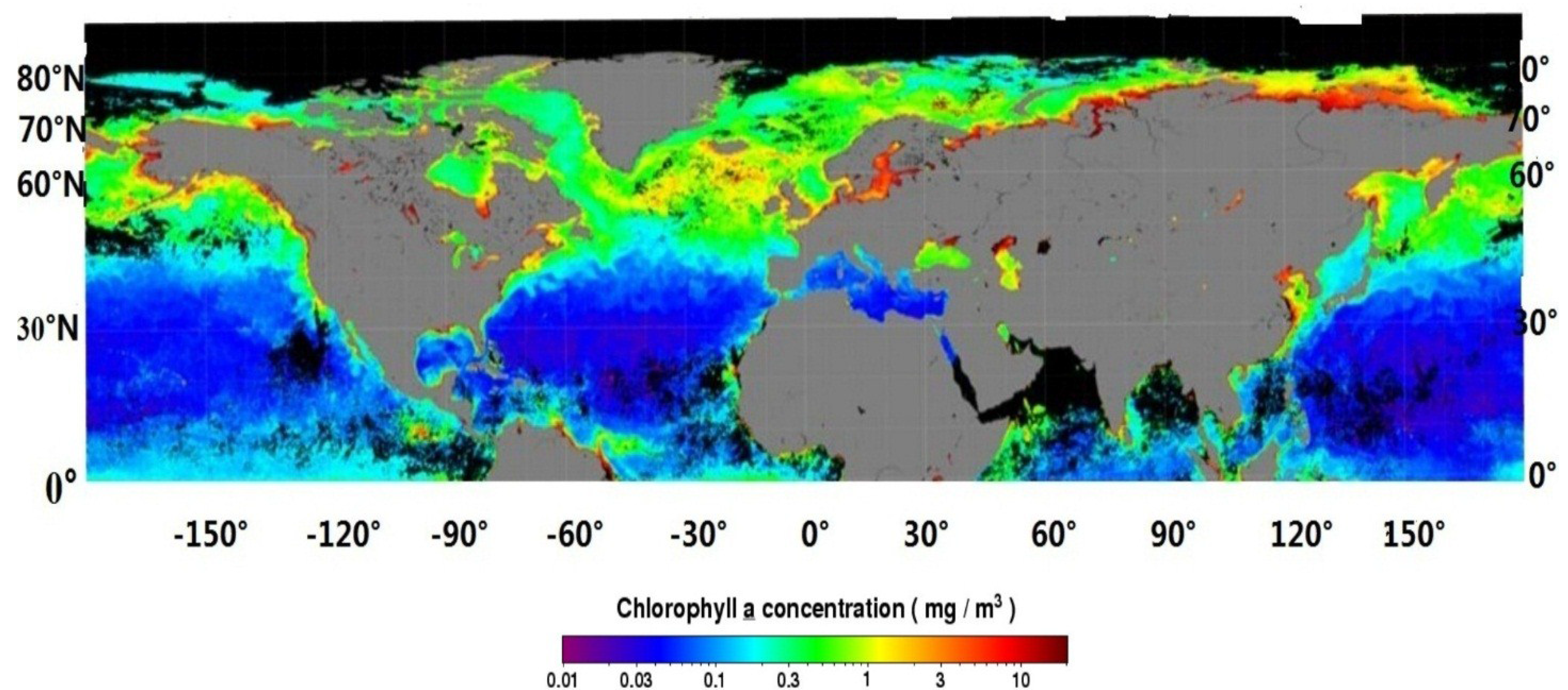
3.3.1. Phytoplankton
3.3.2. Phytoplankton Composition
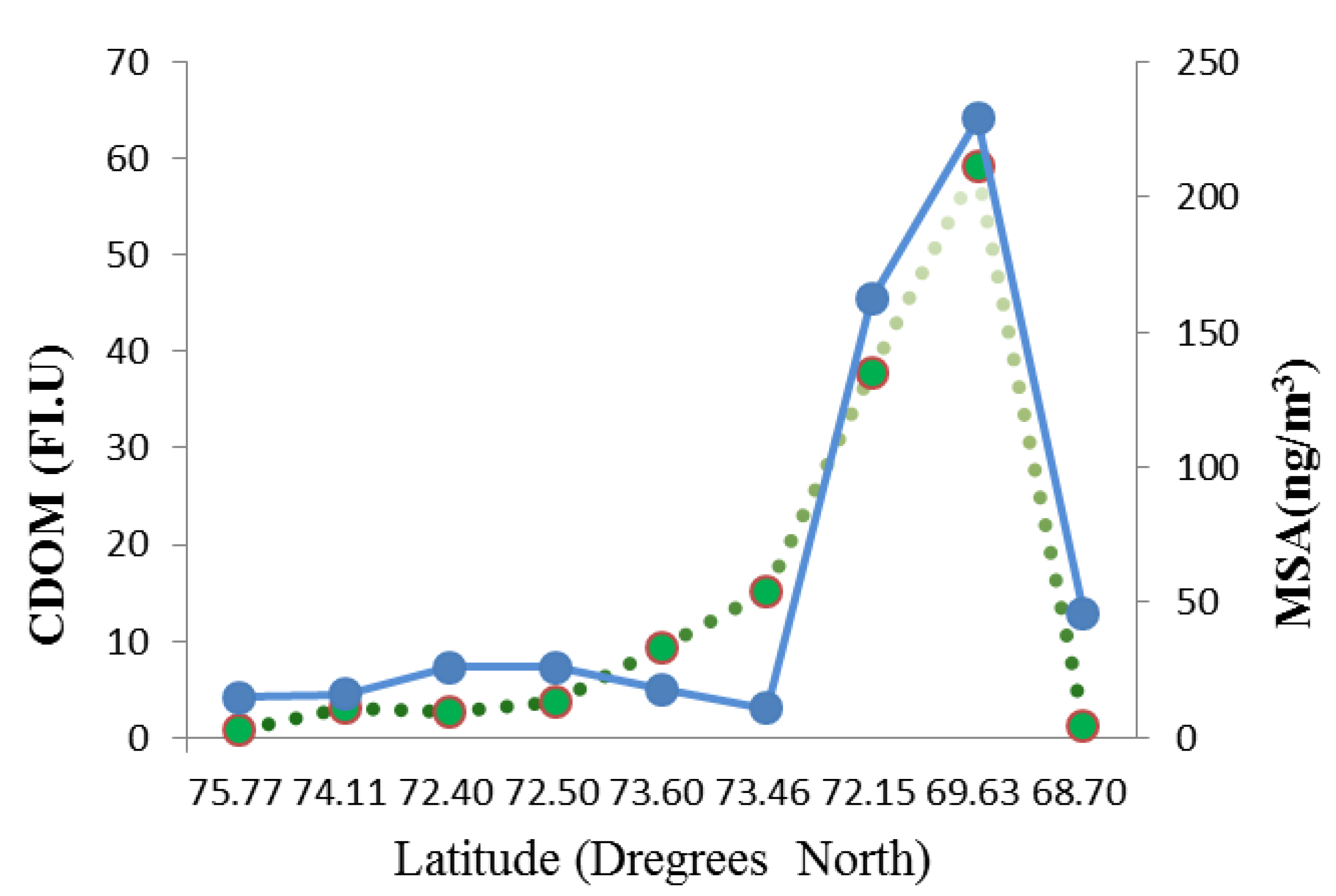
3.3.3. CDOM
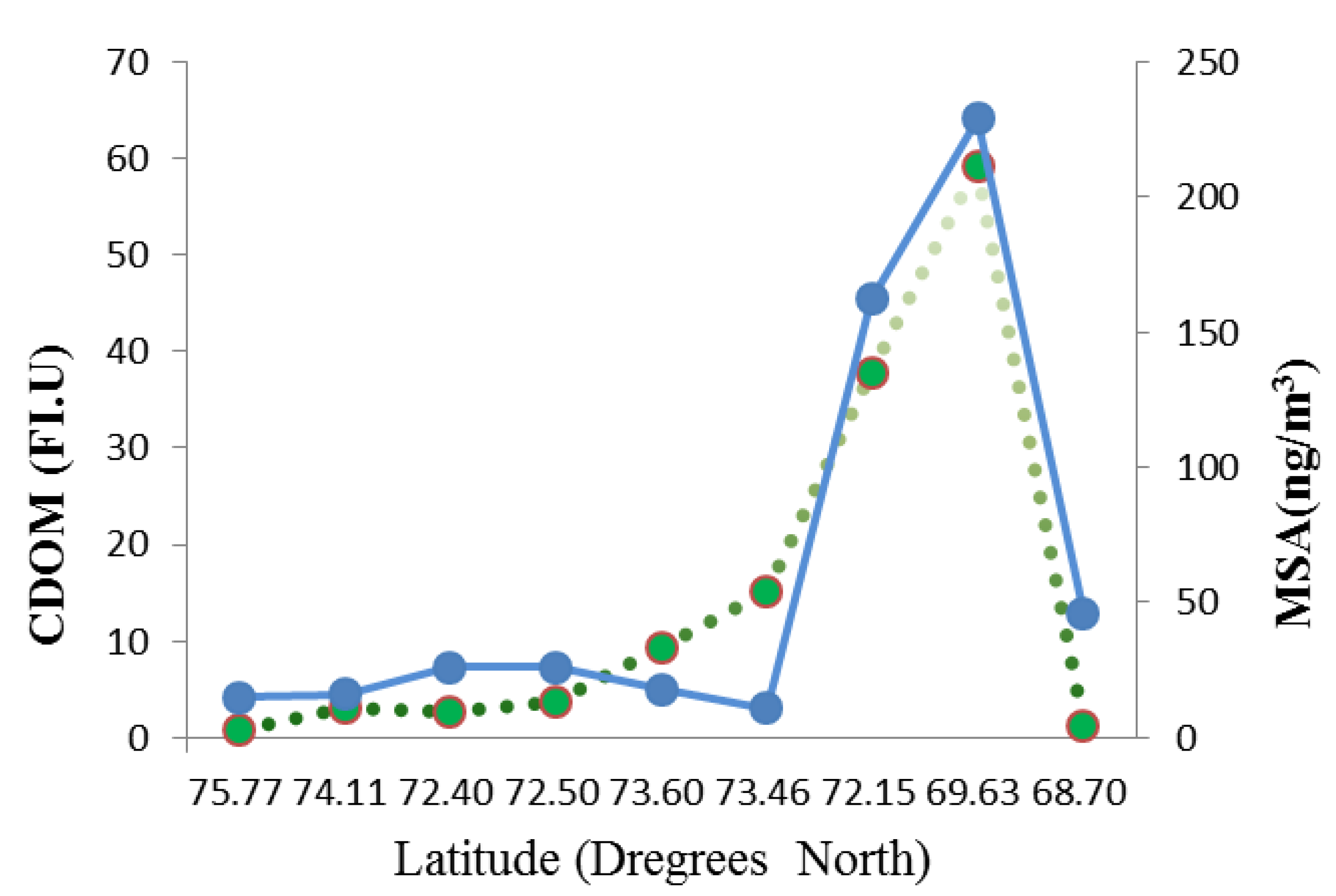
3.4. The Role of Abiotic Factors
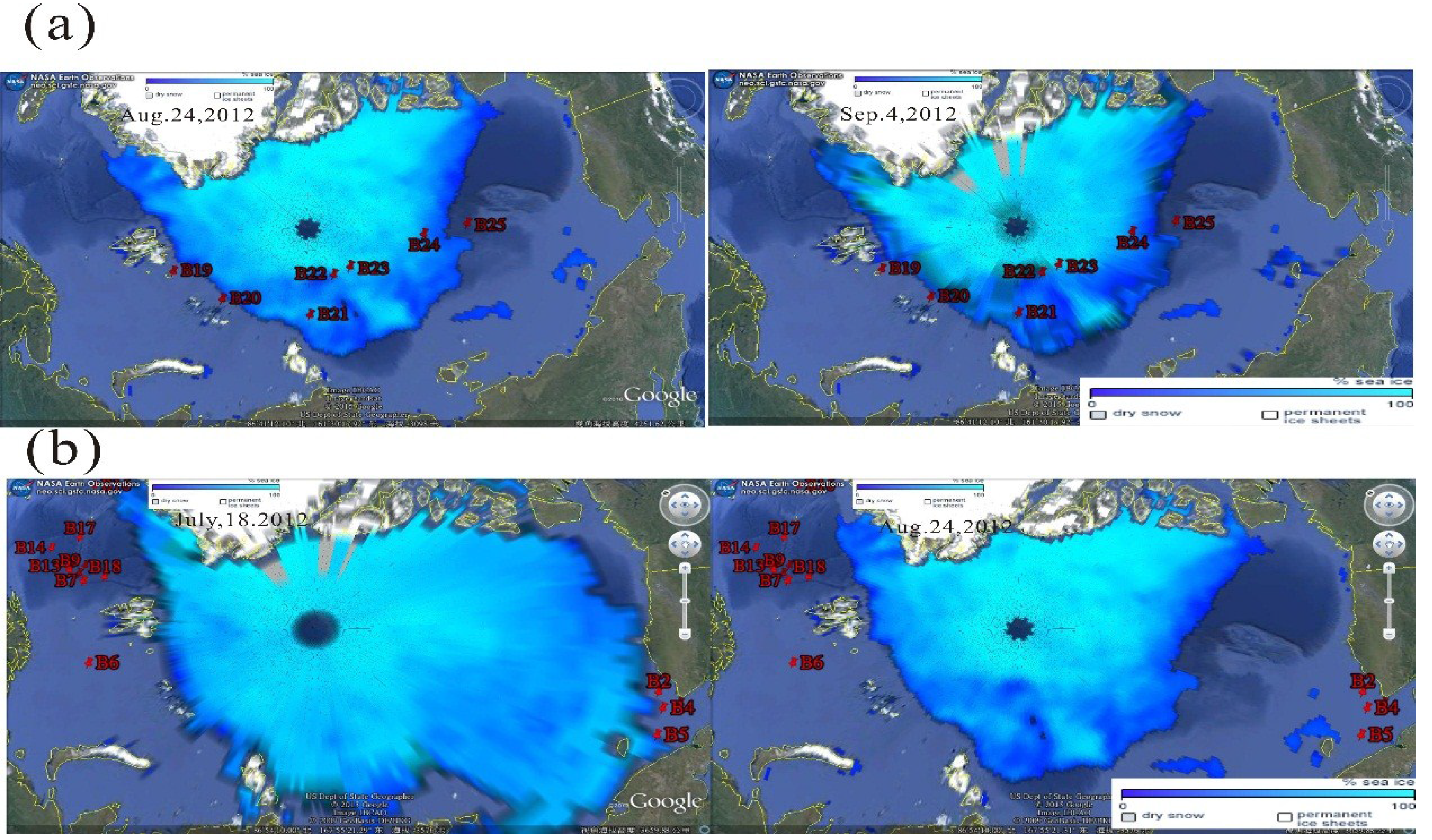
3.4.1. Sea Ice

3.4.2. Sea Surface Temperature
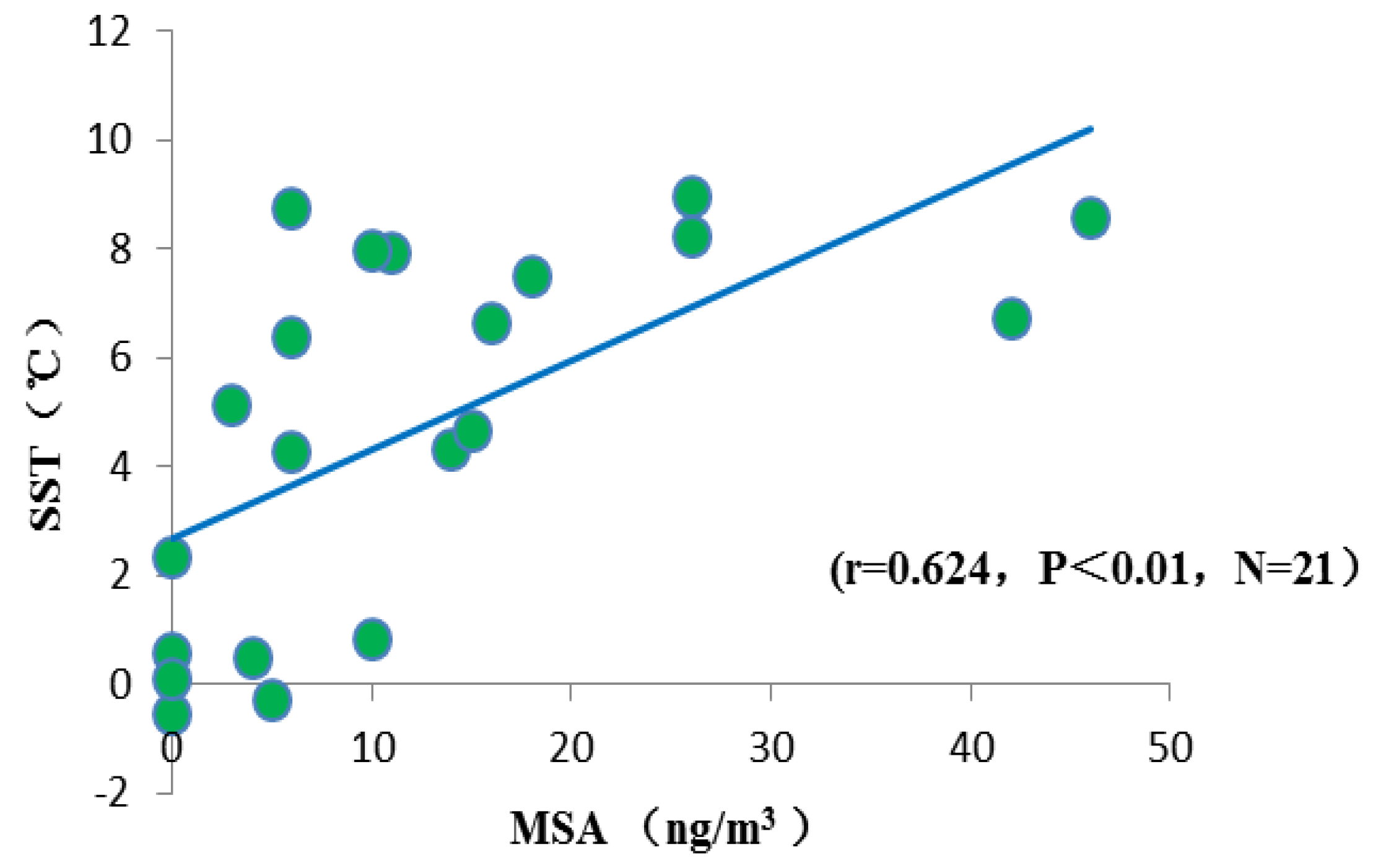
4. Summary
Acknowledgments
Author Contributions
Appendix
| Sample Number | Latitude * | Longitude * | Sampling Date | MSA (ng/m3) | CDOM (FI.U) | SST (°C) | Wind speed (m/s) |
|---|---|---|---|---|---|---|---|
| B01 | 68.63 | −168.85 | 18–19 July 2012 | 42 | 0.899 | 6.74 | 5.6 |
| B02 | 70.16 | −167.67 | 19–20 July 2012 | 6 | 0.572 | 6.38 | 10.55 |
| B04 | 69.66 | −170.62 | 21–22 July 2012 | 14 | 0.381 | 4.325 | 13.5 |
| B05 | 69.52 | −176.00 | 23–24 July 2012 | 10 | 0.626 | 0.855 | 12.7 |
| B06 | 75.77 | 34.31 | 1–2 August 2012 | 15 | 0.890 | 4.68 | 16.4 |
| B07 | 74.11 | 9.76 | 2–3 August 2012 | 16 | 3.082 | 6.655 | 9.9 |
| B08 | 72.40 | 5.95 | 3–4 August 2012 | 26 | 2.700 | 8.24 | 9.4 |
| B09 | 72.50 | 7.38 | 4–5 August 2012 | 26 | 3.664 | 8.96 | 5.9 |
| B11 | 73.60 | 5.39 | 6–7 August 2012 | 18 | 9.386 | 7.515 | 2.95 |
| B12 | 73.46 | 7.82 | 7–8 August 2012 | 11 | 15.108 | 7.92 | 14.58 |
| B13 | 72.15 | 6.54 | 8–9 August 2012 | 162 | 37.667 | 9.00 | 25.35 |
| B14 | 69.63 | 2.91 | 9–10 August 2012 | 229 | 59.226 | 10.17 | 13.35 |
| B16 | 68.70 | −13.93 | 21–22 August 2012 | 46 | 1.141 | - | 14.53 |
| B17 | 71.61 | −1.38 | 22–23 August 2012 | 6 | 2.545 | 8.765 | 13.8 |
| B18 | 75.61 | 7.50 | 23–24 August 2012 | 10 | 2.773 | 7.99 | 13.9 |
| B19 | 79.96 | 21.42 | 24–25 August 2012 | ND | 1.627 | 5.15 | 10.05 |
| B20 | 81.85 | 56.78 | 25–26 August 2012 | ND | 0.644 | 4.255 | 13.65 |
| B21 | 82.97 | 100.80 | 26–28 August 2012 | ND | 0.662 | 0.58 | 7.4 |
| B22 | 85.81 | 121.64 | 28–30 August 2012 | 5 | 0.286 | - | 4.9 |
| B23 | 85.85 | 138.34 | 30 August–1 September 2012 | ND | 0.000 | - | 6.1 |
| B24 | 82.82 | −175.50 | 2 September–4 2012 | 4 | 0.417 | 0.505 | 21.45 |
| B25 | 80.06 | −168.85 | 4–5 September 2012 | ND | 0.226 | −0.555 | 20.8 |
| B26 | 76.96 | −170.47 | 5–6 September 2012 | ND | 0.217 | 0.115 | 16.5 |
| B27 | 70.58 | −168.91 | 7–8 September 2012 | ND | 0.963 | 2.325 | 5.7 |
| B28 | 69.31 | −164.28 | 20–22 July 2010 | 5 | - | - | |
| B29 | 72.37 | −156.58 | 25–27 July 2010 | 4 | - | - | |
| B30 | 75.15 | −156.83 | 28–30 July 2010 | 7 | - | - | |
| B31 | 78.59 | −158.91 | 30 July–Aug.1 2010 | ND | - | - | |
| B32 | 80.69 | −162.07 | 1–2 August 2010 | 12 | - | - | |
| B33 | 86.82 | −179.24 | 7–8 August 2010 | ND | - | - | |
| B34 | 88.16 | −178.38 | 19–20 August 2010 | ND | - | - | |
| B35 | 81.15 | −169.11 | 23–24 August 2010 | ND | - | - | |
| B36 | 78.76 | −170.53 | 21–22 August 2010 | ND | - | - | |
| B37 | 76.26 | −171.90 | 27–28 August 2010 | 43 | - | - | |
| B38 | 72.49 | −169.03 | 29–30 August 2010 | ND | - | - |
Conflicts of Interest
References
- Bates, T.; Lamb, B.; Guenther, A.; Dignon, J.; Stoiber, R. Sulfur emissions to the atmosphere from natural sourees. J. Atmos. Chem. 1992, 14, 315–337. [Google Scholar] [CrossRef]
- Jenkins, A.; Dutrieux, P.; Jacobs, S.S.; McPhail, S.D.; Perrett, J.R.; Webb, A.T.; White, D. Observations beneath Pine Island Glacier in West Antarctica and implications for its retreat. Nat. Geosci. 2010, 3, 468–472. [Google Scholar] [CrossRef]
- Saltzman, E.; Savoie, D.; Zika, R.; Prospero, J. Methane sulfonic acid in the marine atmosphere. J. Geophys. Res. Ocean. 1983, 88, 10897–10902. [Google Scholar] [CrossRef]
- Charlson, R.J.; Lovelock, J.E.; Andreae, M.O.; Warren, S.G. Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate. Nature 1987, 326, 655–661. [Google Scholar] [CrossRef]
- Legrand, M.; Saigne, C.F. Methanesulfonic acid in south polar snow layers: A record of strong El Nino? Geophys. Res. Lett. 1991, 18, 187–190. [Google Scholar] [CrossRef]
- Davis, D.; Chen, G.; Bandy, A.; Thornton, D.; Eisele, F.; Mauldin, L.; Tanner, D.; Lenschow, D.; Fuelberg, H.; Huebert, B. Dimethyl sulfide oxidation in the equatorial Pacific: Comparison of model simulations with field observations for DMS, SO2, H2SO4 (g), MSA (g), MS and NSS. J. Geophys. Res. Atmos. 1999, 104, 5765–5784. [Google Scholar] [CrossRef]
- Savoie, D.L.; Arimoto, R.; Keene, W.C.; Prospero, J.M.; Duce, R.A.; Galloway, J.N. Marine biogenic and anthropogenic contributions to non-sea-salt sulfate in the marine boundary layer over the North Atlantic Ocean. J. Geophys. Res. Atmos. 2002, 107, 4356–4366. [Google Scholar] [CrossRef]
- Curran, M.A.; van Ommen, T.D.; Morgan, V.I.; Phillips, K.L.; Palmer, A.S. Ice core evidence for Antarctic sea ice decline since the 1950s. Science 2003, 302, 1203–1206. [Google Scholar] [CrossRef] [PubMed]
- Isaksson, E.; Kekonen, T.; Moore, J.; Mulvaney, R. The methanesulfonic acid (MSA) record in a Svalbard ice core. Ann. Glaciol. 2005, 42, 345–351. [Google Scholar] [CrossRef]
- Saltzman, E.S.; Dioumaeva, I.; Finley, B.D. Glacial/interglacial variations in methanesulfonate (MSA) in the Siple Dome ice core, West Antarctica. Geophys. Res. Lett. 2006, 33. [Google Scholar] [CrossRef]
- Gabric, A.J.; Qu, B.; Matrai, P.; Hirst, A.C. The simulated response of dimethylsulfide production in the Arctic Ocean to global warming. Tellus B 2005, 57, 391–403. [Google Scholar] [CrossRef]
- Sharma, S.; Chan, E.; Ishizawa, M.; Toom-Sauntry, D.; Gong, S.; Li, S.; Tarasick, D.; Leaitch, W.; Norman, A.; Quinn, P. Influence of transport and ocean ice extent on biogenic aerosol sulfur in the Arctic atmosphere. J. Geophys. Res. Atmos. 2012, 117. [Google Scholar] [CrossRef]
- Leck, C.; Persson, C. Seasonal and short-term variability in dimethyl sulfide, sulfur dioxide and biogenic sulfur and sea salt aerosol particles in the arctic marine boundary layer during summer and autumn. Tellus B 1996, 48, 272–299. [Google Scholar] [CrossRef]
- O’Dwyer, J.; Isaksson, E.; Vinje, T.; Jauhiainen, T.; Moore, J.; Pohjola, V.; Vaikmäe, R.; van de Wal, R.S.W. Methanesulfonic acid in a Svalbard Ice Core as an indicator of ocean climate. Geophys. Res. Lett. 2000, 27, 1159–1162. [Google Scholar] [CrossRef]
- Chen, L.; Wang, J.; Gao, Y.; Xu, G.; Yang, X.; Lin, Q.; Zhang, Y. Latitudinal distributions of atmospheric MSA and MSA/nss-SO42- ratios in summer over the high latitude regions of the Southern and Northern Hemispheres. J. Geophys. Res. 2012, 117. [Google Scholar] [CrossRef]
- ACIA. Arctic Climate Impact Assessment. Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Forster, P.; Ramaswamy, V.; Artaxo, P.; Berntsen, T.; Betts, R.; Fahey, D.W.; Haywood, J.; Lean, J.; Lowe, D.C.; Myhre, G.; et al. Changes in Atmospheric Constituents and in Radiative Forcing. In Climate Change 2007: The Physical Science Basis; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Gao, Y.; Arimoto, R.; Duce, R.; Chen, L.; Zhou, M.; Gu, D. Atmospheric non-sea-salt sulfate, nitrate and methanesulfonate over the China Sea. J. Geophys. Res. Atmos. 1996, 101, 12601–12611. [Google Scholar] [CrossRef]
- Xie, Z.Q.; Sun, L.G.; Wang, J.J.; Liu, B.Z. A potential source of atmospheric sulfur from penguin colony emissions. J. Geophys. Res. Atmos. 2002, 107, ACH 5-1–ACH 5-10. [Google Scholar]
- Berresheim, H. Biogenic sulfur emissions from the Subantarctic and Antarctic Oceans. J. Geophys. Res. Atmos. 1987, 92, 13245–13262. [Google Scholar] [CrossRef]
- Li, S.M.; Barrie, L.A.; Sirois, A. Biogenic sulfur aerosol in the Arctic troposphere: 2. Trends and seasonal variations. J. Geophys. Res. Atmos. 1993, 98, 20623–20631. [Google Scholar] [CrossRef]
- Xie, Z.; Sun, L.; Jihong, C. Non-sea-salt sulfate in the marine boundary layer and its possible impact on chloride depletion. Acta Oceanol. Sinica 2005, 24, 162–171. [Google Scholar]
- Rempillo, O.; Seguin, A.; Norman, A. Shipboard measurement of atmospheric DMS and its oxidation products over the Canadian Arctic. In Proceedings of the AGU Fall Meeting 2009, San Francisco, CA, USA, 14–18 December 2009.
- Li, S.M.; Barrie, L.A. Biogenic sulfur aerosol in the Arctic troposphere: 1. Contributions to total sulfate. J. Geophys. Res. Atmos. 1993, 98, 20613–20622. [Google Scholar] [CrossRef]
- Savoie, D.; Prospero, J.; Larsen, R.; Huang, F.; Izaguirre, M.; Huang, T.; Snowdon, T.; Custals, L.; Sanderson, C. Nitrogen and sulfur species in Antarctic aerosols at Mawson, Palmer station, and Marsh (King George Island). J. Atmos. Chem. 1993, 17, 95–122. [Google Scholar] [CrossRef]
- Udisti, R.; Rugi, F.; Becagli, S.; Bolzacchini, E.; Calzolai, G.; Chiari, M.; Frosini, D.; Ghedini, C.; Marconi, M.; Grazia Perrone, M.; et al. Spatial distribution of biogenic sulphur compounds in the Arctic aerosol collected during the AREX 2011 and 2012 Oceania ship cruises. In Proceedings of the EGU General Assembly Conference 2013, Vienna, Austria, 7–12 April 2013.
- Savoie, D.L.; Prospero, J.M.; Saltzman, E.S. Non-sea-salt sulfate and nitrate in trade wind aerosols at Barbados: Evidence for long-range transport. J. Geophys. Res. Atmos. 1989, 94, 5069–5080. [Google Scholar] [CrossRef]
- Behrenfeld, M.J.; Falkowski, P.G. Photosynthetic rates derived from satellite-based chlorophyll concentration. Limnol. Oceanogr. 1997, 42, 1–20. [Google Scholar] [CrossRef]
- Alvain, S.; Moulin, C.; Dandonneau, Y.; Bréon, F.M. Remote sensing of phytoplankton groups in case 1 waters from global SeaWiFS imagery. Deep Sea Res. 2005, 52, 1989–2004. [Google Scholar] [CrossRef]
- Belviso, S.; Moulin, C.; Bopp, L.; Stefels, J. Assessment of a global climatology of oceanic dimethylsulfide (DMS) concentrations based on SeaWiFS imagery (1998–2001). Can. J. Fish. Aquat. Sci. 2004, 61, 804–816. [Google Scholar] [CrossRef]
- Vogt, M.; Liss, P. Dimethylsulfide and climate. Geophys. Monogr. Ser. 2009, 197, 197–232. [Google Scholar] [CrossRef]
- Degerlund, M.; Eilertsen, H.C. Main species characteristics of phytoplankton spring blooms in NE Atlantic and Arctic waters (68–80 N). Estuaries Coasts 2010, 33, 242–269. [Google Scholar] [CrossRef]
- Trevena, A.; Jones, G. DMS flux over the Antarctic sea ice zone. March Chem. 2012, 134, 47–58. [Google Scholar] [CrossRef]
- McMinn, A.; Hegseth, E. Quantum yield and photosynthetic parameters of marine microalgae from the southern Arctic Ocean, Svalbard. J. March Biol. Assoc. UK 2004, 84, 865–871. [Google Scholar] [CrossRef]
- Rochelle-Newall, E.; Fisher, T. Production of chromophoric dissolved organic matter fluorescence in marine and estuarine environments: an investigation into the role of phytoplankton. March Chem. 2002, 77, 7–21. [Google Scholar] [CrossRef]
- Stedmon, C.A.; Markager, S. Tracing the production and degradation of autochthonous fractions of dissolved organic matter by fluorescence analysis. Limnol. Oceanogr. 2005, 50, 1415–1426. [Google Scholar] [CrossRef]
- Bushaw, K.L.; Zepp, R.G.; Tarr, M.A.; Schulz-Jander, D.; Bourbonniere, R.A.; Hodson, R.E.; Miller, W.L.; Bronk, D.A.; Moran, M.A. Photochemical release of biologically available nitrogen from aquatic dissolved organic matter. Nature 1996, 381, 404–407. [Google Scholar] [CrossRef]
- Kowalczuk, P.; A Stedmon, C.; Markager, S. Modeling absorption by CDOM in the Baltic Sea from season, salinity and chlorophyll. March Chem. 2006, 101, 1–11. [Google Scholar] [CrossRef]
- Gabric, A.J.; Qu, B.; Matrai, P.A.; Murphy, C.; Lu, H.; Lin, D.R.; Qian, F.; Zhao, M. Investigating the coupling between phytoplankton biomass, aerosol optical depth and sea-ice cover in the Greenland Sea. Dyn. Atmos.Ocean. 2014, 66, 94–109. [Google Scholar] [CrossRef]
- Clarke, D.; Ackley, S. Sea ice structure and biological activity in the Antarctic marginal ice zone. J. Geophys. Res. Ocean. 1984, 89, 2087–2095. [Google Scholar] [CrossRef]
- Trevena, A.J.; Jones, G.B. Dimethylsulphide and dimethylsulphoniopropionate in Antarctic sea ice and their release during sea ice melting. March Chem. 2006, 98, 210–222. [Google Scholar] [CrossRef]
- Andres, R.; Kasgnoc, A. A time-averaged inventory of subaerial volcanic sulfur emissions. J. Geophys. Res. Atmos. 1998, 103, 25251–25261. [Google Scholar] [CrossRef]
- Leck, C.; Persson, C. The central Arctic Ocean as a source of dimethyl sulfide Seasonal variability in relation to biological activity. Tellus B 1996, 48, 156–177. [Google Scholar] [CrossRef]
- Tison, J.L.; Brabant, F.; Dumont, I.; Stefels, J. High-resolution dimethyl sulfide and dimethylsulfoniopropionate time series profiles in decaying summer first-year sea ice at Ice Station Polarstern, western Weddell Sea, Antarctica. J. Geophys. Res. Biog. 2010, 115. [Google Scholar] [CrossRef]
- Kuosa, H.; Norrman, B.; Kivi, K.; Brandini, F. Effects of Antarctic sea ice biota on seeding as studied in aquarium experiments. Polar biol. 1992, 12, 333–339. [Google Scholar] [CrossRef]
- Lannuzel, D.; Schoemann, V.; de Jong, J.; Tison, J.L.; Chou, L. Distribution and biogeochemical behaviour of iron in the East Antarctic sea ice. March Chem. 2007, 106, 18–32. [Google Scholar] [CrossRef]
- Bates, T.S.; Calhoun, J.A.; Quinn, P.K. Variations in the methanesulfonate to sulfate molar ratio in submicrometer marine aerosol particles over the South Pacific Ocean. J. Geophys. Res. Atmos. 1992, 97, 9859–9865. [Google Scholar] [CrossRef]
- Prospero, J.; Savoie, D.; Saltzman, E.; Larsen, R. Impact of oceanic sources of biogenic sulphur on sulphate aerosol concentrations at Mawson, Antarctica. Nature. 1991, 350. [Google Scholar] [CrossRef]
- Wagenbach, D. Coastal Antarctica: Atmospheric Chemical Composition and Atmospheric Transport. In Chemical Exchange between the Atmosphere and Polar Snow; Wolff, E., Bales, R.C., Eds.; Springer: Berlin, Heidelberg, Germany, 1996; pp. 173–199. [Google Scholar]
- Laing, J.R.; Hopke, P.K.; Hopke, E.F.; Husain, L.; Dutkiewicz, V.A.; Paatero, J.; Viisanen, Y. Long-term trends of biogenic sulfur aerosol and its relationship with sea surface temperature in Arctic Finland. J. Geophys. Res. Atmos. 2013, 118, 11770–11776. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, P.; Xie, Z.; Yu, J.; Kang, H. Spatial Distribution of Methanesulphonic Acid in the Arctic Aerosol Collected during the Chinese Arctic Research Expedition. Atmosphere 2015, 6, 699-712. https://doi.org/10.3390/atmos6050699
Ye P, Xie Z, Yu J, Kang H. Spatial Distribution of Methanesulphonic Acid in the Arctic Aerosol Collected during the Chinese Arctic Research Expedition. Atmosphere. 2015; 6(5):699-712. https://doi.org/10.3390/atmos6050699
Chicago/Turabian StyleYe, Peipei, Zhouqing Xie, Juan Yu, and Hui Kang. 2015. "Spatial Distribution of Methanesulphonic Acid in the Arctic Aerosol Collected during the Chinese Arctic Research Expedition" Atmosphere 6, no. 5: 699-712. https://doi.org/10.3390/atmos6050699
APA StyleYe, P., Xie, Z., Yu, J., & Kang, H. (2015). Spatial Distribution of Methanesulphonic Acid in the Arctic Aerosol Collected during the Chinese Arctic Research Expedition. Atmosphere, 6(5), 699-712. https://doi.org/10.3390/atmos6050699