

Diversity Analysis of Fungi Distributed in Inhalable and Respirable Size Fractions of Aerosols: A Report from Kuwait

 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling Site
2.2. DNA Isolation and Amplicon Polymerase Chain Reactions
2.3. Targeted Amplicon (ITS3-ITS4) Sequencing and Analysis
2.4. Taxonomic Profiling and Diversity Analysis
3. Results
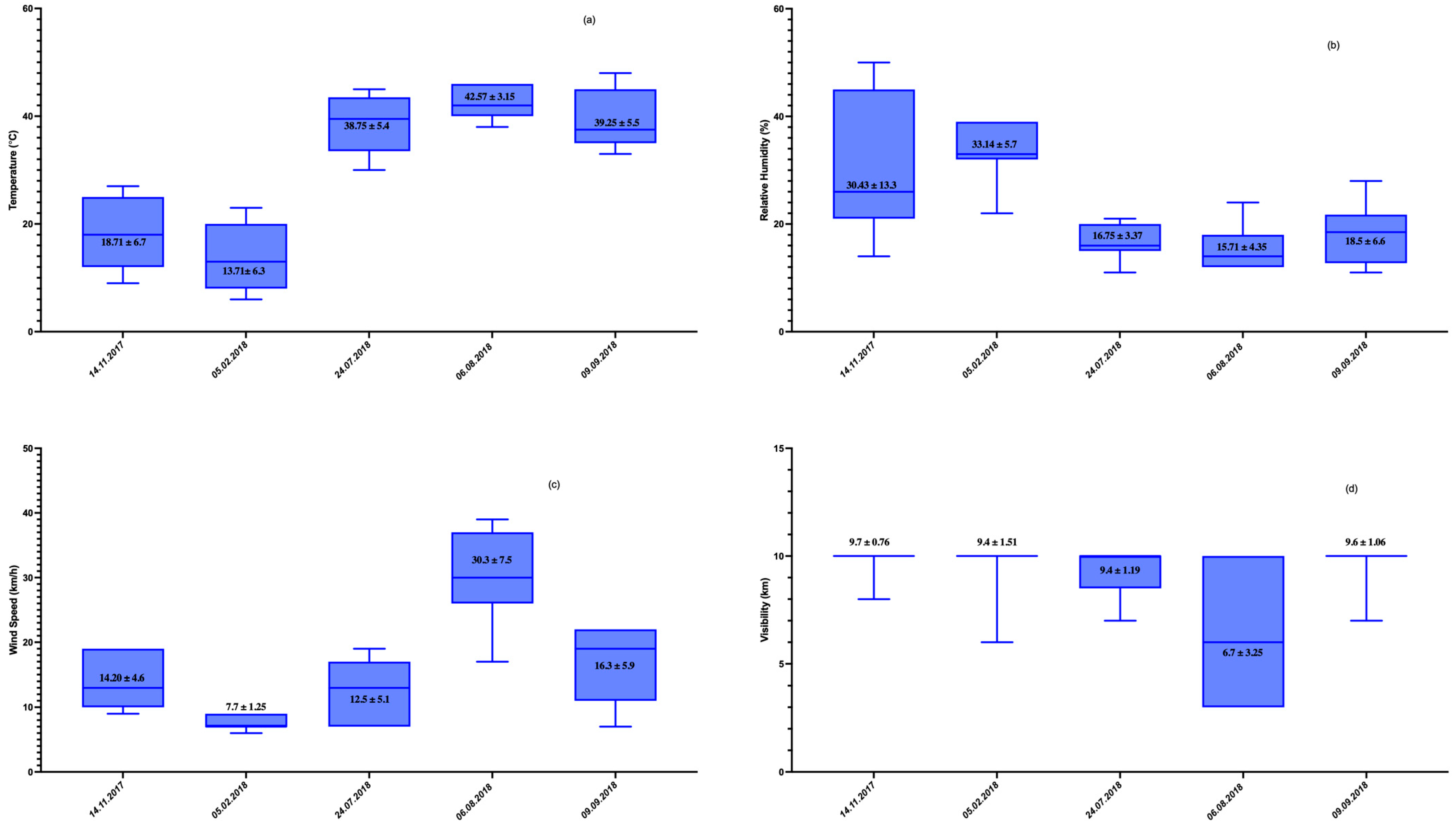
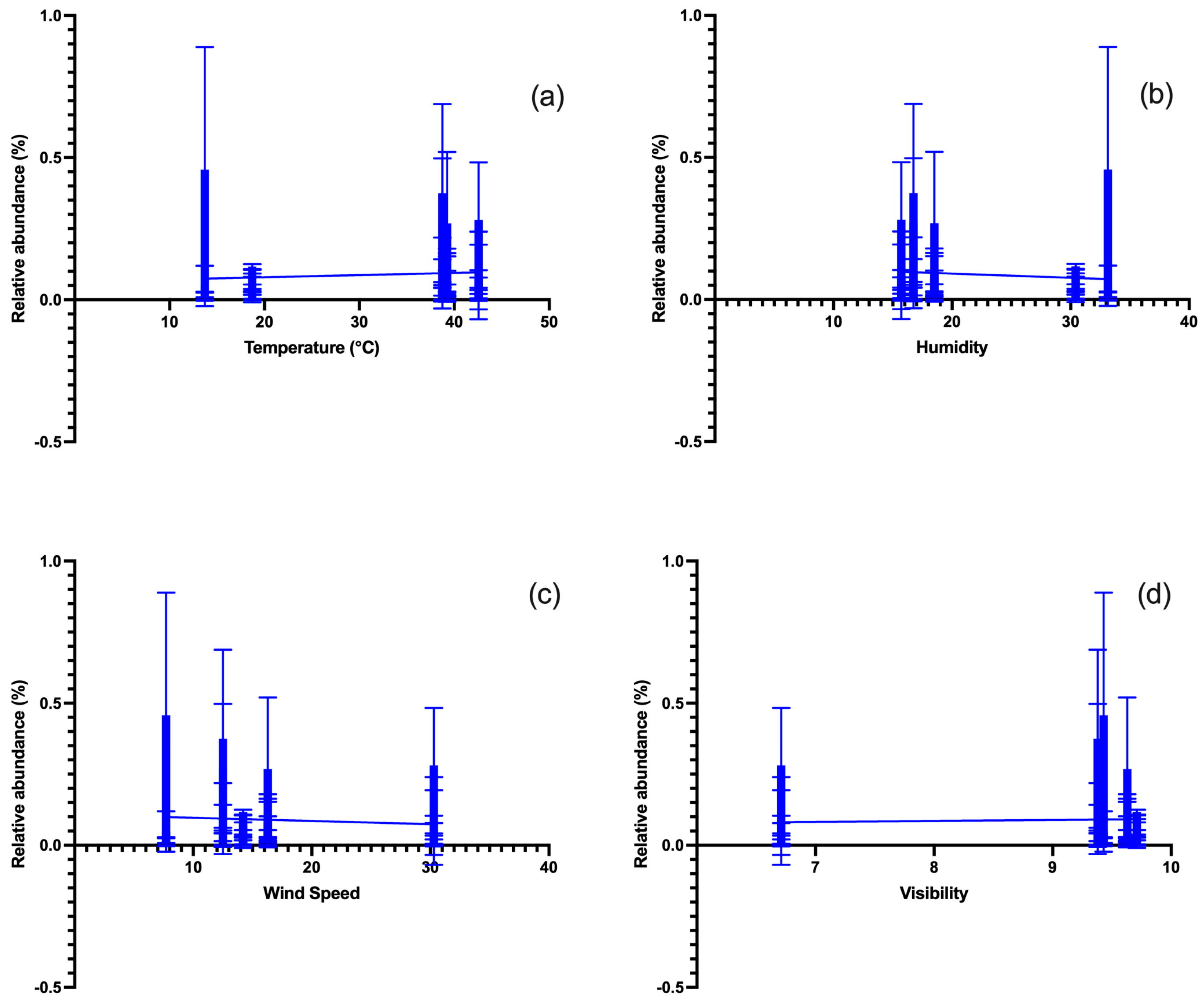
3.1. Meteorological Factors
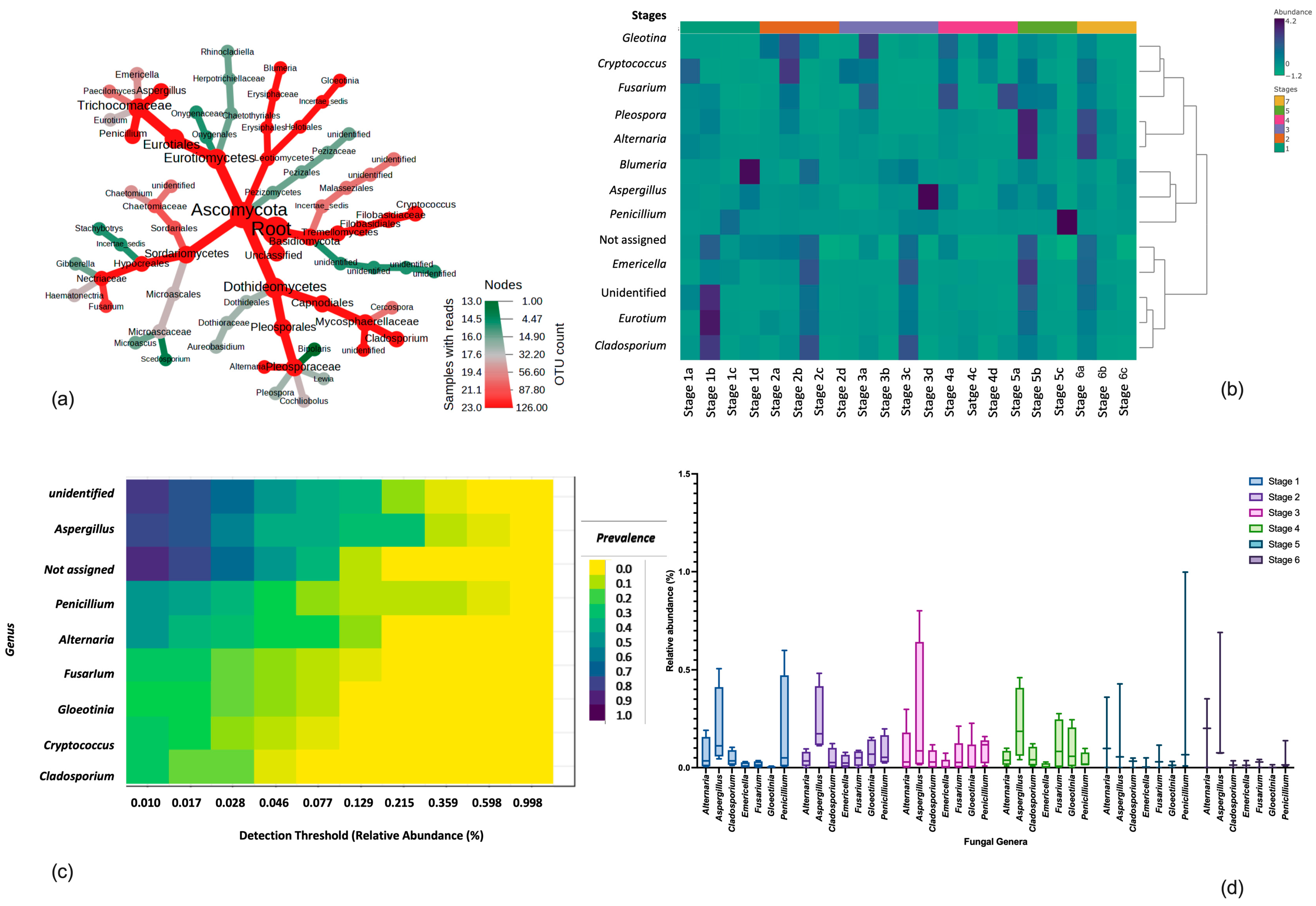
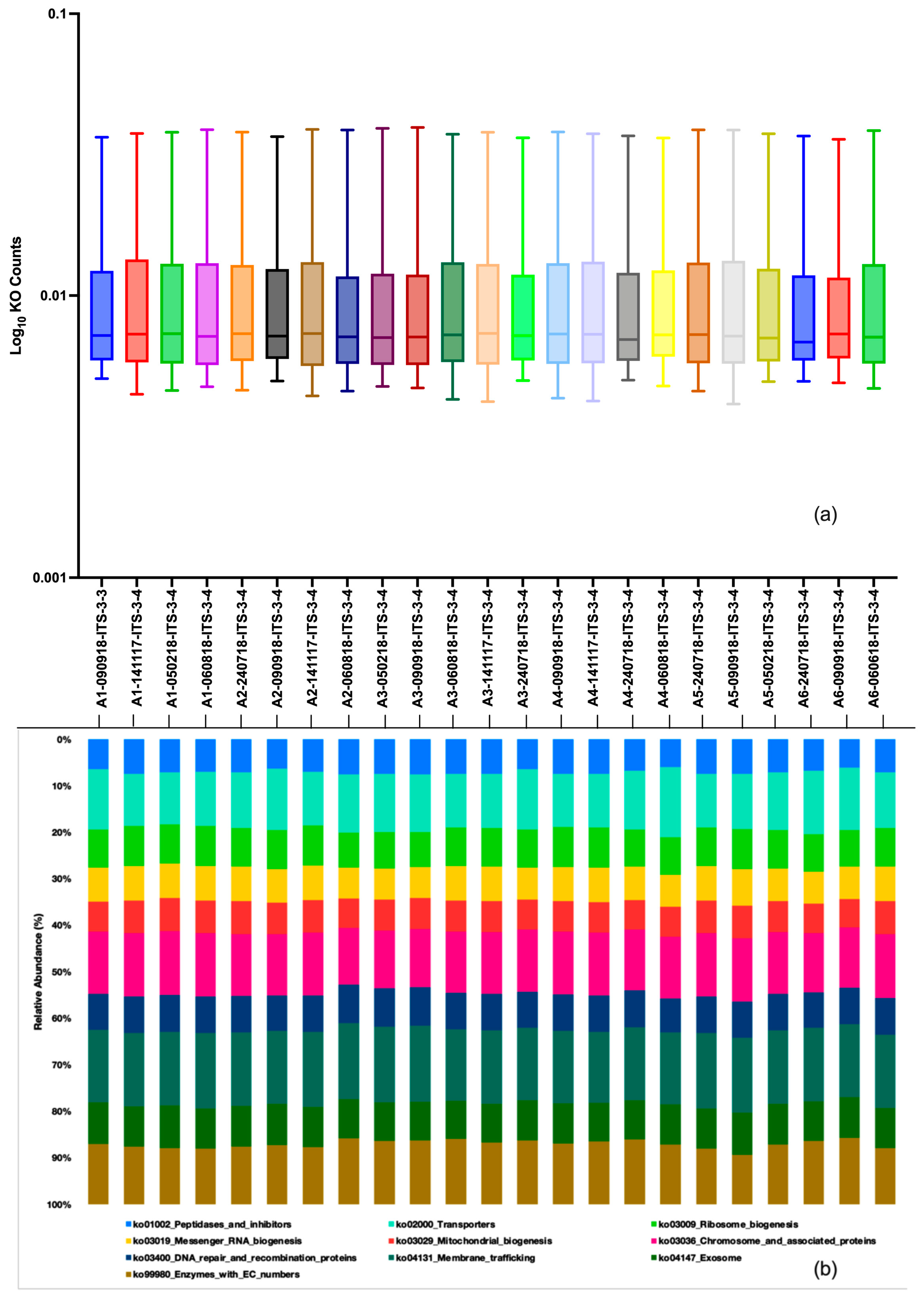
3.2. Taxonomic Composition, Core Fungal Mycobiome and Dominant Fungal Genera in Six-Size Fractions
3.3. Differential Abundance between Different Size Fractions
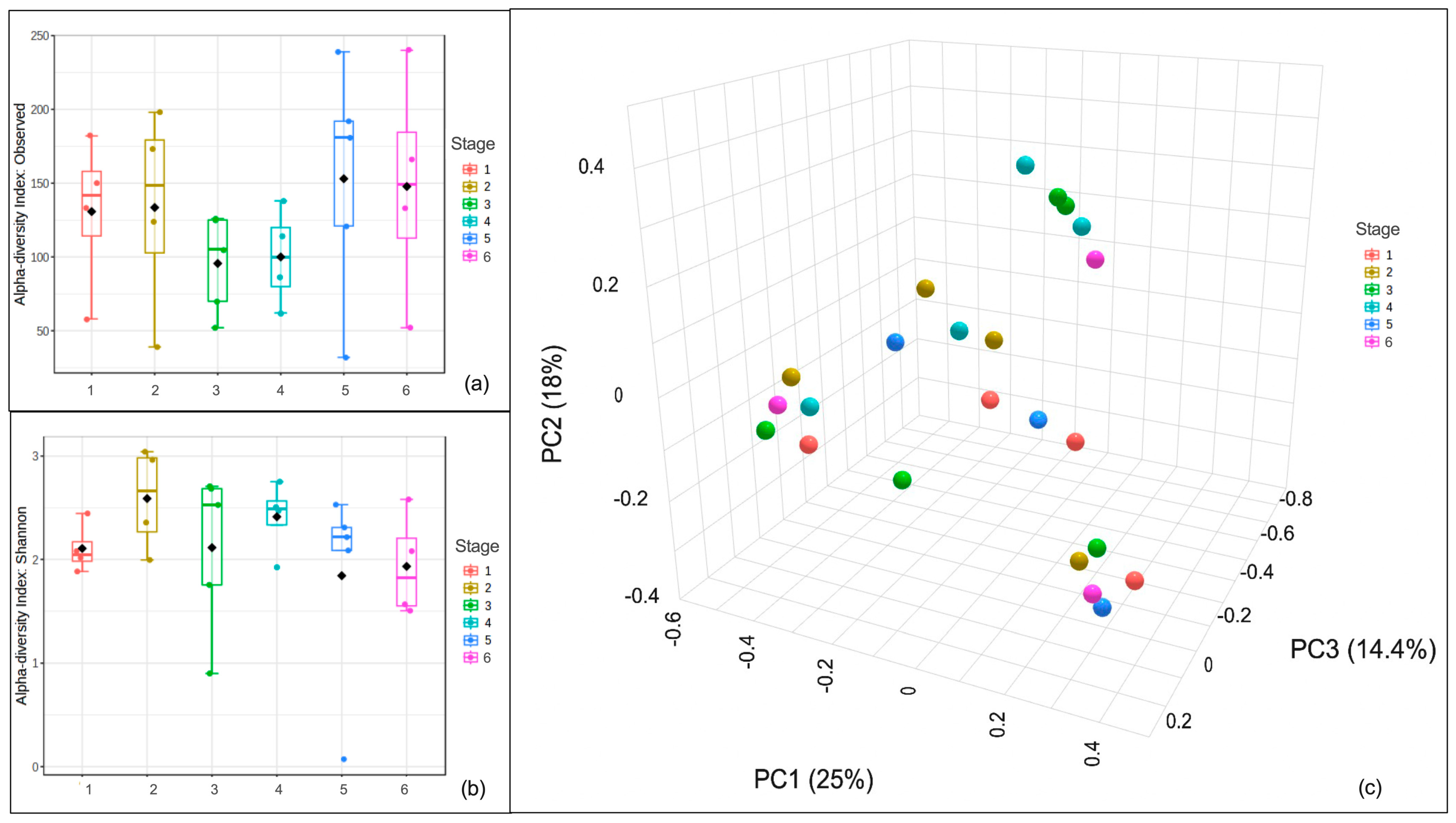
3.4. Species Richness and Community Structure of the Mycobiomes
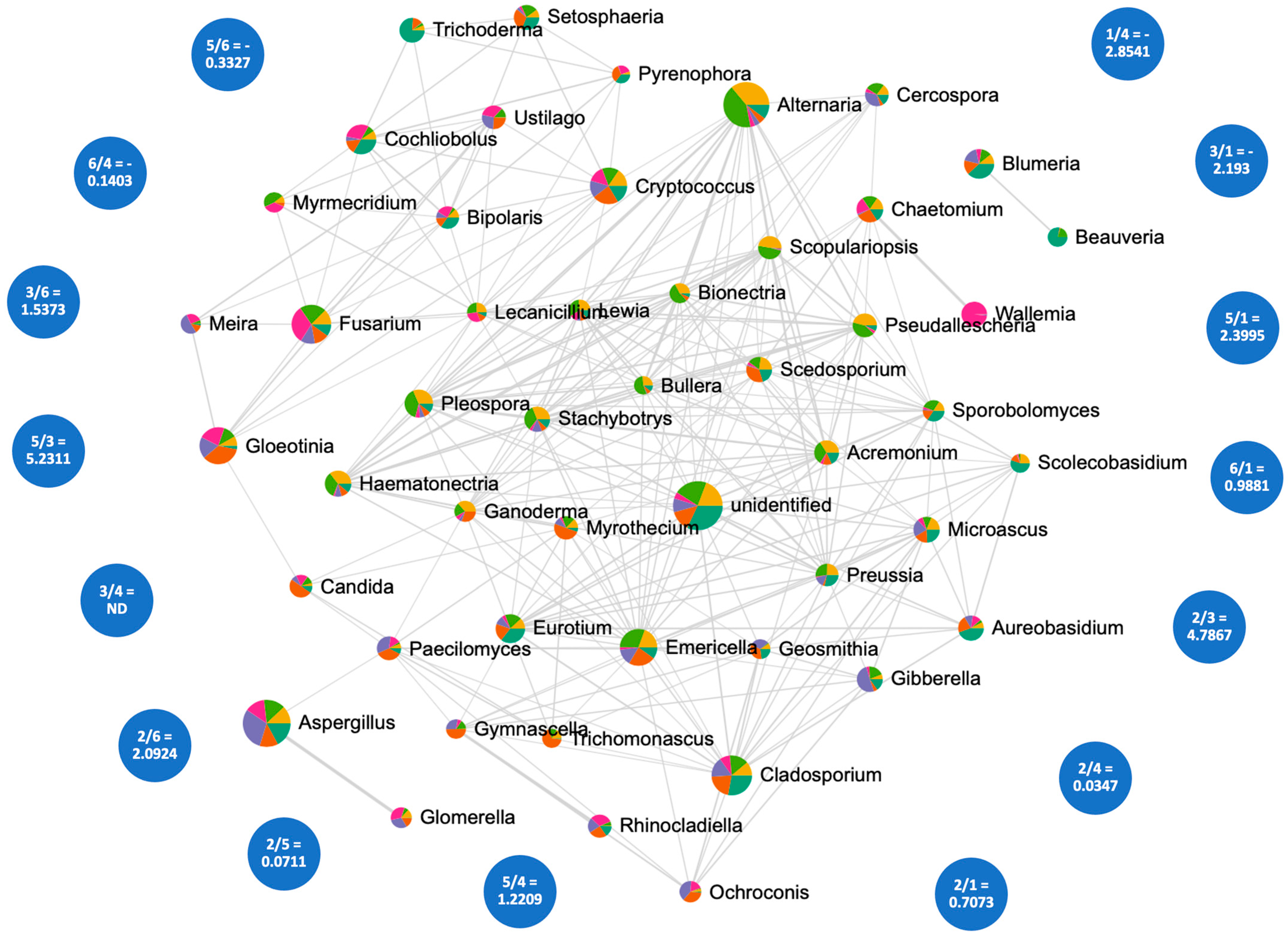
3.5. Correlation Analysis
3.6. Comparisons between Respirable and Inhalable Fractions of Air
3.7. Functional Annotations of Fungal Forms
3.8. Multivariate Analysis between Environmental Parameters and Fungal OTUs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adhikari, A.; Sen, M.M.; Guptabhattacharya, S.; Chanda, S. Airborne viable, non-viable, and allergenic fungi in a rural agricultural area of India: A 2-year study at five outdoor sampling stations. Sci. Total Environ. 2004, 326, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.S.; Wathes, C.M. Bioaerosols Handbook; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- D’Amato, G. Effects of climatic changes and urban air pollution on the rising trends of respiratory allergy and asthma. Multidiscip. Respir. Med. 2019, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Barberán, A.; Ladau, J.; Leff, J.W.; Pollard, K.S.; Menninger, H.L.; Dunn, R.R.; Fierer, N. Continental-scale distributions of dust-associated bacteria and fungi. Proc. Natl. Acad. Sci. USA 2015, 112, 5756–5761. [Google Scholar] [CrossRef] [PubMed]
- Sadyś, M.; Skjøth, C.; Kennedy, R. Back-trajectories show export of airborne fungal spores (Ganoderma sp.) from forests to agricultural and urban areas in England. Atmos. Environ. 2014, 84, 88–99. [Google Scholar] [CrossRef]
- Singh, A.B.; Singh, A.; Pandit, T. Respiratory diseases among agricultural industry workers in India: A cross-sectional epidemiological study. Ann. Agric. Environ. Med. 1999, 6, 115–126. [Google Scholar] [PubMed]
- Younis, F.; Salem, E. Respiratory health disorders associated with occupational exposure to bioaerosols among workers in poultry breeding farms. Environ. Sci. Pollut. Res. 2020, 27, 19869–19876. [Google Scholar] [CrossRef] [PubMed]
- Hai, V.D.; Hoang, S.M.T.; Hung, N.T.Q.; Ky, N.M.; Gwi-Nam, B.; Ki-hong, P.; Chang, S.W.; Bach, Q.-V.; Nhu-Trang, T.-T.; Nguyen, D.D. Characteristics of airborne bacteria and fungi in the atmosphere in Ho Chi Minh City, Vietnam—A case study over three years. Int. Biodeterior. Biodegrad. 2019, 145, 104819. [Google Scholar] [CrossRef]
- Chow, J.C.; Yang, X.; Wang, X.; Kohl, S.D.; Hurbain, P.R.; Chen, L.A.; Watson, J.G. Characterization of Ambient PM10 Bioaerosols in a California Agricultural Town. Aerosol Air Qual. Res. 2015, 15, 1433–1447. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Al Salameen, F.; Habibi, N.; Uddin, S.; Al Mataqi, K.; Kumar, V.; Al Doaij, B.; Al Amad, S.; Al Ali, E.; Shirshikhar, F. Spatio-temporal variations in bacterial and fungal community associated with dust aerosol in Kuwait. PLoS ONE 2020, 15, e0241283. [Google Scholar] [CrossRef]
- Habibi, N.; Uddin, S.; Al Salameen, F.; Behbehani, M.; Shirshikhar, F.; Razzack, N.A.; Shajan, A.; Hussain, F.Z. Collection of Bacterial Community Associated with Size Fractionated Aerosols from Kuwait. Data 2021, 6, 123. [Google Scholar] [CrossRef]
- Habibi, N.; Uddin, S.; Behbehani, M.; Al Salameen, F.; Razzack, N.A.; Zakir, F.; Shajan, A.; Alam, F. Bacterial and Fungal Communities in Indoor Aerosols from Two Kuwaiti Hospitals. Front. Microbiol. 2022, 13, 955913. [Google Scholar] [CrossRef] [PubMed]
- Núñez, A.; Amo de Paz, G.; Rastrojo, A.; García, A.M.; Alcamí, A.; Gutiérrez-Bustillo, A.M.; Moreno, D.A. Monitoring of airborne biological particles in outdoor atmosphere. Part 2: Metagenomics applied to urban environments. Int. Microbiol. 2016, 19, 69–80. [Google Scholar] [PubMed]
- Núñez, A.; de Paz, G.A.; Rastrojo, A.; Ferencova, Z.; Gutiérrez-Bustillo, A.M.; Alcamí, A.; Moreno, D.A.; Guantes, R. Temporal patterns of variability for prokaryotic and eukaryotic diversity in the urban air of Madrid (Spain). Atmos. Environ. 2019, 217, 116972. [Google Scholar] [CrossRef]
- Uddin, S.; Habibi, N.; Fowler, S.W.; Behbehani, M.; Gevao, B.; Faizuddin, M.; Gorgun, A.U. Aerosols as Vectors for Contaminants: A Perspective Based on Outdoor Aerosol Data from Kuwait. Atmosphere 2023, 14, 470. [Google Scholar] [CrossRef]
- Thalib, L.; Al-Taiar, A. Dust storms and the risk of asthma admissions to hospitals in Kuwait. Sci. Total Environ. 2012, 433, 347–351. [Google Scholar] [CrossRef]
- Al-Awadhi, J.M. Dust fallout characteristics in Kuwait: A case study. Kuwait J. Sci. Eng. 2005, 32, 135. [Google Scholar]
- Khadadah, M. The cost of asthma in Kuwait. Med. Princ. Pract. 2013, 22, 87–91. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J.; Walsh, D.C.I. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol. Monogr. 2013, 83, 557–574. [Google Scholar] [CrossRef]
- Watts, S.C.; Ritchie, S.; Inouye, M.; Holt, K.E. FastSpar: Rapid and scalable correlation estimation for compositional data. Bioinformatics 2018, 35, 1064–1066. [Google Scholar] [CrossRef] [PubMed]
- Soares, R.C.; Camargo-Penna, P.H.; de Moraes, V.C.S.; De Vecchi, R.; Clavaud, C.; Breton, L.; Braz, A.S.K.; Paulino, L.C. Dysbiotic bacterial and fungal communities not restricted to clinically affected skin sites in Dandruff. Front. Cell. Infect. Microbiol. 2016, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Krivonos, D.V.; Konanov, D.N.; Ilina, E.N. FunFun: ITS-based functional annotator of fungal communities. Ecol. Evol. 2023, 13, e9874. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, M. The Fungi: 1, 2, 3… 5.1 million species? Am. J. Bot. 2011, 98, 426–438. [Google Scholar] [CrossRef]
- Huffnagle, G.B.; Noverr, M.C. The emerging world of the fungal microbiome. Trends Microbiol. 2013, 21, 334–341. [Google Scholar] [CrossRef]
- Baxi, S.N.; Portnoy, J.M.; Larenas-Linnemann, D.; Phipatanakul, W.; Barnes, C.; Baxi, S.; Grimes, C.; Horner, W.E.; Kennedy, K.; Larenas-Linnemann, D.; et al. Exposure and health effects of fungi on humans. J. Allergy Clin. Immunol. Pract. 2016, 4, 396–404. [Google Scholar] [CrossRef]
- Fröhlich-Nowoisky, J.; Pickersgill, D.A.; Després, V.R.; Pöschl, U. High diversity of fungi in air particulate matter. Proc. Natl. Acad. Sci. USA 2009, 106, 12814–12819. [Google Scholar] [CrossRef]
- Egidi, E.; Delgado-Baquerizo, M.; Plett, J.M.; Wang, J.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K. A few Ascomycota taxa dominate soil fungal communities worldwide. Nat. Commun. 2019, 10, 2369. [Google Scholar] [CrossRef]
- Szoboszlay, M.; Schramm, L.; Pinzauti, D.; Scerri, J.; Sandionigi, A.; Biazzo, M. Nanopore is preferable over Illumina for 16S amplicon sequencing of the gut microbiota when species-level taxonomic classification, accurate estimation of richness, or focus on rare taxa Is required. Microorganisms 2023, 11, 804. [Google Scholar] [CrossRef] [PubMed]
- Abu-Dieyeh, M.H.; Barham, R.; Abu-Elteen, K.; Al-Rashidi, R.; Shaheen, I. Seasonal variation of fungal spore populations in the atmosphere of Zarqa area, Jordan. Aerobiologia 2010, 26, 263–276. [Google Scholar] [CrossRef]
- Al-Barakah, F.; Radwan, S.; Modaihsh, A. Seasonal and spatial variation of microbial contents in falling dust in Riyadh city, Saudi Arabia. Int. J. Curr. Microbiol. Appl. Sci. 2014, 3, 647–656. [Google Scholar]
- Vitte, J.; Michel, M.; Malinovschi, A.; Caminati, M.; Odebode, A.; Annesi-Maesano, I.; Caimmi, D.P.; Cassagne, C.; Demoly, P.; Heffler, E.; et al. Fungal exposome, human health, and unmet needs: A 2022 update with special focus on allergy. Allergy 2022, 77, 3199–3216. [Google Scholar] [CrossRef]
- Dunaevsky, Y.; Popova, V.; Semenova, T.; Beliakova, G.; Belozersky, M. Fungal inhibitors of proteolytic enzymes: Classification, properties, possible biological roles, and perspectives for practical use. Biochimie 2014, 101, 10–20. [Google Scholar] [CrossRef]
- Habibi, N.; Uddin, S.; Al-Sarawi, H.; Aldhameer, A.; Shajan, A.; Zakir, F.; Abdul Razzack, N.; Alam, F. Metagenomes from coastal sediments of Kuwait: Insights into the microbiome, metabolic functions and resistome. Microorganisms 2023, 11, 531. [Google Scholar] [CrossRef]
- Habibi, N.; Uddin, S.; Behbehani, M.; Khan, M.W.; Razzack, N.A.; Shirshikhar, F. The transcriptome profile of the marine Calanoid copepod Parvocalanus crassirostris isolated from Kuwait territorial waters and generations cultured under different ocean acidification scenarios. Reg. Stud. Mar. Sci. 2023, 67, 103231. [Google Scholar] [CrossRef]
- Gahoi, S.; Singh, S.; Gautam, B. Genome-wide identification and comprehensive analysis of Excretory/Secretory proteins in nematodes provide potential drug targets for parasite control. Genomics 2019, 111, 297–309. [Google Scholar] [CrossRef]
- Barney, B.M.; Vogel, E.; Mancipe, N.C. Algal Strain and Methods for Producing Simple Sugars. U.S. Patent 11932889, 19 March 2024. [Google Scholar]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Niessing, D. RNA-Binding proteins in fungi and their role in mRNA localization. In RNA-Binding Proteins; Lorkovic, Z., Ed.; Landes Bioscience: Austin, TX, USA, 2012; Volume 81, pp. 81–94. [Google Scholar]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Al-Ghadban, A.N.; Gevao, B.; Al-Shamroukh, D.; Al-Khabbaz, A. Estimation of suspended particulate matter in Gulf using MODIS data. Aquat. Ecosyst. Health Manag. 2012, 15, 41–44. [Google Scholar] [CrossRef]
- Neelamani, S.; Al-Dousari, A. A study on the annual fallout of the dust and the associated elements into the Kuwait Bay, Kuwait. Arab. J. Geosci. 2016, 9, 210. [Google Scholar] [CrossRef]
- Qasem, J.A.; Nasrallah, H.; Al-Khalaf, B.N.; Al-Sharifi, F.; Al-Sherafyee, A.; Almathkouri, S.A.; Al-Saraf, H. Meteorological factors, aeroallergens and asthma-related visits in Kuwait: A 12-month retrospective study. Ann. Saudi Med. 2008, 28, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Al-Salameen, F.; Habibi, N.; Uddin, S.; Al-Mataqi, K.; Al-Doaij, B.; Al-Amad, S.; Ali, E. Characterization and Identification of Microorganisms Associated with Airborne Dust in Kuwait; Kuwait Institute for Scientific Research: Safat, Kuwait, 2020. [Google Scholar] [CrossRef]
- Behbehani, M.; Carvalho, F.P.; Uddin, S.; Habibi, N. Enhanced Polonium Concentrations in Aerosols from the Gulf Oil Producing Region and the Role of Microorganisms. Int. J. Environ. Res. Public Health 2021, 18, 13309. [Google Scholar] [CrossRef] [PubMed]
- Habibi, N.; Uddin, S.; Al-Salameen, F.; Al-Amad, S.; Kumar, V.; Al-Otaibi, M.; Razzack, N.A.; Shajan, A.; Shirshikar, F. SARS-CoV-2, other respiratory viruses and bacteria in aerosols: Report from Kuwait’s hospitals. Indoor Air 2021, 31, 1815–1825. [Google Scholar] [CrossRef]
- Habibi, N.; Uddin, S.; Behbehani, M.; Abdul Razzack, N.; Hussain, F.Z.; Shajan, A. SARS-CoV-2 in hospital air as revealed by comprehensive respiratory viral panel sequencing. Infect. Prev. Pract. 2022, 4, 100199. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa Rank | Taxa Name | Stages | Log2 Median Ratio | Median diff RA % | Mean diff RA % | Wilcoxon (p) | |
|---|---|---|---|---|---|---|---|
| Order | Incertae sedis | 1 | 4 | −7.04 | −0.002 | −0.002 | 0.029 |
| Family | Glomerellaceae | 1 | 4 | −5.71 | −0.001 | −0.001 | 0.029 |
| Genus | Glomerella | 1 | 4 | −5.71 | −0.001 | −0.001 | 0.029 |
| Species | Colletotrichum gloeosporioides | 1 | 4 | −5.71 | −0.001 | −0.001 | 0.029 |
| Species | Candida sp_F15 | 1 | 4 | -Inf | −0.000 | −0.000 | 0.021 |
| Species | Cercospora capsici | 6 | 4 | Inf | 0.000 | 0.000 | 0.044 |
| Genus | Pseudallescheria | 3 | 1 | -Inf | −0.000 | −0.000 | 0.042 |
| Species | Pseudallescheria fimeti | 3 | 1 | -Inf | −0.000 | −0.000 | 0.042 |
| Species | Chaetomium sp_CBS_123294 | 3 | 1 | -Inf | −0.000 | −0.000 | 0.042 |
| Family | Glomerellaceae | 5 | 4 | -Inf | −0.001 | −0.001 | 0.050 |
| Family | Clavicipitaceae | 5 | 4 | Inf | 0.000 | 0.000 | 0.032 |
| Genus | Glomerella | 5 | 4 | -Inf | −0.001 | −0.001 | 0.050 |
| Genus | Beauveria | 5 | 4 | Inf | 0.000 | 0.000 | 0.032 |
| Species | Colletotrichum gloeosporioides | 5 | 4 | -Inf | −0.001 | −0.001 | 0.050 |
| Species | Beauveria bassiana | 5 | 4 | Inf | 0.000 | 0.000 | 0.032 |
| Family | Clavicipitaceae | 5 | 3 | Inf | 0.000 | 0.000 | 0.017 |
| Genus | Beauveria | 5 | 3 | Inf | 0.000 | 0.000 | 0.017 |
| Species | Beauveria bassiana | 5 | 3 | Inf | 0.000 | 0.000 | 0.017 |
| Class | Saccharomycetes | 2 | 4 | 1.07 | 0.002 | 0.003 | 0.029 |
| Order | Saccharomycetales | 2 | 4 | 1.07 | 0.002 | 0.003 | 0.029 |
| Family | Hypocreaceae | 2 | 4 | Inf | 0.001 | 0.001 | 0.027 |
| Genus | Trichoderma | 2 | 4 | Inf | 0.001 | 0.001 | 0.027 |
| Class | Saccharomycetes | 2 | 1 | 3.05 | 0.003 | 0.003 | 0.029 |
| Order | Saccharomycetales | 2 | 1 | 3.05 | 0.003 | 0.003 | 0.029 |
| Family | Incertae sedis | 2 | 1 | 4.30 | 0.002 | 0.003 | 0.029 |
| Genus | Candida | 2 | 1 | 4.30 | 0.002 | 0.003 | 0.029 |
| Family | Incertae sedis | 2 | 3 | 2.84 | 0.001 | 0.002 | 0.037 |
| Genus | Candida | 2 | 3 | 2.84 | 0.001 | 0.002 | 0.037 |
| Family | Hypocreaceae | 2 | 5 | Inf | 0.001 | 0.001 | 0.050 |
| Genus | Trichoderma | 2 | 5 | Inf | 0.001 | 0.001 | 0.050 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habibi, N.; Uddin, S.; Behbehani, M.; Kishk, M.; Khan, M.W.; Al-Fouzan, W.A. Diversity Analysis of Fungi Distributed in Inhalable and Respirable Size Fractions of Aerosols: A Report from Kuwait. Atmosphere 2024, 15, 806. https://doi.org/10.3390/atmos15070806
Habibi N, Uddin S, Behbehani M, Kishk M, Khan MW, Al-Fouzan WA. Diversity Analysis of Fungi Distributed in Inhalable and Respirable Size Fractions of Aerosols: A Report from Kuwait. Atmosphere. 2024; 15(7):806. https://doi.org/10.3390/atmos15070806
Chicago/Turabian StyleHabibi, Nazima, Saif Uddin, Montaha Behbehani, Mohammad Kishk, Mohd. Wasif Khan, and Wadha A. Al-Fouzan. 2024. "Diversity Analysis of Fungi Distributed in Inhalable and Respirable Size Fractions of Aerosols: A Report from Kuwait" Atmosphere 15, no. 7: 806. https://doi.org/10.3390/atmos15070806
APA StyleHabibi, N., Uddin, S., Behbehani, M., Kishk, M., Khan, M. W., & Al-Fouzan, W. A. (2024). Diversity Analysis of Fungi Distributed in Inhalable and Respirable Size Fractions of Aerosols: A Report from Kuwait. Atmosphere, 15(7), 806. https://doi.org/10.3390/atmos15070806