Regional Air Quality Model Application of the Aqueous-Phase Photo Reduction of Atmospheric Oxidized Mercury by Dicarboxylic Acids

Abstract
:1. Introduction
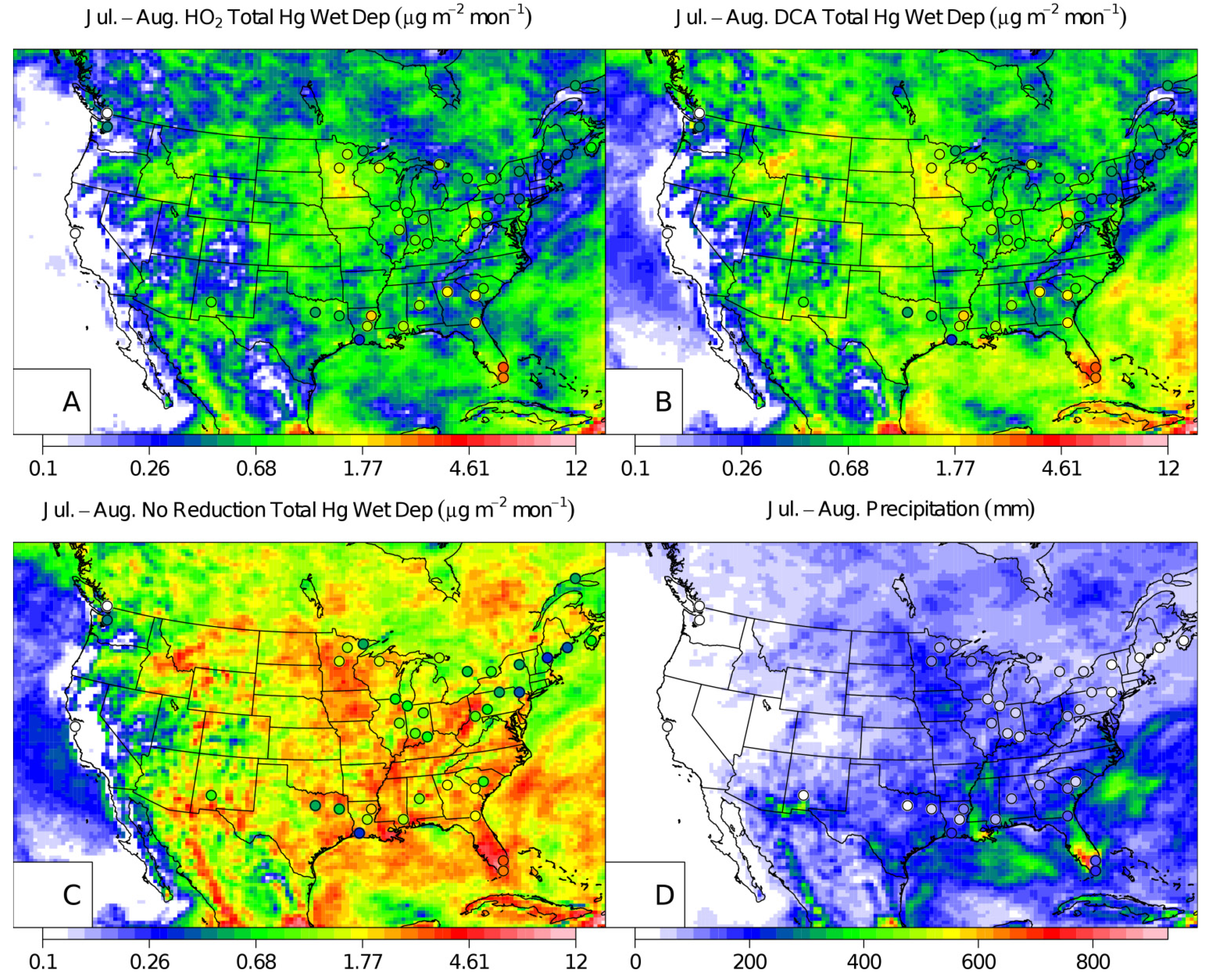
2. Results and Discussion
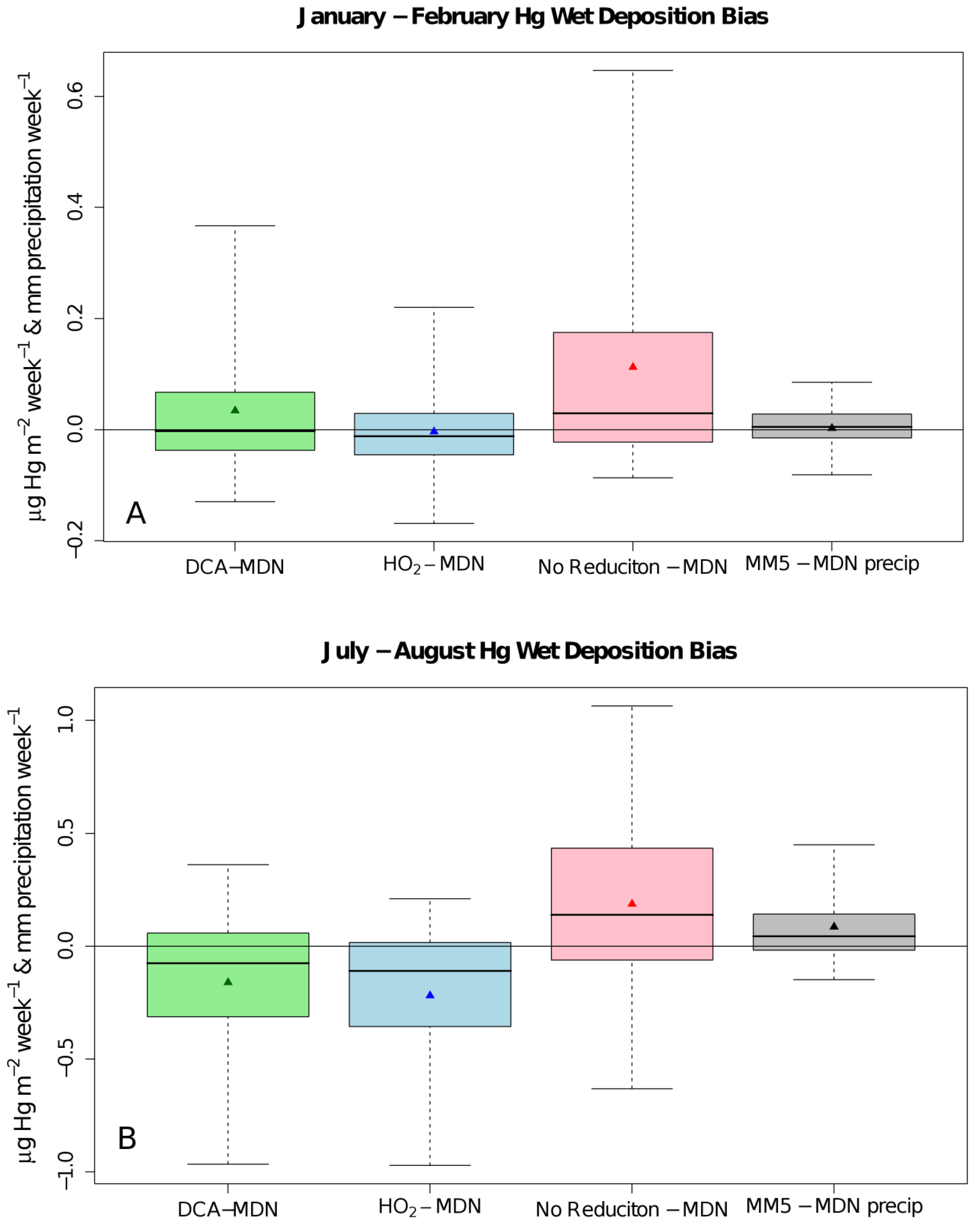
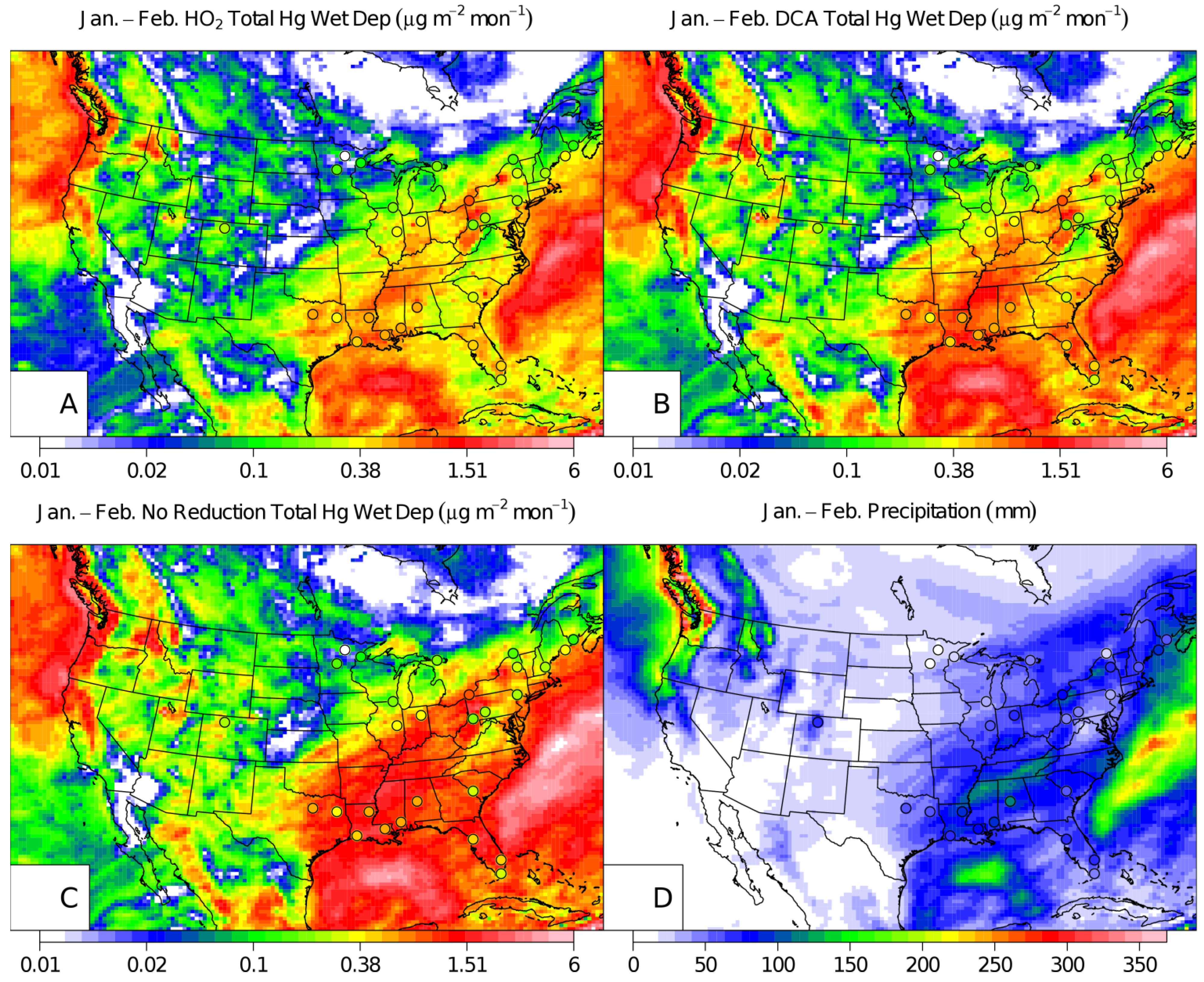

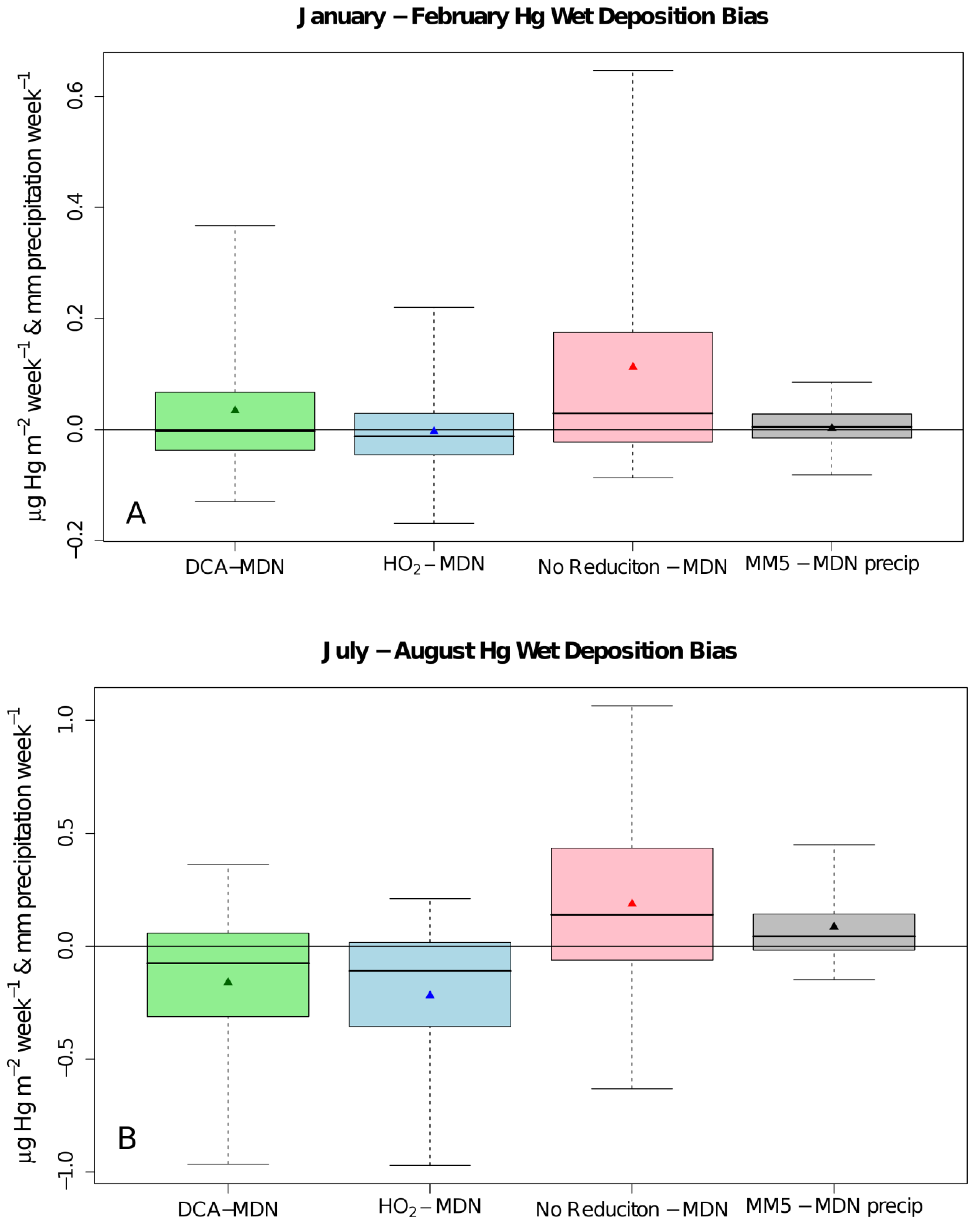{kind=link}
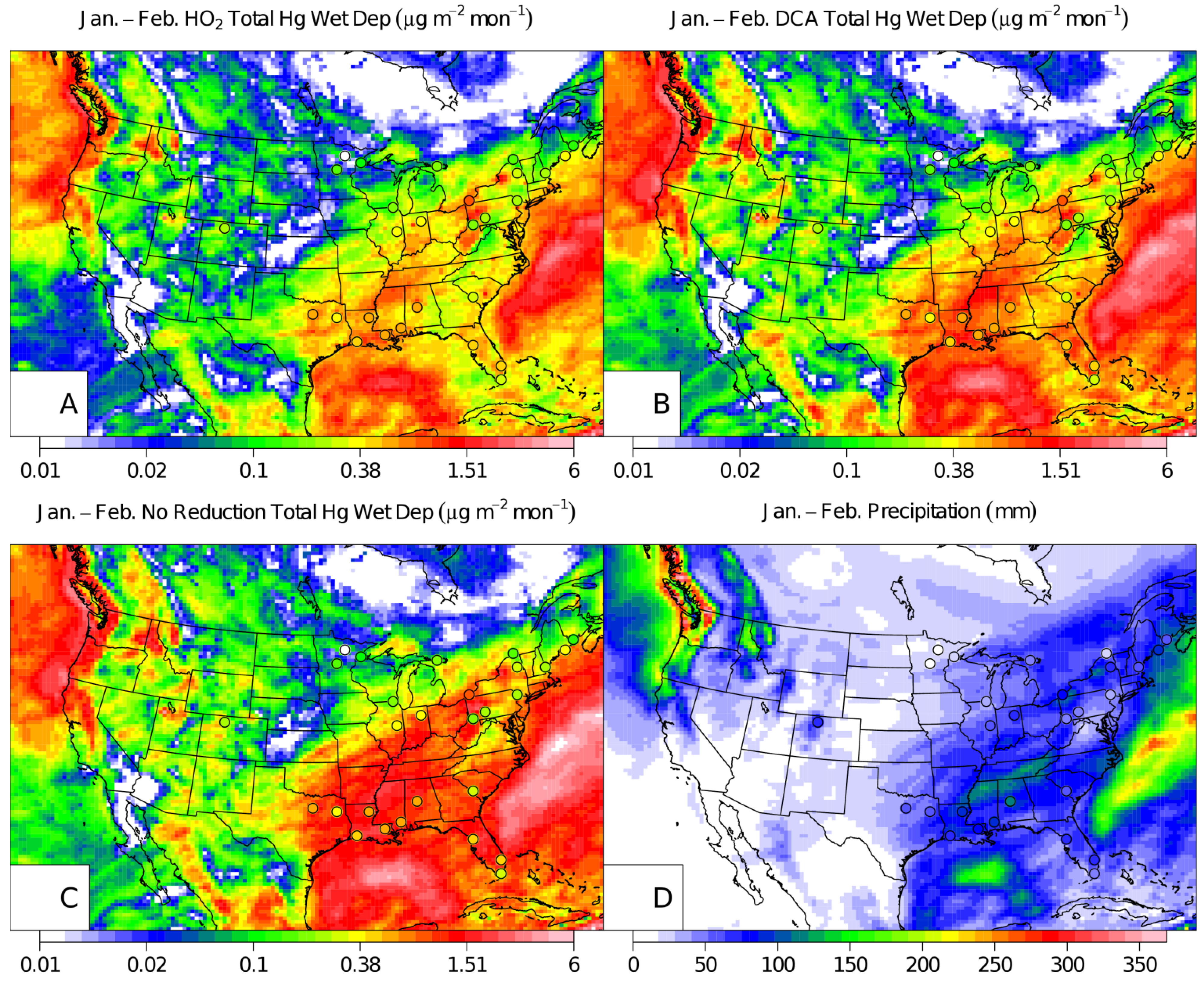{kind=link}
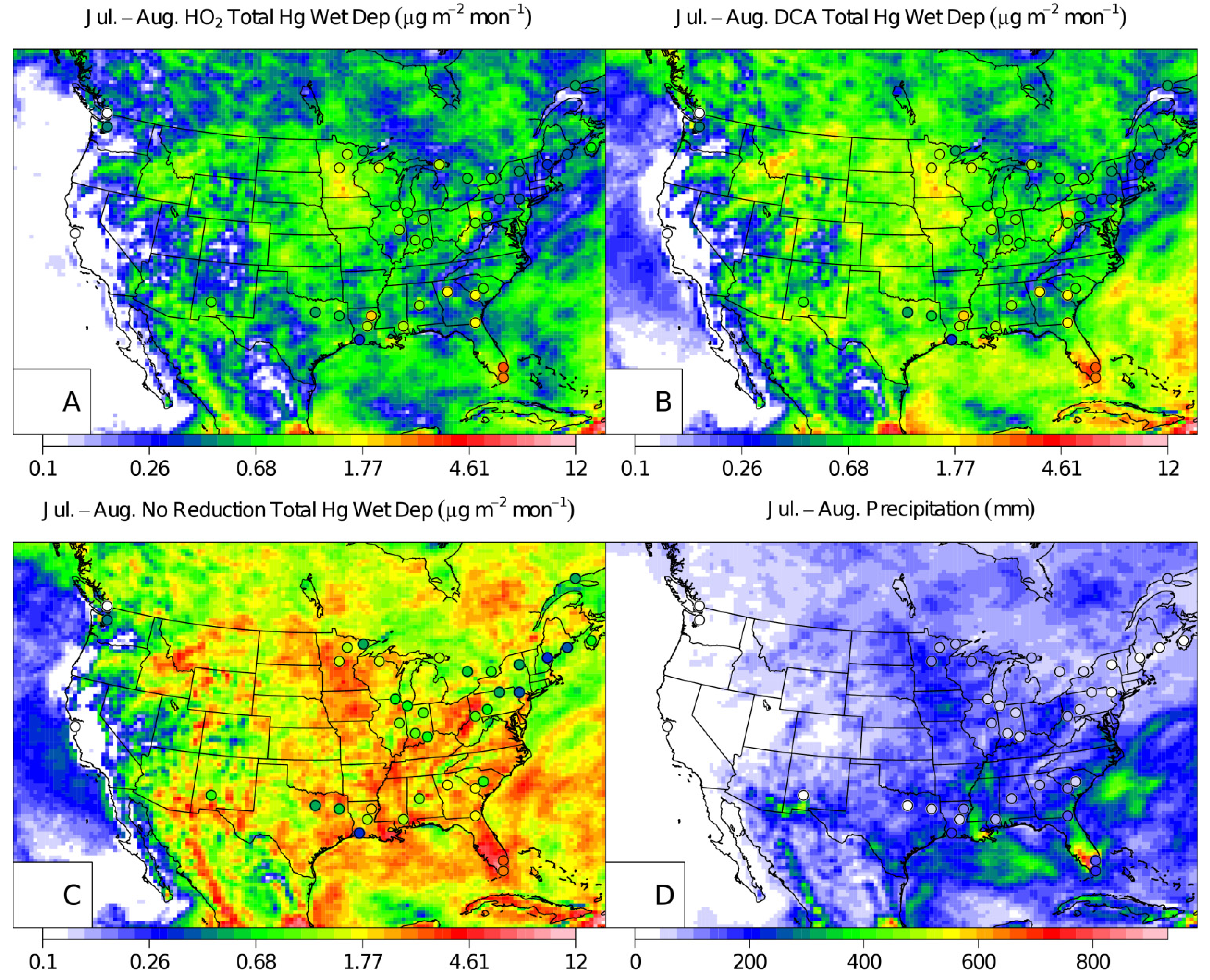{kind=link}
{kind=link}
| ρ | MdnB | MndE | NMndB | NMndE | ||
|---|---|---|---|---|---|---|
| MM5
Precipitation | January–February | 0.807 | 1.0 mm∙week−1 | 4.5 mm∙week−1 | 7.2% | 33.2% |
| July–August | 0.424 | 8.5 mm∙week−1 | 16.2 mm∙week−1 | 53.4% | 98.9% | |
| No
Reduction | January–February | 0.586 | 29 ng∙m−2∙week−1 | 61 ng∙m−2∙week−1 | 43.7% | 91.8% |
| July–August | 0.338 | 138 ng∙m−2∙week−1 | 269 ng∙m−2∙week−1 | 57.3% | 111.3% | |
| HO2 Reduction | January–February | 0.570 | −12 ng∙m−2∙week−1 | 41 ng∙m−2∙week−1 | −17.5% | 61.5% |
| July–August | 0.241 | −110 ng∙m−2∙week−1 | 163 ng∙m−2∙week−1 | −45.7% | 67.5% | |
| DCA
Reduction | January–February | 0.586 | −3 ng∙m−2∙week−1 | 46 ng∙m−2∙week−1 | −4.3% | 69.4% |
| July–August | 0.252 | −76 ng∙m−2∙week−1 | 175 ng∙m−2∙week−1 | −31.4% | 72.3% |
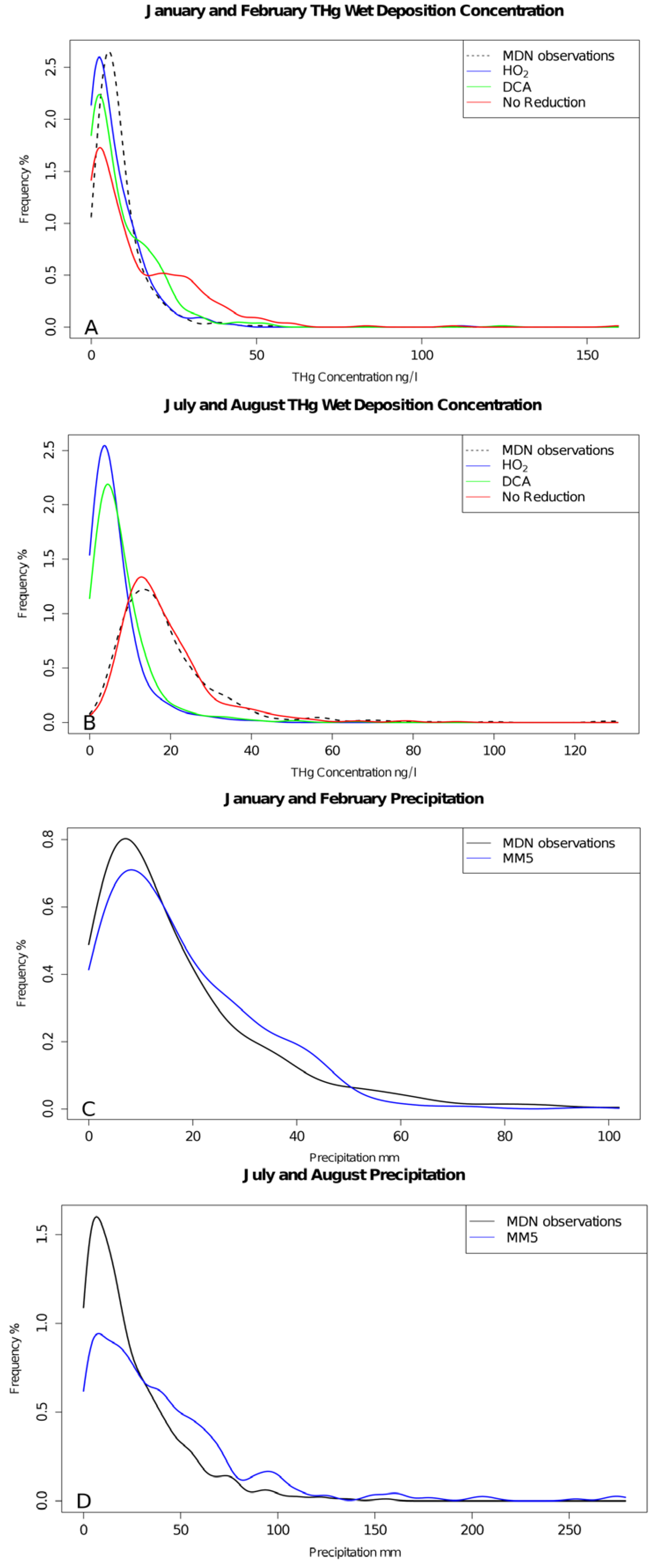
3. Experimental Section
3.1. Application of an Aqueous-Phase Hg2+aq DCA Reduction Scheme in CMAQ
3.2. CMAQ Model Simulations
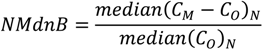
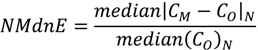
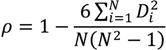
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Streets, D.G.; Devane, M.K.; Lu, Z.F.; Bond, T.C.; Sunderland, E.M.; Jacob, D.J. All-time releases of mercury to the atmosphere from human activities. Environ. Sci. Tech. 2011, 45, 10485–10491. [Google Scholar] [CrossRef]
- Sunderland, E.M.; Krabbenhoft, D.P.; Moreau, J.W.; Strode, S.A.; Landing, W.M. Mercury sources, distribution, and bioavailability in the North Pacific Ocean: Insights from data and models. Glob. Biogeochem. Cy. 2009, 23, GB2010. [Google Scholar]
- Goulet, R.R.; Holmes, J.; Page, B.; Poissant, L.; Siciliano, S.D.; Lean, D.R.S.; Wang, F.; Amyot, M.; Tessier, A. Mercury transformations and fluxes in sediments of a riverine wetland. Geochim. Cosmochim. Acta 2007, 71, 3393–3406. [Google Scholar] [CrossRef]
- Wolfe, M.F.; Schwarzbach, S.; Sulaiman, R.A. Effects of mercury on wildlife: A comprehensive review. Environ. Toxicol. Chem. 1998, 17, 146–160. [Google Scholar] [CrossRef]
- Lin, C.J.; Pongprueksa, P.; Lindberg, S.E.; Pehkonen, S.O.; Byun, D.; Jang, C. Scientific uncertainties in atmospheric mercury models I: Model science evaluation. Atmos. Environ. 2006, 40, 2911–2928. [Google Scholar] [CrossRef]
- Schroeder, W.H.; Munthe, J. Atmospheric mercury—An overview. Atmos. Environ. 1998, 32, 809–822. [Google Scholar] [CrossRef]
- Fain, X.; Obrist, D.; Hallar, A.G.; McCubbin, I.; Rahn, T. High levels of reactive gaseous mercury observed at a high elevation research laboratory in the Rocky Mountains. Atmos. Chem. Phys. 2009, 9, 8049–8060. [Google Scholar] [CrossRef]
- Selin, N.E. Global Biogeochemical Cycling of Mercury: A Review. Annu. Rev. Environ. Resour. 2009, 34, 43–63. [Google Scholar] [CrossRef]
- Pacyna, E.G.; Pacyna, J.M.; Steenhuisen, F.; Wilson, S. Global anthropogenic mercury emission inventory for 2000. Atmos. Environ. 2006, 40, 4048–4063. [Google Scholar] [CrossRef]
- Pongprueksa, P.; Lin, C.J.; Lindberg, S.E.; Jang, C.; Braverman, T.; Bullock, O.R.; Ho, T.C.; Chu, H.W. Scientific uncertainties in atmospheric mercury models III: Boundary and initial conditions, model grid resolution, and Hg(II) reduction mechanism. Atmos. Environ. 2008, 42, 1828–1845. [Google Scholar] [CrossRef]
- Si, L.; Ariya, P.A. Reduction of oxidized mercury species by dicarboxylic acids (C(2)–C(4)): Kinetic and product studies. Environ. Sci. Tech. 2008, 42, 5150–5155. [Google Scholar] [CrossRef]
- Van Loon, L.; Mader, E.; Scott, S.L. Reduction of the aqueous mercuric ion by sulfite: UV spectrum of HgSO3 and its intramolecular redox reaction. J. Phys. Chem. A 2000, 104, 1621–1626. [Google Scholar] [CrossRef]
- Pehkonen, S.O.; Lin, C.J. Aqueous photochemistry of mercury with organic acids. J. Air Waste Manag. Assoc. 1998, 48, 144–150. [Google Scholar] [CrossRef]
- Xiao, Z.F.; Munthe, J.; Strömberg, D.; Lindqvist, O. Photochemical Behaviour of Inorganic Mercury Compounds in Aqueous Solution; Lewis Publishers: London, UK, 1994. [Google Scholar]
- Seigneur, C.; Vijayaraghavan, K.; Lohman, K. Atmospheric mercury chemistry: Sensitivity of global model simulations to chemical reactions. J. Geophys. Res.: Atmos. 2006, 111. [Google Scholar] [CrossRef]
- Selin, N.E.; Jacob, D.J.; Park, R.J.; Yantosca, R.M.; Strode, S.; Jaegle, L.; Jaffe, D. Chemical cycling and deposition of atmospheric mercury: Global constraints from observations. J. Geophys. Res.: Atmos. 2007, 112. [Google Scholar] [CrossRef]
- Gardfeldt, K.; Jonsson, M. Is bimolecular reduction of Hg(II) complexes possible in aqueous systems of environmental importance. J. Phys. Chem. A 2003, 107, 4478–4482. [Google Scholar] [CrossRef]
- Sorooshian, A.; Lu, M.L.; Brechtel, F.J.; Jonsson, H.; Feingold, G.; Flagan, R.C.; Seinfeld, J.H. On the source of organic acid aerosol layers above clouds. Environ. Sci. Tech. 2007, 41, 4647–4654. [Google Scholar] [CrossRef]
- Liu, J.; Horowitz, L.W.; Fan, S.; Carlton, A.G.; Levy, H. Global in-cloud production of secondary organic aerosols: Implementation of a detailed chemical mechanism in the GFDL atmospheric model AM3. J. Geophys. Res.: Atmos. 2012, 117, D15303. [Google Scholar] [CrossRef]
- Lindberg, S.; Vermette, S. Workshop on sampling mercury in precipitation for the national atmospheric deposition program. Atmos. Environ. 1995, 29, 1219–1220. [Google Scholar] [CrossRef]
- Foley, K.M.; Roselle, S.J.; Appel, K.W.; Bhave, P.V.; Pleim, J.E.; Otte, T.L.; Mathur, R.; Sarwar, G.; Young, J.O.; Gilliam, R.C.; et al. Incremental testing of the Community Multiscale Air Quality (CMAQ) modeling system version 4.7. Geosci. Model. Dev. 2010, 3, 205–226. [Google Scholar] [CrossRef]
- Myers, T.; Atkinson, R.D.; Bullock, O.R.; Bash, J.O. Investigation of effects of varying model inputs on mercury deposition estimates in the Southwest US. Atmos. Chem. Phys. 2013, 13, 997–1009. [Google Scholar] [CrossRef]
- Carlton, A.G.; Bhave, P.V.; Napelenok, S.L.; Edney, E.D.; Sarwar, G.; Pinder, R.W.; Pouliot, G.A.; Houyoux, M. Model representation of secondary organic aerosol in CMAQv4.7. Environ. Sci. Tech. 2010, 44, 8553–8560. [Google Scholar] [CrossRef]
- Perri, M.J.; Seitzinger, S.; Turpin, B.J. Secondary organic aerosol production from aqueous photooxidation of glycolaldehyde: Laboratory experiments. Atmos. Environ. 2009, 43, 1487–1497. [Google Scholar] [CrossRef]
- El Haddad, I.; Liu, Y.; Nieto-Gligorovski, L.; Michaud, V.; Temime-Roussel, B.; Quivet, E.; Marchand, N.; Sellegri, K.; Monod, A. In-cloud processes of methacrolein under simulated conditions-Part 2: Formation of secondary organic aerosol. Atmos. Chem. Phys. 2009, 9, 5107–5117. [Google Scholar] [CrossRef]
- Baker, K.R.; Bash, J.O. Regional scale photochemical model evaluation of total mercury wet deposition and speciated ambient mercury. Atmos. Environ. 2012, 49, 151–162. [Google Scholar] [CrossRef]
- Holloway, T.; Voigt, C.; Morton, J.; Spak, S.N.; Rutter, A.P.; Schauer, J.J. An assessment of atmospheric mercury in the Community Multiscale Air Quality (CMAQ) model at an urban site and a rural site in the Great Lakes Region of North America. Atmos. Chem. Phys. 2012, 12, 7117–7133. [Google Scholar] [CrossRef]
- Huang, J.Y.; Miller, M.B.; Weiss-Penzias, P.; Gustin, M.S. Comparison of gaseous oxidized Hg measured by KCl-coated denuders, and nylon and cation exchange membranes. Environ. Sci. Tech. 2013, 47, 7307–7316. [Google Scholar]
- Ambrose, J.L.; Lyman, S.N.; Huang, J.Y.; Gustin, M.S.; Jaffe, D.A. Fast time resolution oxidized mercury measurements during the Reno Atmospheric Mercury Intercomparison Experiment (RAMIX). Environ. Sci. Tech. 2013, 47, 7285–7294. [Google Scholar]
- Gustin, M.S.; Huang, J.Y.; Miller, M.B.; Peterson, C.; Jaffe, D.A.; Ambrose, J.; Finley, B.D.; Lyman, S.N.; Call, K.; Talbot, R.; et al. Do we understand what the mercury speciation instruments are actually measuring? results of RAMIX. Environ. Sci. Tech. 2013, 47, 7295–7306. [Google Scholar]
- Lyman, S.N.; Jaffe, D.A.; Gustin, M.S. Release of mercury halides from KCl denuders in the presence of ozone. Atmos. Chem. Phys. 2010, 10, 8197–8204. [Google Scholar] [CrossRef]
- Hynes, A.J.; Donohoue, D.L.; Goodsite, M.E.; Hedgecock, I.M. Our Current Understanding of Major Chemical and Physical Processes Affecting Mercury Dynamics in the Atmosphere and at the Air-Water/Terrestrial Iterfaces. In Mercury Fate and Transport in the Global Atmosphere; Springer: Berlin, Germany, 2009; pp. 427–457. [Google Scholar]
- Subir, M.; Ariya, P.A.; Dastoor, A.P. A review of the sources of uncertainties in atmospheric mercury modeling II. Mercury surface and heterogeneous chemistry—A missing link. Atmos. Environ. 2012, 46, 1–10. [Google Scholar] [CrossRef]
- Kolker, A.; Olson, M.L.; Krabbenhoft, D.P.; Tate, M.T.; Engle, M.A. Patterns of mercury dispersion from local and regional emission sources, rural central wisconsin, USA. Atmos. Chem. Phys. 2010, 10, 4467–4476. [Google Scholar] [CrossRef]
- Timonen, H.; Ambrose, J.L.; Jaffe, D.A. Oxidation of elemental Hg in anthropogenic and marine airmasses. Atmos. Chem. Phys. 2013, 13, 2827–2836. [Google Scholar] [CrossRef]
- Kos, G.; Ryzhkov, A.; Dastoor, A.; Narayan, J.; Steffen, A.; Ariya, P.A.; Zhang, L. Evaluation of discrepancy between measured and modelled oxidized mercury species. Atmos. Chem. Phys. 2013, 13, 4839–4863. [Google Scholar]
- Carlton, A.G.; Turpin, B.J.; Altieri, K.E.; Seitzinger, S.P.; Mathur, R.; Roselle, S.J.; Weber, R.J. CMAQ model performance enhanced when in-cloud secondary organic aerosol is included: Comparisons of organic carbon predictions with measurements. Environ. Sci. Tech. 2008, 42, 8798–8802. [Google Scholar] [CrossRef]
- Carlton, A.G.; Turpin, B.J.; Lim, H.J.; Altieri, K.E.; Seitzinger, S. Link between isoprene and secondary organic aerosol (SOA): Pyruvic acid oxidation yields low volatility organic acids in clouds. Geophys. Res. Lett. 2006, 33, L06822. [Google Scholar]
- Carlton, A.G.; Turpin, B.J.; Altieri, K.E.; Seitzinger, S.; Reff, A.; Lim, H.J.; Ervens, B. Atmospheric oxalic acid and SOA production from glyoxal: Results of aqueous photooxidation experiments. Atmos. Environ. 2007, 41, 7588–7602. [Google Scholar] [CrossRef]
- Altieri, K.E.; Carlton, A.G.; Lim, H.-J.; Turpin, B.J.; Seitzinger, S.P. Evidence for oligomer formation in clouds: Reactions of isoprene oxidation products. Environ. Sci. Tech. 2006, 40, 4956–4960. [Google Scholar] [CrossRef]
- Altieri, K.E.; Seitzinger, S.P.; Carlton, A.G.; Turpin, B.J.; Klein, G.C.; Marshall, A.G. Oligomers formed through in-cloud methylglyoxal reactions: Chemical composition, properties, and mechanisms investigated by ultra-high resolution FT-ICR mass spectrometry. Atmos. Environ. 2008, 42, 1476–1490. [Google Scholar] [CrossRef]
- Tan, Y.; Carlton, A.G.; Seitzinger, S.P.; Turpin, B.J. SOA from methylglyoxal in clouds and wet aerosols: Measurement and prediction of key products. Atmos. Environ. 2010, 44, 5218–5226. [Google Scholar] [CrossRef]
- Legrand, M.; Preunkert, S.; Oliveira, T.; Pio, C.A.; Hammer, S.; Gelencser, A.; Kasper-Giebl, A.; Laj, P. Origin of C-2-C-5 dicarboxylic acids in the European atmosphere inferred from year-round aerosol study conducted at a west-east transect. J. Geophys. Res.: Atmos. 2007, 112, D23S07. [Google Scholar] [CrossRef]
- Sorooshian, A.; Varutbangkul, V.; Brechtel, F.J.; Ervens, B.; Feingold, G.; Bahreini, R.; Murphy, S.M.; Holloway, J.S.; Atlas, E.L.; Buzorius, G.; et al. Oxalic acid in clear and cloudy atmospheres: Analysis of data from international consortium for atmospheric research on transport and transformation 2004. J. Geophys. Res.: Atmos. 2006, 111, D23S45. [Google Scholar]
- Ervens, B.; Feingold, G.; Frost, G.J.; Kreidenweis, S.M. A modeling study of aqueous production of dicarboxylic acids: 1. Chemical pathways and speciated organic mass production. J. Geophys. Res.: Atmos. 2004, 109, D15205. [Google Scholar] [CrossRef]
- Sarwar, G.; Luecken, D.; Yarwood, G.; Whitten, G.Z.; Carter, W.P.L. Impact of an updated carbon bond mechanism on predictions from the CMAQ modeling system: Preliminary assessment. J. Appl. Meteorol. Clim. 2008, 47, 3–14. [Google Scholar] [CrossRef]
- Strode, S.A.; Jaegle, L.; Selin, N.E.; Jacob, D.J.; Park, R.J.; Yantosca, R.M.; Mason, R.P.; Slemr, F. Air-sea exchange in the global mercury cycle. Glob. Biogeochem. Cy. 2007, 21, GB1017. [Google Scholar]
- Grell, G.; Dudhia, J.; Stouffer, D. A Description of the Fifth-Generation PENN State/NCAR Mesoscale Model (MM5); National Center for Atmospheric Research: Boulder, CO, USA, 1994. [Google Scholar]
- Pleim, J.E.; Xiu, A. Development and testing of a surface flux and planetary boundary-layer model for application in mesoscale models. J. Appl. Meteorol. 1995, 34, 16–32. [Google Scholar] [CrossRef]
- Bullock, O.R.; Atkinson, D.; Braverman, T.; Civerolo, K.; Dastoor, A.; Davignon, D.; Ku, J.Y.; Lohman, K.; Myers, T.C.; Park, R.J.; et al. The North American Mercury Model Intercomparison Study (NAMMIS): Study description and model-to-model comparisons. J. Geophys. Res.: Atmos. 2008, 113, D17310. [Google Scholar] [CrossRef]
- Appel, K.W.; Roselle, S.J.; Gilliam, R.C.; Pleim, J.E. Sensitivity of the Community Multiscale Air Quality (CMAQ) model v4.7 results for the eastern United States to MM5 and WRF meteorological drivers. Geosci. Model. Dev. 2010, 3, 169–188. [Google Scholar] [CrossRef]
- Simon, H.; Baker, K.R.; Phillips, S. Compilation and interpretation of photochemical model performance statistics published between 2006 and 2012. Atmos. Environ. 2012, 61, 124–139. [Google Scholar] [CrossRef]
- Zheng, W.; Hintelmann, H. Mercury isotope fractionation during photoreduction in natural water is controlled by its Hg/DOC ratio. Geochim. Cosmochim. Acta 2009, 73, 6704–6715. [Google Scholar] [CrossRef]
- Amos, H.M.; Jacob, D.J.; Holmes, C.D.; Fisher, J.A.; Wang, Q.; Yantosca, R.M.; Corbitt, E.S.; Galarneau, E.; Rutter, A.P.; Gustin, M.S.; et al. Gas-particle partitioning of atmospheric Hg(II) and its effect on global mercury deposition. Atmos. Chem. Phys. 2012, 12, 591–603. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bash, J.O.; Carlton, A.G.; Hutzell, W.T.; Bullock Jr., O.R. Regional Air Quality Model Application of the Aqueous-Phase Photo Reduction of Atmospheric Oxidized Mercury by Dicarboxylic Acids. Atmosphere 2014, 5, 1-15. https://doi.org/10.3390/atmos5010001
Bash JO, Carlton AG, Hutzell WT, Bullock Jr. OR. Regional Air Quality Model Application of the Aqueous-Phase Photo Reduction of Atmospheric Oxidized Mercury by Dicarboxylic Acids. Atmosphere. 2014; 5(1):1-15. https://doi.org/10.3390/atmos5010001
Chicago/Turabian StyleBash, Jesse O., Annmarie G. Carlton, William T. Hutzell, and O. Russell Bullock Jr. 2014. "Regional Air Quality Model Application of the Aqueous-Phase Photo Reduction of Atmospheric Oxidized Mercury by Dicarboxylic Acids" Atmosphere 5, no. 1: 1-15. https://doi.org/10.3390/atmos5010001
APA StyleBash, J. O., Carlton, A. G., Hutzell, W. T., & Bullock Jr., O. R. (2014). Regional Air Quality Model Application of the Aqueous-Phase Photo Reduction of Atmospheric Oxidized Mercury by Dicarboxylic Acids. Atmosphere, 5(1), 1-15. https://doi.org/10.3390/atmos5010001