Aerosol Composition in a Semi-Urban Environment in Central Mexico: Influence of Local and Regional Processes on Overall Composition and First Quantification of Nitroaromatics

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Site
2.2. Online Measurements
2.3. Offline Measurements
2.4. Retro-Trajectories
3. Results and Discussion
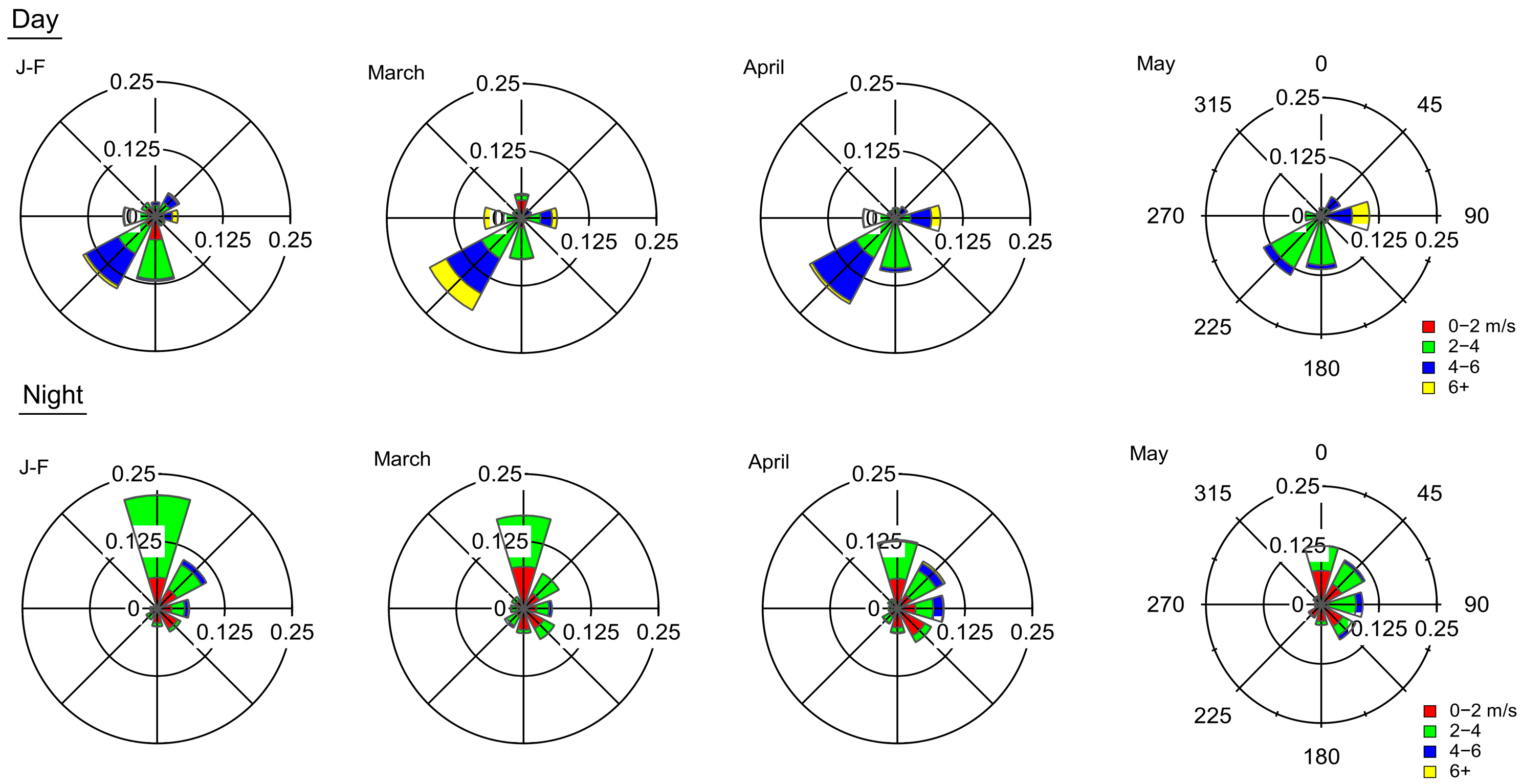
3.1. Meteorology
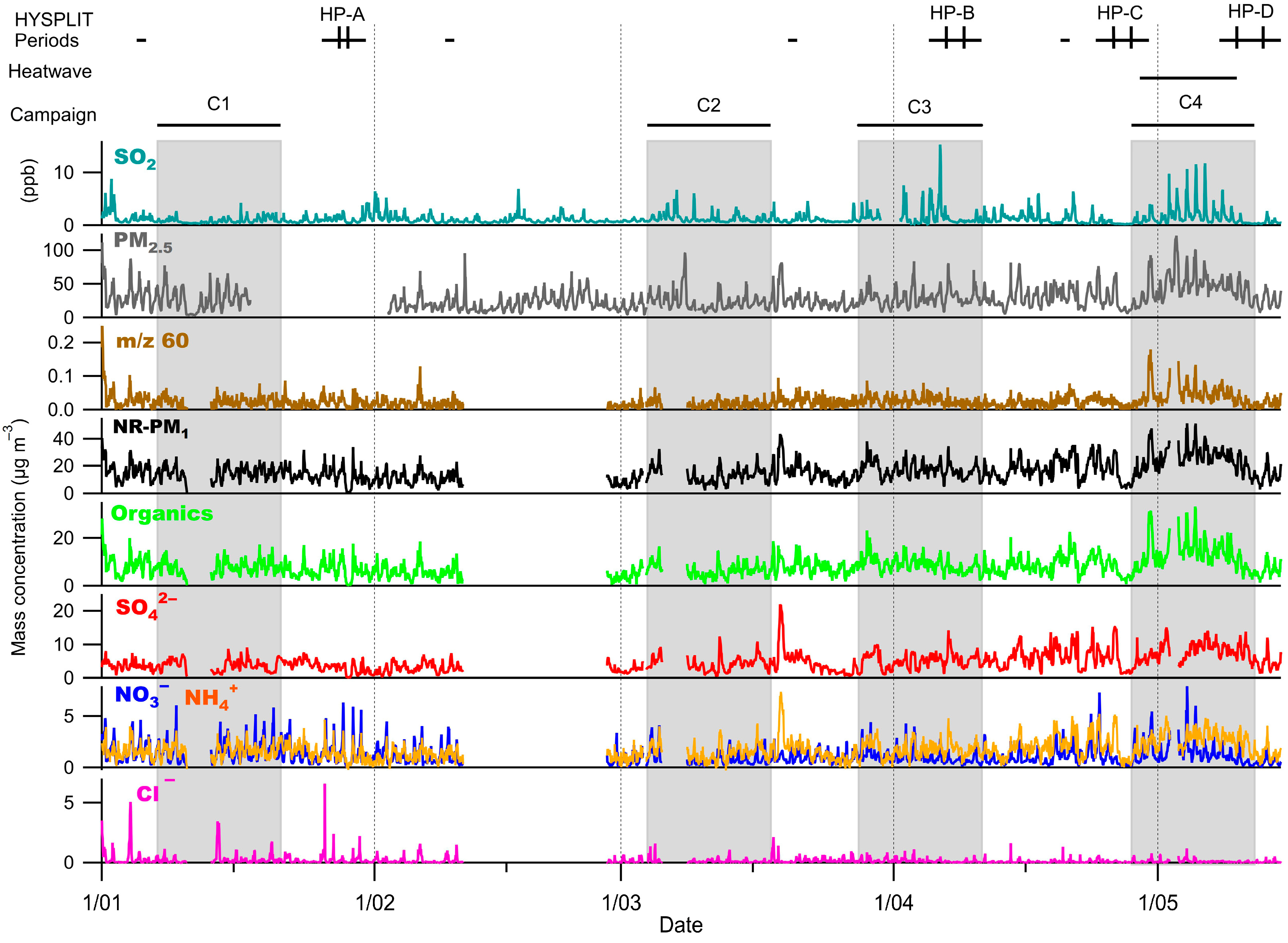
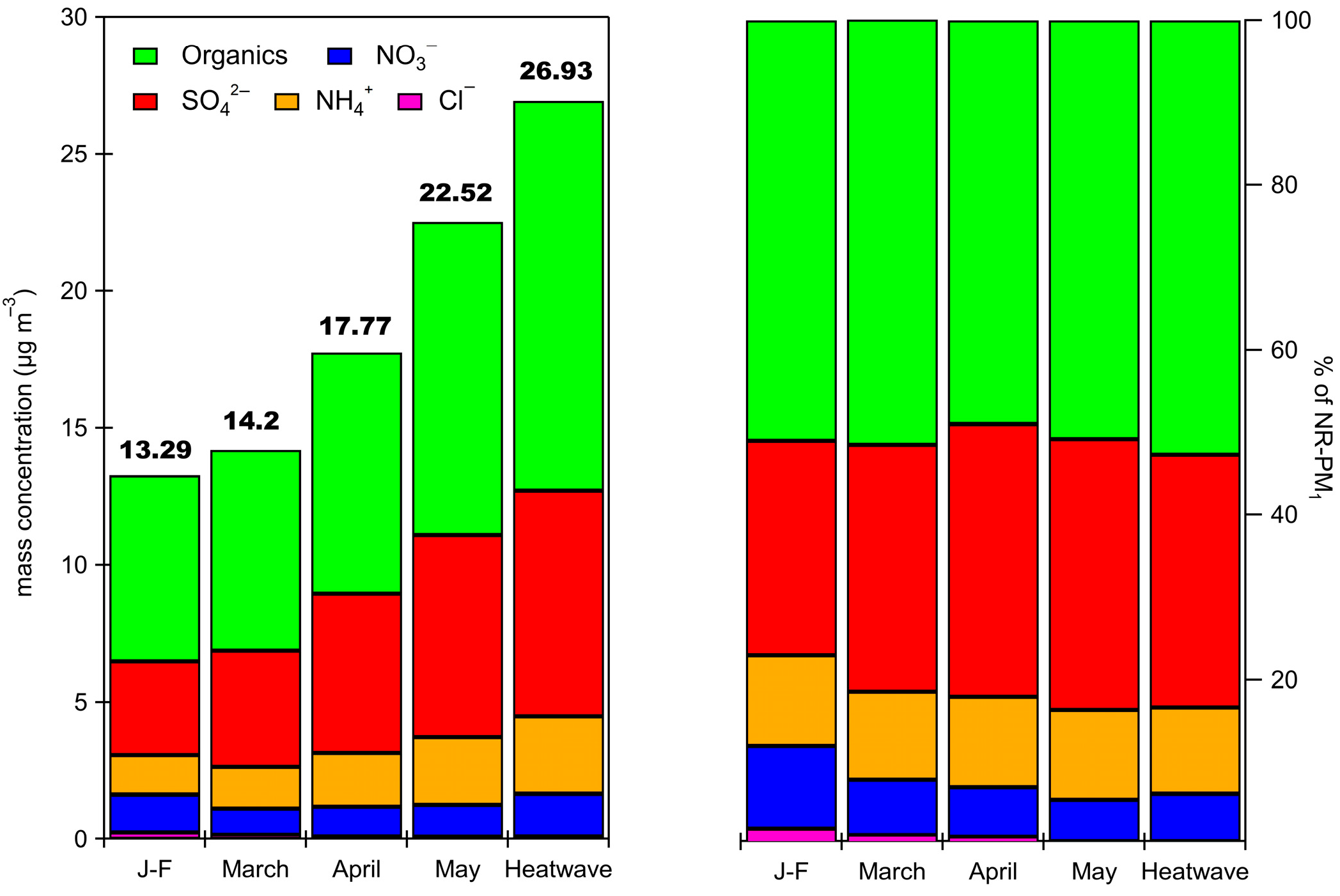
3.2. NR-PM1 Concentrations and Chemical Composition
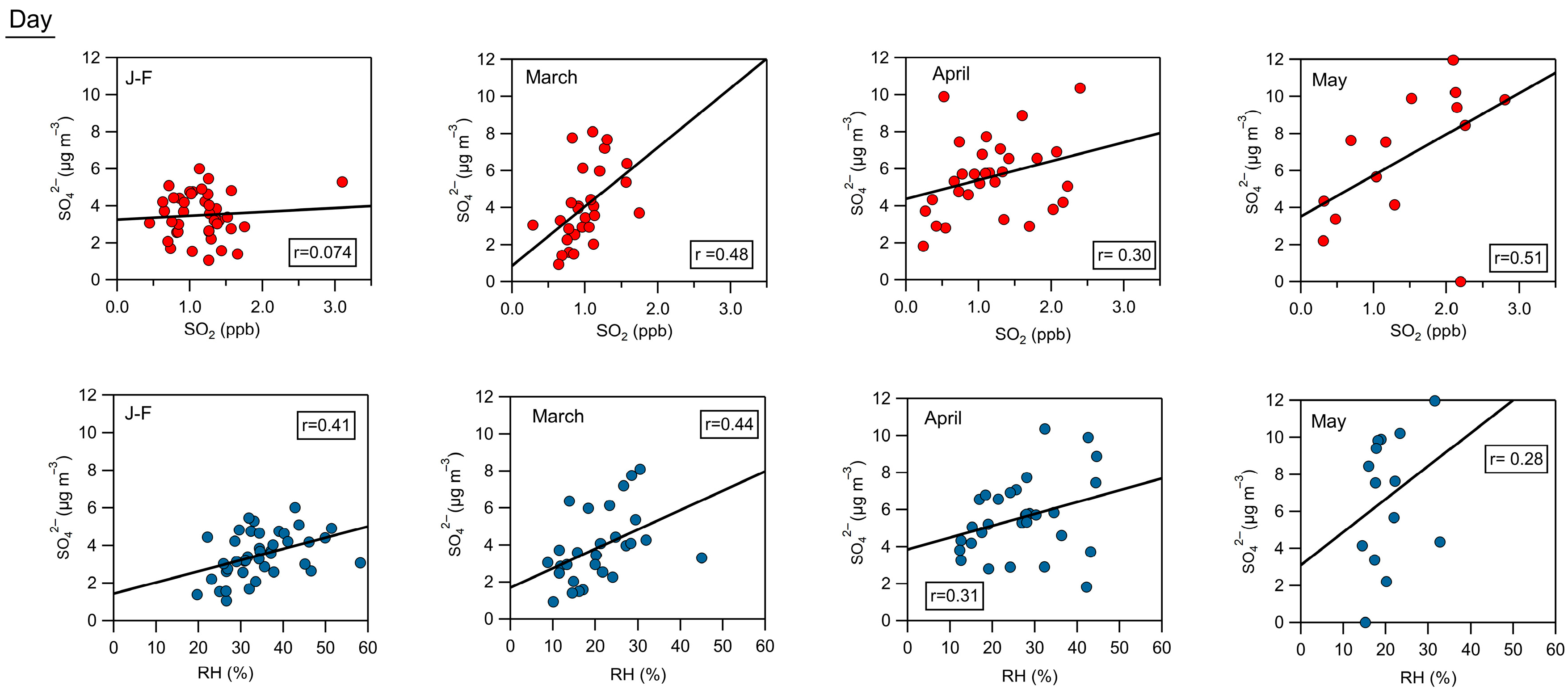
3.3. Inorganic Fraction of the NR-PM1
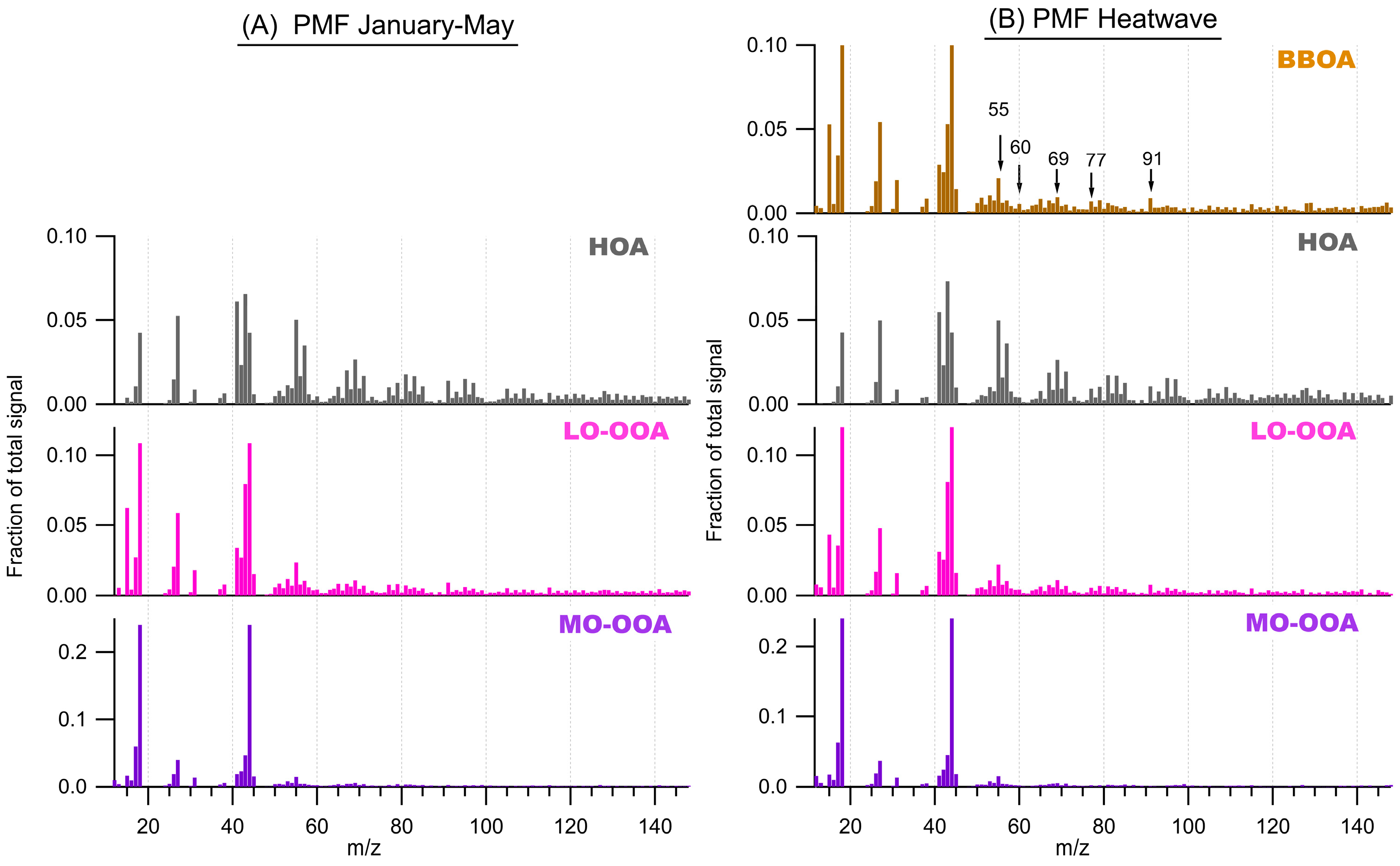
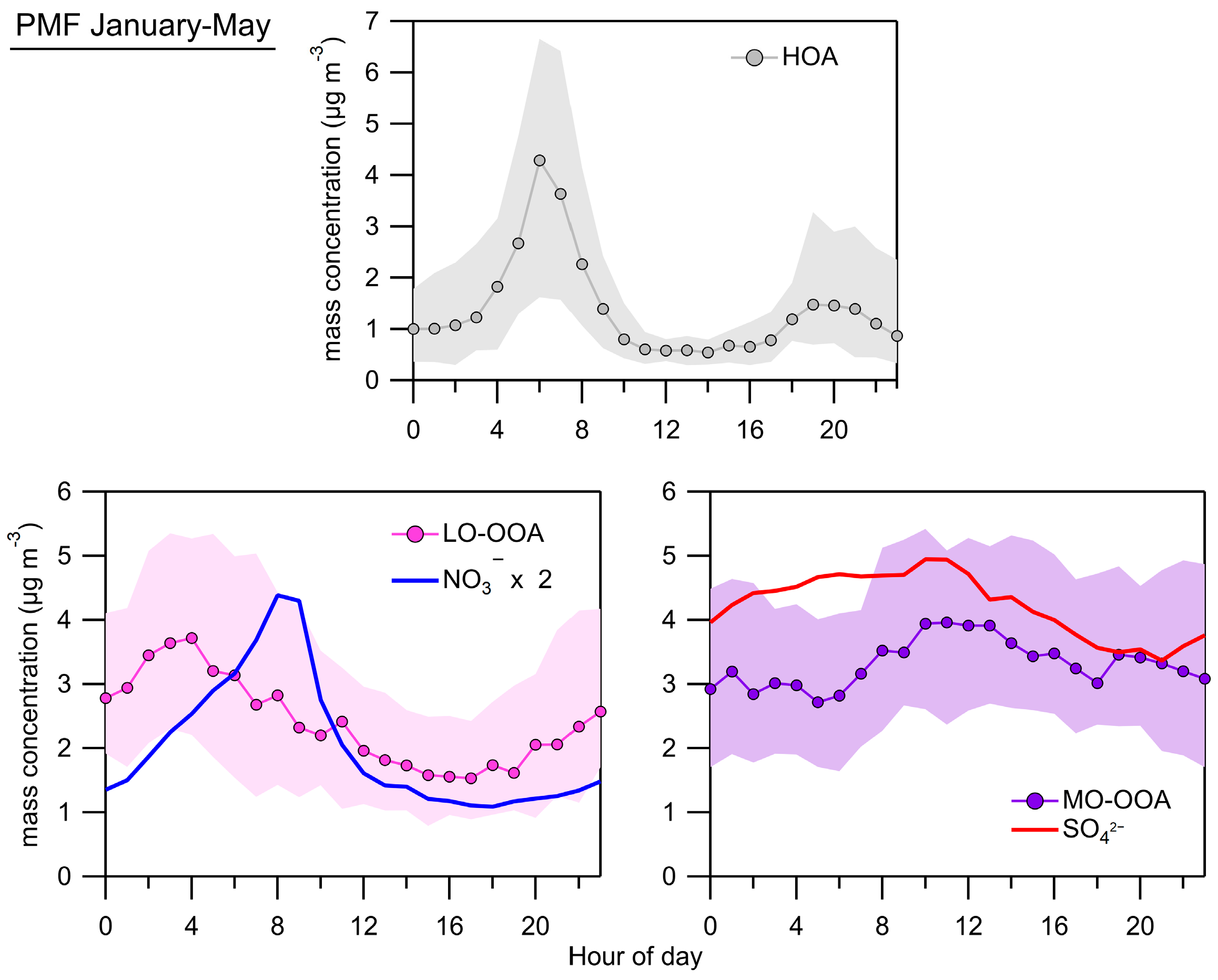
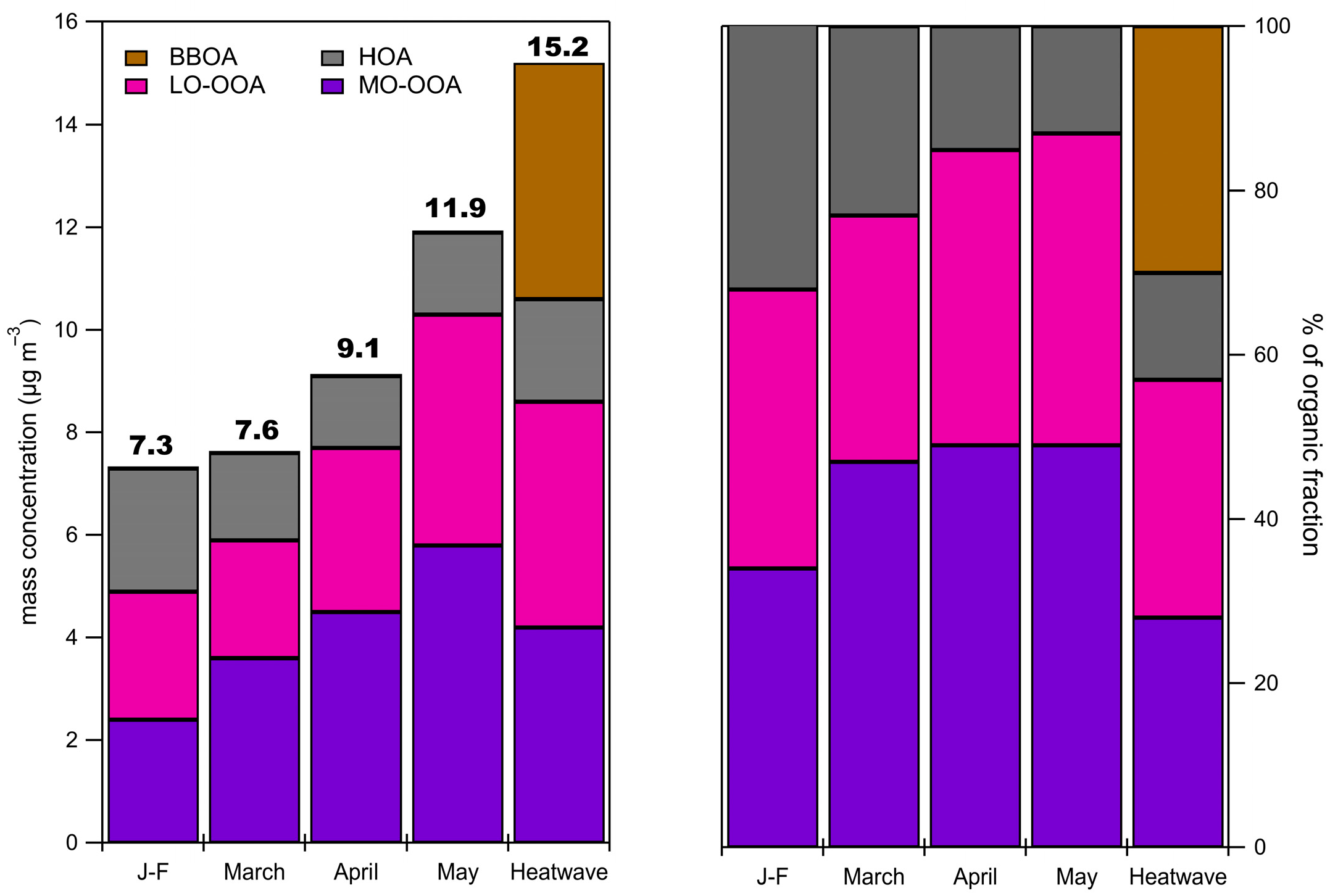
3.4. Organic Fraction of the NR-PM1
3.5. Organic Fraction During the Heatwave Period
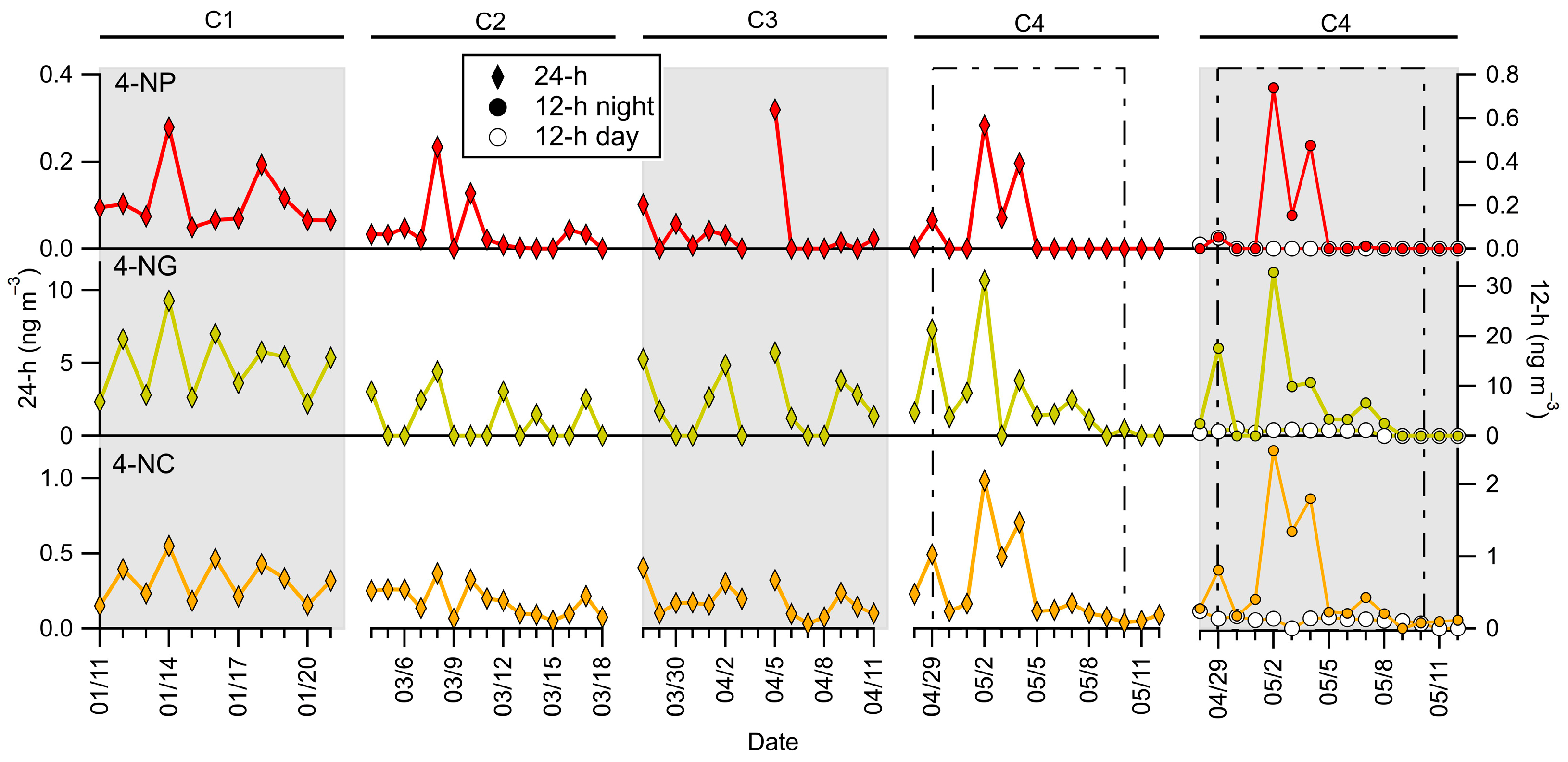
3.6. Nitroaromatic Compounds
3.7. HYSPLIT Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MAQ | Metropolitan Area of Queretaro |
| PM | Particulate Matter |
| NACs | Nitroaromatic Compounds |
| MCMA | Mexico City Metropolitan Area |
| NPs | Nitrophenols |
| NCs | Nitrocatechols |
| NSAs | Nitrosalicylic Acids |
| NGs | Nitroguaiacols |
| ACSM | Aerosol Speciation Chemical Monitor |
| HYSPLIT | Hybrid Single Particle Lagrangian Integrated Trajectory |
| GDL | Guadalajara |
| SR | Salamanca |
| UPLC-ESI-HR-QTOF MS | Ultra-performance liquid chromatography with electrospray ionization and high-resolution quadrupole time of flight mass spectrometry. |
| BLD | Below Limit Detection |
| RUOA | Atmospheric Observatories University Network (Spanish acronym) |
| JUR | Juriquilla |
| NR-PM1 | Non-refractory submicron particles |
| PMF | Positive Matrix Factorization |
| T | Temperature |
| RH | Relative Humidity |
| WS | Wind Speed |
| MDL | Method Detection Limit |
| NAM | North American Mesoscale |
| NCEP | National Center for Environmental Prediction |
| NOAA | National Oceanic and Atmospheric Agency |
| DC | Dry-Cold |
| DW | Dry-Warm |
| HOA | Hydrocarbon-like organic aerosol |
| LO-OOA | Less-oxidized oxygenated organic aerosol |
| MO-OOA | More-oxidized oxygenated organic aerosol |
| BBOA | Biomass burning organic aerosol |
| COA | Cooking organic aerosol |
| SMN | National Meteorological Service (Spanish acronym) |
| SD | Standard deviation |
References
- WHO. Ambient (Outdoor) Air Pollution. Available online: https://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health (accessed on 20 February 2025).
- Wu, S.; Deng, F.; Wei, H.; Huang, J.; Wang, X.; Hao, Y.; Zheng, C.; Qin, Y.; Lv, H.; Shima, M.; et al. Association of Cardiopulmonary Health Effects with Source-Appointed Ambient Fine Particulate in Beijing, China: A Combined Analysis from the Healthy Volunteer Natural Relocation (HVNR) Study. Environ. Sci. Technol. 2014, 48, 3438–3448. [Google Scholar] [CrossRef]
- Terzano, C.; Di Stefano, F.; Conti, V.; Graziani, E.; Petroianni, A. Air Pollution Ultrafine Particles: Toxicity beyond the Lung. Eur. Rev. Med. Pharmacol. Sci. 2010, 14, 809–821. [Google Scholar]
- Brook, R.D.; Rajagopalan, S.; Pope, C.A.; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. Particulate Matter Air Pollution and Cardiovascular Disease: An Update to the Scientific Statement from the American Heart Association. Circulation 2010, 121, 2331–2378. [Google Scholar] [CrossRef]
- Cachon, B.F.; Firmin, S.; Verdin, A.; Ayi-Fanou, L.; Billet, S.; Cazier, F.; Martin, P.J.; Aissi, F.; Courcot, D.; Sanni, A.; et al. Proinflammatory Effects and Oxidative Stress within Human Bronchial Epithelial Cells Exposed to Atmospheric Particulate Matter (PM2.5 and PM>2.5) Collected from Cotonou, Benin. Environ. Pollut. 2014, 185, 340–351. [Google Scholar] [CrossRef]
- IPCC. Clouds and Aerosols. In Climate Change 2013—The Physical Science Basis; Intergovernmental Panel on Climate Change, Ed.; Cambridge University Press: Cambridge, UK, 2013; pp. 571–658. [Google Scholar] [CrossRef]
- Koplik, E.V.; Umriukhin, P.E.; Konorova, I.L.; Terechina, O.L.; Michaleva, I.I.; Gannushkina, I.V.; Sudakov, K.V. Atmospheric Aerosols: Composition, Transformation, Climate and Health Effects. Zhurnal Nevrol. I Psihiatr. Im. S S Korsakova 2007, 107, 50–54. [Google Scholar] [CrossRef]
- Molina, L.T.; Madronich, S.; Gaffney, J.S.; Apel, E.; De Foy, B.; Fast, J.; Ferrare, R.; Herndon, S.; Jimenez, J.L.; Lamb, B.; et al. An Overview of the MILAGRO 2006 Campaign: Mexico City Emissions and Their Transport and Transformation. Atmos. Chem. Phys. 2010, 10, 8697–8760. [Google Scholar] [CrossRef]
- Rozanes-Valenzuela, D.A.; Magaldi, A.V.; Salcedo, D. Regional Flow Climatology for Central Mexico (Querétaro): A First Case Study. Atmosfera 2023, 36, 239–252. [Google Scholar] [CrossRef]
- Olivares-Salazar, S.E.; Alvarez-Ospina, H.; Aguillon-Vazquez, C.; Salcedo, D. Source Apportionment of Particulate Matter in the Metropolitan Area of Querétaro (Central Mexico): First Case Study. ACS Earth Space Chem. 2021, 5, 2347–2355. [Google Scholar] [CrossRef]
- Salcedo, D.; Alvarez-Ospina, H.; Olivares-Salazar, S.E.; Liñan-Abanto, R.N.; Castro, T. PM Chemical Characterization at a Semi-Arid Urban Environment in Central Mexico. Urban Clim. 2023, 52, 101723. [Google Scholar] [CrossRef]
- Bejan, I.G.; Olariu, R.I.; Wiesen, P. Secondary Organic Aerosol Formation from Nitrophenols Photolysis under Atmospheric Conditions. Atmosphere 2020, 11, 1346. [Google Scholar] [CrossRef]
- Tremp, J.; Mattrel, P.; Fingler, S.; Giger, W. Phenols and Nitrophenols as Tropospheric Pollutants: Emissions from Automobile Exhausts and Phase Transfer in the Atmosphere. Water Air Soil Pollut. 1993, 68, 113–123. [Google Scholar] [CrossRef]
- Chow, K.S.; Huang, X.H.H.; Yu, J.Z. Quantification of Nitroaromatic Compounds in Atmospheric Fine Particulate Matter in Hong Kong over 3 Years: Field Measurement Evidence for Secondary Formation Derived from Biomass Burning Emissions. Environ. Chem. 2016, 13, 665–673. [Google Scholar] [CrossRef]
- Ikemori, F.; Nakayama, T.; Hasegawa, H. Characterization and Possible Sources of Nitrated Mono- and Di-Aromatic Hydrocarbons Containing Hydroxyl and/or Carboxyl Functional Groups in Ambient Particles in Nagoya, Japan. Atmos. Environ. 2019, 211, 91–102. [Google Scholar] [CrossRef]
- Kahnt, A.; Behrouzi, S.; Vermeylen, R.; Safi Shalamzari, M.; Vercauteren, J.; Roekens, E.; Claeys, M.; Maenhaut, W. One-Year Study of Nitro-Organic Compounds and Their Relation to Wood Burning in PM10 Aerosol from a Rural Site in Belgium. Atmos. Environ. 2013, 81, 561–568. [Google Scholar] [CrossRef]
- Malagón, A. Directorio Empresarial PIQ. 2025. Available online: https://www.piq.com.mx/files/DIRECTORIO_EMPRESAS_PIQ_FEBRERO_2025.pdf (accessed on 18 May 2025).
- Red Universitaria De Observatorio Atmosféricos. RUOA. Available online: https://ruoa.unam.mx/estaciones/jqro/ (accessed on 18 May 2025).
- Jimenez, J.L.; Jayne, J.T.; Shi, Q.; Kolb, C.E.; Worsnop, D.R.; Yourshaw, I.; Seinfeld, J.H.; Flagan, R.C.; Zhang, X.; Smith, K.A.; et al. Ambient Aerosol Sampling Using the Aerodyne Aerosol Mass Spectrometer. J. Geophys. Res. Atmos. 2003, 108, 1–13. [Google Scholar] [CrossRef]
- Ng, N.L.; Herndon, S.C.; Trimborn, A.; Canagaratna, M.R.; Croteau, P.L.; Onasch, T.B.; Sueper, D.; Worsnop, D.R.; Zhang, Q.; Sun, Y.L. An aerosol chemical speciation monitor (ACSM) for routine monitoring of the composition and mass concentrations of ambient aerosol. Aerosol. Sci. Technol. 2011, 45, 780–794. [Google Scholar] [CrossRef]
- Canagaratna, M.R.; Jayne, J.T.; Jimenez, J.L.; Allan, J.D.; Alfarra, M.R.; Zhang, Q.; Onasch, T.B.; Drewnick, F.; Coe, H.; Middlebrook, A.; et al. Chemical and Microphysical Characterization of Ambient Aerosols with the Aerodyne Aerosol Mass Spectrometer. Mass Spectrom. Rev. 2007, 2, 185–222. [Google Scholar] [CrossRef]
- Paatero, P.; Tappert, U. Positive Matrix Factorization: A Non-Negative Factor Model with Optimal Utilization of Error Estimates of Data Values. Environmetrics 1994, 5, 111–126. [Google Scholar] [CrossRef]
- Ulbrich, I.M.; Canagaratna, M.R.; Zhang, Q.; Worsnop, D.R.; Jimenez, J.L. Interpretation of Organic Components from Positive Matrix Factorization of Aerosol Mass Spectrometric Data. Atmos. Chem. Phys. 2009, 9, 2891–2918. [Google Scholar] [CrossRef]
- Hopke, P.K. Review of Receptor Modeling Methods for Source Apportionment. J. Air Waste Manag. Assoc. 2016, 66, 237–259. [Google Scholar] [CrossRef] [PubMed]
- Paatero, P. User’s Guide for the Multilinear Engine Program “ME2” for Fitting Multilinear and Quasi-Multilinear Models; University of Helsinki, Department of Physics: Helsinki, Finland, 2000; pp. 1–24. [Google Scholar]
- Zhang, Q.; Jimenez, J.L.; Canagaratna, M.R.; Ulbrich, I.M.; Ng, N.L.; Worsnop, D.R.; Sun, Y. Understanding Atmospheric Organic Aerosols via Factor Analysis of Aerosol Mass Spectrometry: A Review. Anal. Bioanal. Chem. 2011, 401, 3045–3067. [Google Scholar] [CrossRef]
- Jiang, H.; Frie, A.L.; Lavi, A.; Chen, J.Y.; Zhang, H.; Bahreini, R.; Lin, Y.H. Brown Carbon Formation from Nighttime Chemistry of Unsaturated Heterocyclic Volatile Organic Compounds. Environ. Sci. Technol. Lett. 2019, 6, 184–190. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, K.; Zhang, H.; Lin, Y.-H.; Bahreini, R. Chemical Composition and Optical Properties of Secondary Organic Aerosol from Photooxidation of Volatile Organic Compound Mixtures. ACS EST Air 2024, 1, 247–258. [Google Scholar] [CrossRef]
- Kitanovski, Z.; Grgić, I.; Vermeylen, R.; Claeys, M.; Maenhaut, W. Liquid Chromatography Tandem Mass Spectrometry Method for Characterization of Monoaromatic Nitro-Compounds in Atmospheric Particulate Matter. J. Chromatogr. A 2012, 1268, 35–43. [Google Scholar] [CrossRef]
- Gegenschatz, S.A.; Chiappini, F.A.; Teglia, C.M.; de la Peña, A.M.; Goicoechea, H.C. Binding the Gap between Experiments, Statistics, and Method Comparison: A Tutorial for Computing Limits of Detection and Quantification in Univariate Calibration for Complex Samples. Anal. Chim. Acta 2021, 1209, 339342. [Google Scholar] [CrossRef]
- Rolph, G.; Stein, A.; Stunder, B. Real-Time Environmental Applications and Display SYstem: READY. Environ. Model. Softw. 2017, 95, 210–228. [Google Scholar] [CrossRef]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics from Air Pollution to Climate Change, 2nd ed.; Publication; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Zhang, Q.; Canagaratna, M.R.; Jayne, J.T.; Worsnop, D.R.; Jimenez, J.L. Time- and Size-Resolved Chemical Composition of Submicron Particles in Pittsburgh: Implications for Aerosol Sources and Processes. J. Geophys. Res. Atmos. 2005, 110, 1–19. [Google Scholar] [CrossRef]
- Lanz, V.A.; Alfarra, M.R.; Baltensperger, U.; Buchmann, B.; Hueglin, C.; Prévôt, A.S.H. Source Apportionment of Submicron Organic Aerosols at an Urban Site by Factor Analytical Modelling of Aerosol Mass Spectra. Atmos. Chem. Phys. 2007, 7, 1503–1522. [Google Scholar] [CrossRef]
- Atabakhsh, S.; Poulain, L.; Chen, G.; Canonaco, F.; Prévôt, A.S.H.; Pöhlker, M.; Wiedensohler, A.; Herrmann, H. A 1-Year Aerosol Chemical Speciation Monitor (ACSM) Source Analysis of Organic Aerosol Particle Contributions from Anthropogenic Sources after Long-Range Transport at the TROPOS Research Station Melpitz. Atmos. Chem. Phys. 2023, 23, 6963–6988. [Google Scholar] [CrossRef]
- Ng, N.L.; Canagaratna, M.R.; Zhang, Q.; Jimenez, J.L.; Tian, J.; Ulbrich, I.M.; Kroll, J.H.; Docherty, K.S.; Chhabra, P.S.; Bahreini, R.; et al. Organic Aerosol Components Observed in Northern Hemispheric Datasets from Aerosol Mass Spectrometry. Atmos. Chem. Phys. 2010, 10, 4625–4641. [Google Scholar] [CrossRef]
- Simoneit, B.R.T.; Schauer, J.J.; Nolte, C.G.; Oros, D.R.; Elias, V.O.; Fraser, M.P.; Rogge, W.F.; Cass, G.R. Levoglucosan, a Tracer for Cellulose in Biomass Burning and Atmospheric Particles. Atmos. Environ. 1999, 33, 173–182. [Google Scholar] [CrossRef]
- Jeon, S.; Walker, M.J.; Sueper, D.T.; Day, D.A.; Handschy, A.V.; Jimenez, J.L.; Williams, B.J. A Searchable Database and Mass Spectral Comparison Tool for Aerosol Mass Spectrometry (AMS) and Aerosol Chemical Speciation Monitor (ACSM). EGUsphere 2023, 16, 6075–6095. [Google Scholar] [CrossRef]
- Al-Naiema, I.M.; Stone, E.A. Evaluation of Anthropogenic Secondary Organic Aerosol Tracersfrom Aromatic Hydrocarbons. Atmos. Chem. Phys. 2017, 17, 2053–2065. [Google Scholar] [CrossRef]
- Xie, M.; Chen, X.; Holder, A.L.; Hays, M.D.; Lewandowski, M.; Offenberg, J.H.; Kleindienst, T.E.; Jaoui, M.; Hannigan, M.P. Light Absorption of Organic Carbon and Its Sources at a Southeastern U.S. Location in Summer. Environ. Pollut. 2018, 244, 38–46. [Google Scholar] [CrossRef]
- Lauraguais, A.; Coeur-Tourneur, C.; Cassez, A.; Deboudt, K.; Fourmentin, M.; Choël, M. Atmospheric Reactivity of Hydroxyl Radicals with Guaiacol (2-Methoxyphenol), a Biomass Burning Emitted Compound: Secondary Organic Aerosol Formation and Gas-Phase Oxidation Products. Atmos. Environ. 2014, 86, 155–163. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, S.; Wang, G.; Lv, S.; Li, D.; Liu, L.; Li, J.; Liu, S.; Du, W.; Meng, J.; et al. Efficient Heterogeneous Formation of Ammonium Nitrate on the Saline Mineral Particle Surface in the Atmosphere of East Asia during Dust Storm Periods. Environ. Sci. Technol. 2020, 54, 15622–15630. [Google Scholar] [CrossRef]
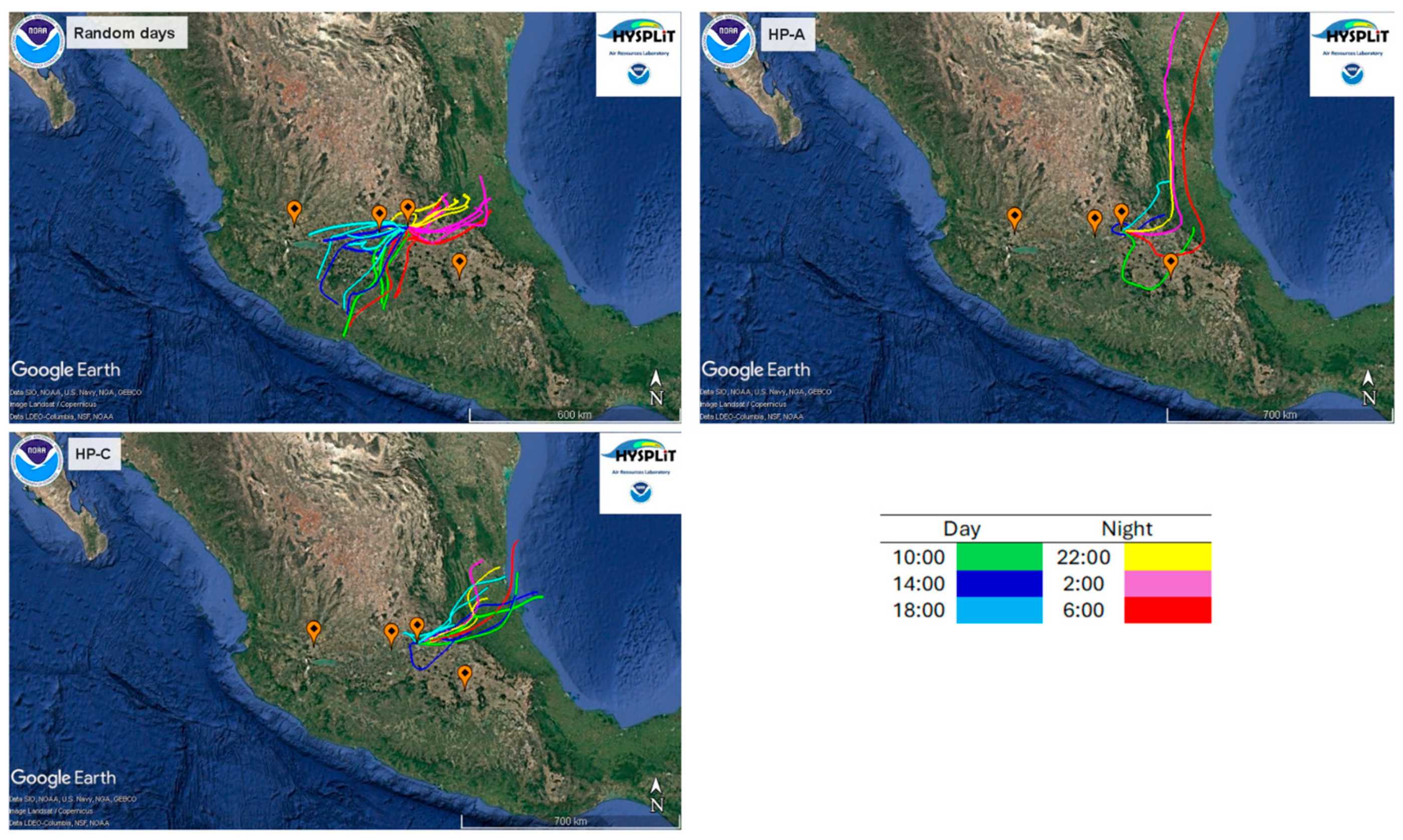

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Campaign | Sampling Periods | Number of Samples |
|---|---|---|
| C1 | 7–21 January 2022 | 11 24 h samples |
| C2 | 4–18 March 2022 | 15 24 h samples |
| C3 | 28 March–11 April 2022 | 14 24 h samples |
| C4 | 28 April–12 May 2022 | 15 24 h samples 15 12 h day samples 15 12 h night samples |
| Compounds | M (g mol−1) | Measured m/z [M − H]− | MDL (ng mL−1) | Retention Time (min) |
|---|---|---|---|---|
| 4-Nitrocatechol (4-NC) | 155 | 154 | 11.15 | 7.5–9.5 |
| 4-Nitrophenol (4-NP) | 139 | 138 | 3.6 | 8.5–10 |
| 4-Nitroguaicol (4-NG) | 169 | 168 | 16.47 | 9–10 |
| 5-Nitrosalicilyc acid (5-NSA) | 183 | 182 | 4.49 | 9–9.7 |
| JUR–2022 | JUR–2015 | ||||||
|---|---|---|---|---|---|---|---|
| 1 January–15 May 2022 | Heatwave | 1 March 2015–29 February 2016 | |||||
| DC | DW | ||||||
| Mean (SD) (µg m−3) | % | Mean (µg m−3) | Mean (SD) (µg m−3) | (%) | Mean (SD) (µg m−3) | % | |
| Organics | 8.0 (4.4) | 51 | 14.20 (5.8) | 5.4 (3.9) | 50 | 3.3 (2.1) | 41 |
| SO42− | 4.8 (2.9) | 30 | 8.24 (2.6) | 2.7 (1.9) | 25 | 2.9 (1.9) | 37 |
| NO3− | 1.2 (1.0) | 11 | 1.55 (1.1) | 1.2 (1.2) | 11 | 0.6 (0.6) | 7.5 |
| NH4+ | 1.7 (1.0) | 7 | 2.83 (0.9) | 1.3 (0.9) | 12 | 1.1 (0.7) | 14 |
| Cl− | 0.2 (0.4) | 1 | 0.11 (0.2) | 0.19 (0.5) | 2 | 0.05 (0.1) | 0.6 |
| NR-PM1 | 15.9 (7.8) | 100 | 26.93 (7.9) | 10.8 (6.9) | 100 | 7.9 (4.6) | 100 |
| Other Sites | ||||||||
|---|---|---|---|---|---|---|---|---|
| C1 | C2 | C3 | C4 | Heatwave | NC-USA [41] | Iowa-USA [40] | Nagoya-Japan [15] | |
| ng m−3 | ng m−3 | |||||||
| 4-NP | 0.10 (0.07) | 0.05 (0.06) | 0.07 (0.1) | 0.12 (0.1) | 0.15 (0.1) | 0.018–0.12 | 0.63 | -- |
| 4-NC | 0.31 (0.1) | 0.18 (0.1) | 0.18 (0.1) | 0.26 (0.2) | 0.29 (0.3) | 0.057–0.16 | 1.6 | 0.74–6.8 |
| 4-NG | 4.83 (2.6) | 2.82 (0.9) | 3.26 (1.7) | 3.13 (3.1) | 3.29 (3.2) | - | 0.08 | 0.037–0.55 |
| 5-NSA | BDL | BDL | BDL | BDL | BDL | - | - | - |
| Campaign 1 | Campaign 2 | ||||||||||||||
| 4-NC | 4-NG | 4-NP | LO-OOA | m/z 60 | NO3− | HOA | 4-NC | 4-NG | 4-NP | LO-OOA | m/z 60 | NO3− | HOA | BBOA | |
| 4-NC | 1 | 1 | |||||||||||||
| 4-NG | 0.98 | 1 | 0.44 | 1 | |||||||||||
| 4-NP | 0.71 | 0.70 | 1 | 0.78 | 0.45 | 1 | |||||||||
| LO-OOA | 0.71 | 0.67 | 0.68 | 1 | 0.25 | 0.08 | 0.23 | 1 | |||||||
| m/z 60 | 0.72 | 0.64 | 0.55 | 0.67 | 1 | −0.07 | 0.10 | −0.27 | 0.56 | 1 | |||||
| NO3 | 0.47 | 0.53 | 0.49 | 0.56 | 0.59 | 1 | 0.02 | 0.27 | −0.18 | 0.66 | 0.85 | 1 | |||
| HOA | 0.22 | 0.17 | 0.08 | −0.19 | 0.39 | −0.1 | 1 | 0.28 | 0.50 | 0.09 | 0.32 | 0.48 | 0.43 | 1 | |
| Campaign 3 | Campaign 4/Heatwave | ||||||||||||||
| 4-NC | 4-NG | 4-NP | LO-OOA | m/z 60 | NO3− | HOA | 4-NC | 4-NG | 4-NP | LO-OOA | m/z 60 | NO3− | HOA | BBOA | |
| 4-NC | 1 | 1 | |||||||||||||
| 4-NG | 0.80 | 1 | 0.82 | 1 | |||||||||||
| 4-NP | 0.60 | 0.63 | 1 | 0.97 | 0.78 | 1 | |||||||||
| LO-OOA | 0.58 | 0.42 | 0.28 | 1 | 0.87 | 0.79 | 0.82 | 1 | |||||||
| m/z 60 | 0.65 | 0.42 | 0.50 | 0.80 | 1 | 0.66 | 0.65 | 0.52 | 0.83 | 1 | |||||
| NO3 | 0.82 | 0.59 | 0.30 | 0.84 | 0.73 | 1 | 0.90 | 0.78 | 0.80 | 0.95 | 0.86 | 1 | |||
| HOA | 0.74 | 0.42 | 0.30 | 0.74 | 0.33 | 0.86 | 1 | 0.91 | 0.83 | 0.83 | 0.92 | 0.74 | 0.95 | 1 | |
| BBOA | 0.82 | 0.91 | 0.72 | 0.84 | 0.85 | 0.87 | 0.86 | 1 | |||||||
| HYSPLIT Modeling Period | Reference Periods | ||
|---|---|---|---|
| Previous Days | Subsequent Days | ||
| HP-A | 28 January, 13:00 to 29, 12:00 | 26–27 January | 30–31 January |
| HP-B | 7 April, 06:00 to 9, 23:00 | 4–5 April | 10–11 April |
| HP-C | 26 April, 08:00 to 28, 09:00 | 24–25 April | 29–30 April |
| HP-D | 10 May, 12:00 to 13, 05:00 | 8–9 May | 14–15 May |
| Random days | 5 January, 9 February, 20 March, 20 April, 4 May | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivares-Salazar, S.E.; Bahreini, R.; Lin, Y.-H.; Castro, T.; Alvarez-Ospina, H.; Salcedo, D. Aerosol Composition in a Semi-Urban Environment in Central Mexico: Influence of Local and Regional Processes on Overall Composition and First Quantification of Nitroaromatics. Atmosphere 2025, 16, 827. https://doi.org/10.3390/atmos16070827
Olivares-Salazar SE, Bahreini R, Lin Y-H, Castro T, Alvarez-Ospina H, Salcedo D. Aerosol Composition in a Semi-Urban Environment in Central Mexico: Influence of Local and Regional Processes on Overall Composition and First Quantification of Nitroaromatics. Atmosphere. 2025; 16(7):827. https://doi.org/10.3390/atmos16070827
Chicago/Turabian StyleOlivares-Salazar, Sara E., Roya Bahreini, Ying-Hsuan Lin, Telma Castro, Harry Alvarez-Ospina, and Dara Salcedo. 2025. "Aerosol Composition in a Semi-Urban Environment in Central Mexico: Influence of Local and Regional Processes on Overall Composition and First Quantification of Nitroaromatics" Atmosphere 16, no. 7: 827. https://doi.org/10.3390/atmos16070827
APA StyleOlivares-Salazar, S. E., Bahreini, R., Lin, Y.-H., Castro, T., Alvarez-Ospina, H., & Salcedo, D. (2025). Aerosol Composition in a Semi-Urban Environment in Central Mexico: Influence of Local and Regional Processes on Overall Composition and First Quantification of Nitroaromatics. Atmosphere, 16(7), 827. https://doi.org/10.3390/atmos16070827