
Atmospheric Particle Number Concentrations and New Particle Formation over the Southern Ocean and Antarctica: A Critical Review



Abstract
1. Introduction
2. Instrumentation
3. Progress on Research of Atmospheric PNCs over the SO and Antarctica
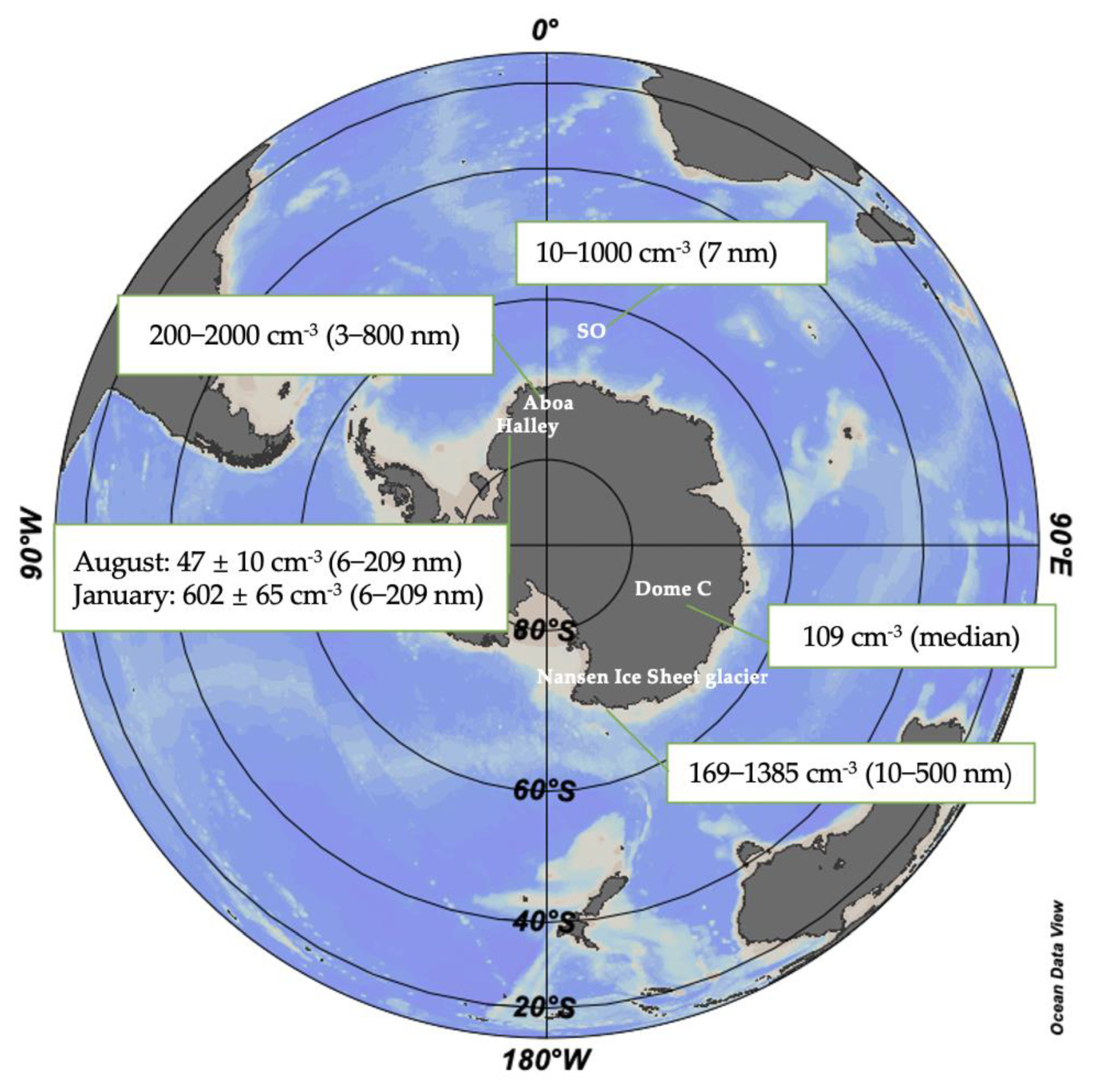
3.1. Spatial Variation
3.2. Seasonal Variation
4. Progress on Research of NPF Evens over the SO and Antarctica
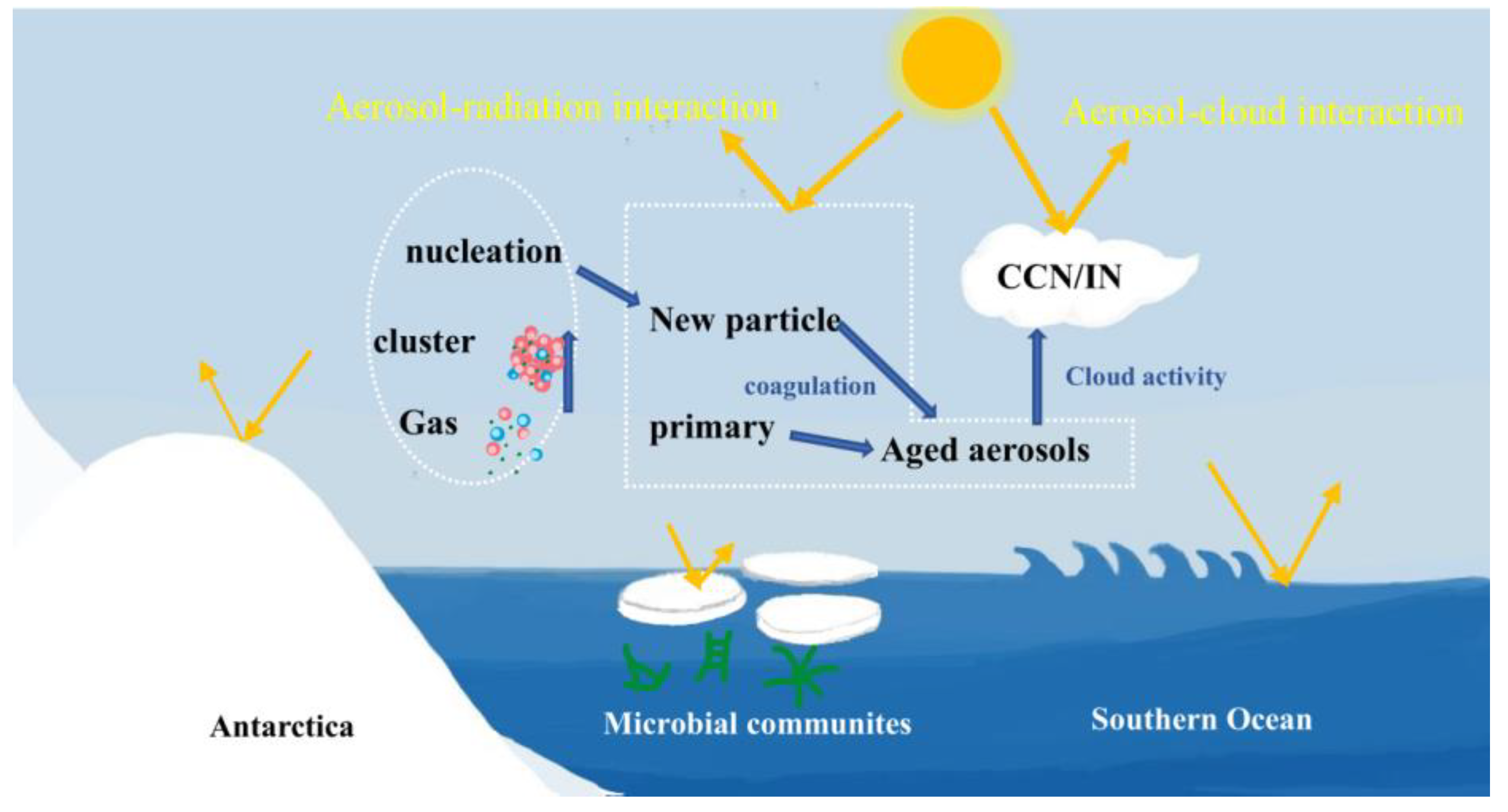
4.1. Precursors and Nucleation Mechanism
4.2. Environmental Influencing Factors
4.3. NPF Contribution to CCN
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lim, S.; Lee, M.; Rhee, T.S. Chemical characteristics of submicron aerosols observed at the King Sejong Station in the northern Antarctic Peninsula from fall to spring. Sci. Total Environ. 2019, 668, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Kyrö, E.M.; Kerminen, V.M.; Virkkula, A.; Dal Maso, M.; Parshintsev, J.; Ruíz-Jimenez, J.; Forsström, L.; Manninen, H.E.; Riekkola, M.L.; Heinonen, P.; et al. Antarctic new particle formation from continental biogenic precursors. Atmos. Chem. Phys. 2013, 13, 3527–3546. [Google Scholar] [CrossRef]
- Kim, J.; Yoon, Y.J.; Gim, Y.; Choi, J.H.; Kang, H.J.; Park, K.-T.; Park, J.; Lee, B.Y. New particle formation events observed at King Sejong Station, Antarctic Peninsula—Part 1: Physical characteristics and contribution to cloud condensation nuclei. Atmos. Chem. Phys. 2019, 19, 7583–7594. [Google Scholar] [CrossRef]
- Hara, K.; Nishita-Hara, C.; Osada, K.; Yabuki, M.; Yamanouchi, T. Characterization of aerosol number size distributions and their effect on cloud properties at Syowa Station, Antarctica. Atmos. Chem. Phys. 2021, 21, 12155–12172. [Google Scholar] [CrossRef]
- Teinilä, K.; Frey, A.; Hillamo, R.; Tülp, H.C.; Weller, R. A study of the sea-salt chemistry using size-segregated aerosol measurements at coastal Antarctic station Neumayer. Atmos. Environ. 2014, 96, 11–19. [Google Scholar] [CrossRef]
- Kammer, J.; Perraudin, E.; Flaud, P.M.; Lamaud, E.; Bonnefond, J.M.; Villenave, E. Observation of nighttime new particle formation over the French Landes forest. Sci. Total Environ. 2018, 621, 1084–1092. [Google Scholar] [CrossRef]
- Charron, A.; Birmili, W.; Harrison, R.M. Factors influencing new particle formation at the rural site, Harwell, United Kingdom. J. Geophys. Res. 2007, 112, D14210. [Google Scholar] [CrossRef]
- Hong, J.; Tang, M.; Wang, Q.; Ma, N.; Zhu, S.; Zhang, S.; Pan, X.; Xie, L.; Li, G.; Kuhn, U.; et al. Measurement Report: Wintertime new particle formation in the rural area of North China Plain: Influencing factors and possible formation mechanism. Atmos. Chem. Phys. Discuss. 2022, 2022, 1–32. [Google Scholar] [CrossRef]
- Qiao, X.; Yan, C.; Li, X.; Guo, Y.; Yin, R.; Deng, C.; Li, C.; Nie, W.; Wang, M.; Cai, R.; et al. Contribution of Atmospheric Oxygenated Organic Compounds to Particle Growth in an Urban Environment. Environ. Sci. Technol. 2021, 55, 13646–13656. [Google Scholar] [CrossRef]
- Bousiotis, D.; Pope, F.D.; Beddows, D.C.S.; Dall’Osto, M.; Massling, A.; Nøjgaard, J.K.; Nordstrøm, C.; Niemi, J.V.; Portin, H.; Petäjä, T.; et al. A phenomenology of new particle formation (NPF) at 13 European sites. Atmos. Chem. Phys. 2021, 21, 11905–11925. [Google Scholar] [CrossRef]
- Zheng, G.; Wang, Y.; Wood, R.; Jensen, M.P.; Kuang, C.; McCoy, I.L.; Matthews, A.; Mei, F.; Tomlinson, J.M.; Shilling, J.E.; et al. New particle formation in the remote marine boundary layer. Nat. Commun. 2021, 12, 527. [Google Scholar] [CrossRef]
- Huang, R.J.; Hoffmann, T.; Ovadnevaite, J.; Laaksonen, A.; Kokkola, H.; Xu, W.; Xu, W.; Ceburnis, D.; Zhang, R.; Seinfeld, J.H.; et al. Heterogeneous iodine-organic chemistry fast-tracks marine new particle formation. Proc. Natl. Acad. Sci. USA 2022, 119, e2201729119. [Google Scholar] [CrossRef]
- Lachlan-Cope, T.; Beddows, D.C.S.; Brough, N.; Jones, A.E.; Harrison, R.M.; Lupi, A.; Yoon, Y.J.; Virkkula, A.; Dall’Osto, M. On the annual variability of Antarctic aerosol size distributions at Halley Research Station. Atmos. Chem. Phys. 2020, 20, 4461–4476. [Google Scholar] [CrossRef]
- Herenz, P.; Wex, H.; Mangold, A.; Laffineur, Q.; Gorodetskaya, I.V.; Fleming, Z.L.; Panagi, M.; Stratmann, F. CCN measurements at the Princess Elisabeth Antarctica research station during three austral summers. Atmos. Chem. Phys. 2019, 19, 275–294. [Google Scholar] [CrossRef]
- Alroe, J.; Cravigan, L.T.; Miljevic, B.; Johnson, G.R.; Selleck, P.; Humphries, R.S.; Keywood, M.D.; Chambers, S.D.; Williams, A.G.; Ristovski, Z.D. Marine productivity and synoptic meteorology drive summer-time variability in Southern Ocean aerosols. Atmos. Chem. Phys. 2020, 20, 8047–8062. [Google Scholar] [CrossRef]
- Saliba, G.; Sanchez, K.J.; Russell, L.M.; Twohy, C.H.; Roberts, G.C.; Lewis, S.; Dedrick, J.; McCluskey, C.S.; Moore, K.; DeMott, P.J.; et al. Organic composition of three different size ranges of aerosol particles over the Southern Ocean. Aerosp. Sci. Technol. 2020, 55, 268–288. [Google Scholar] [CrossRef]
- O’Dowd, C.D.; Jimenez, J.L.; Bahreini, R.; Flagan, R.C.; Seinfeld, J.H.; Hämeri, K.; Pirjola, L.; Kulmala, M.; Jennings, S.G.; Hoffmann, T. Marine aerosol formation from biogenic iodine emissions. Nature 2002, 417, 632–636. [Google Scholar] [CrossRef]
- Koponen, I.K.; Virkkula, A.; Hillamo, R.; Kerminen, V.M.; Kulmala, M. Number size distributions and concentrations of marine aerosols: Observations during a cruise between the English Channel and the coast of Antarctica. J. Geophys. Res. 2002, 107, D24. [Google Scholar] [CrossRef]
- Cai, M.; Liang, B.; Sun, Q.; Liu, L.; Yuan, B.; Shao, M.; Huang, S.; Peng, Y.; Wang, Z.; Tan, H.; et al. The important roles of surface tension and growth rate in the contribution of new particle formation (NPF) to cloud condensation nuclei (CCN) number concentration: Evidence from field measurements in southern China. Atmos. Chem. Phys. 2021, 21, 8575–8592. [Google Scholar] [CrossRef]
- Wang, J.; Li, M.; Li, L.; Zheng, R.; Fan, X.; Hong, Y.; Xu, L.; Chen, J.; Hu, B. Particle number size distribution and new particle formation in Xiamen, the coastal city of Southeast China in wintertime. Sci. Total Environ. 2022, 826, 154208. [Google Scholar] [CrossRef]
- Holmes, N.S. A review of particle formation events and growth in the atmosphere in the various environments and discussion of mechanistic implications. Atmos. Environ. 2007, 41, 2183–2201. [Google Scholar] [CrossRef]
- Lee, S.H.; Gordon, H.; Yu, H.; Lehtipalo, K.; Haley, R.; Li, Y.; Zhang, R. New Particle Formation in the Atmosphere: From Molecular Clusters to Global Climate. J. Geophys. Res. 2019, 124, 7098–7146. [Google Scholar] [CrossRef]
- Kulmala, M.; Petäjä, T.; Nieminen, T.; Sipilä, M.; Manninen, H.E.; Lehtipalo, K.; Dal Maso, M.; Aalto, P.P.; Junninen, H.; Paasonen, P.; et al. Measurement of the nucleation of atmospheric aerosol particles. Nat. Protoc. 2012, 7, 1651–1667. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, M.; Wu, Z.; Yue, D. Reasearch on the Formation Mechanisms of New Particles in the Atmosphere. Acta Chim. Sin. 2013, 71, 519–527. (In Chinese) [Google Scholar] [CrossRef]
- Vu, T.V.; Delgado-Saborit, J.M.; Harrison, R.M. Review: Particle number size distributions from seven major sources and implications for source apportionment studies. Atmos. Environ. 2015, 122, 114–132. [Google Scholar] [CrossRef]
- Su, P.; Joutsensaari, J.; Dada, L.; Zaidan, M.A.; Nieminen, T.; Li, X.; Wu, Y.; Decesari, S.; Tarkoma, S.; Petäjä, T.; et al. New particle formation event detection with Mask R-CNN. Atmos. Chem. Phys. 2022, 22, 1293–1309. [Google Scholar] [CrossRef]
- Kulmala, M.; Kerminen, V.-M. On the formation and growth of atmospheric nanoparticles. Atmos. Res. 2008, 90, 132–150. [Google Scholar] [CrossRef]
- Brean, J.; Dall’Osto, M.; Simó, R.; Shi, Z.; Beddows, D.C.S.; Harrison, R.M. Open ocean and coastal new particle formation from sulfuric acid and amines around the Antarctic Peninsula. Nat. Geosci. 2021, 14, 383–388. [Google Scholar] [CrossRef]
- Quéléver, L.L.J.; Dada, L.; Asmi, E.; Lampilahti, J.; Chan, T.; Ferrara, J.E.; Copes, G.E.; Pérez-Fogwill, G.; Barreira, L.; Aurela, M.; et al. Investigation of new particle formation mechanisms and aerosol processes at Marambio Station, Antarctic Peninsula. Atmos. Chem. Phys. 2022, 22, 8417–8437. [Google Scholar] [CrossRef]
- Arquero, K.D.; Gerber, R.B.; Finlayson-Pitts, B.J. The Role of Oxalic Acid in New Particle Formation from Methanesulfonic Acid, Methylamine, and Water. Environ. Sci. Technol. 2017, 51, 2124–2130. [Google Scholar] [CrossRef]
- Yu, S. Role of organic acids (formic, acetic, pyruvic and oxalic) in the formation of cloud condensation nuclei (CCN): A review. Atmos. Res. 2000, 53, 185–217. [Google Scholar] [CrossRef]
- Sipilä, M.; Sarnela, N.; Neitola, K.; Laitinen, T.; Kemppainen, D.; Beck, L.; Duplissy, E.-M.; Kuittinen, S.; Lehmusjärvi, T.; Lampilahti, J.; et al. Wintertime subarctic new particle formation from Kola Peninsula sulfur emissions. Atmos. Chem. Phys. 2021, 21, 17559–17576. [Google Scholar] [CrossRef]
- Kulmala, M.; Laaksonen, A. Binary nucleation of water–sulfuric acid system: Comparison of classical theories with different H2SO4 saturation vapor pressures. J. Chem. Phys. 1990, 93, 696–701. [Google Scholar] [CrossRef]
- Korhonen, P.; Kulmala, M.; Laaksonen, A.; Viisanen, Y.; McGraw, R.; Seinfeld, J.H. Ternary nucleation of H2SO4, NH3, and H2O in the atmosphere. J. Geophys. Res. 1999, 104, 26349–26353. [Google Scholar] [CrossRef]
- Yu, F.; Luo, G.; Nair, A.A.; Eastham, S.; Williamson, C.J.; Kupc, A.; Brock, C.A. Particle number concentrations and size distributions in the stratosphere: Implications of nucleation mechanisms and particle microphysics. Atmos. Chem. Phys. 2023, 23, 1863–1877. [Google Scholar] [CrossRef]
- Yu, F.; Luo, G. Simulation of particle size distribution with a global aerosol model: Contribution of nucleation to aerosol and CCN number concentrations. Atmos. Chem. Phys. 2009, 9, 7691–7710. [Google Scholar] [CrossRef]
- Sipilä, M.; Lehtipalo, K.; Kulmala, M. Atmospheric Particle Nucleation. Aerosol Sci. 2013, 7, 153–180. [Google Scholar] [CrossRef]
- Spracklen, D.V.; Carslaw, K.S.; Kulmala, M.; Kerminen, V.M.; Mann, G.W.; Sihto, S.L. The contribution of boundary layer nucleation events to total particle concentrations on regional and global scales. Atmos. Chem. Phys. 2006, 6, 5631–5648. [Google Scholar] [CrossRef]
- Yu, F.; Luo, G.; Bates, T.S.; Anderson, B.; Clarke, A.; Kapustin, V.; Yantosca, R.M.; Wang, Y.; Wu, S. Spatial distributions of particle number concentrations in the global troposphere: Simulations, observations, and implications for nucleation mechanisms. J. Geophys. Res. 2010, 115, D17205. [Google Scholar] [CrossRef]
- Yao, L.; Garmash, O.; Bianchi, F.; Zheng, J.; Yan, C.; Kontkanen, J.; Junninen, H.; Mazon, S.B.; Ehn, M.; Paasonen, P.; et al. Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity. Science 2018, 361, 278–281. [Google Scholar] [CrossRef]
- Ning, A.; Liu, L.; Ji, L.; Zhang, X. Molecular-level nucleation mechanism of iodic acid and methanesulfonic acid. Atmos. Chem. Phys. 2022, 22, 6103–6114. [Google Scholar] [CrossRef]
- Ramachandran, G.; Park, J.Y.; Raynor, P.C. Assessing Exposures to Nanomaterials in the Occupational Environment; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Kangasluoma, J.; Ahonen, L.R.; Laurila, T.M.; Cai, R.; Enroth, J.; Mazon, S.B.; Korhonen, F.; Aalto, P.P.; Kulmala, M.; Attoui, M.; et al. Laboratory verification of a new high flow differential mobility particle sizer, and field measurements in Hyytiälä. J. Aerosol Sci. 2018, 124, 1–9. [Google Scholar] [CrossRef]
- Volckens, J.; Peters, T.M. Counting and particle transmission efficiency of the aerodynamic particle sizer. J. Aerosol Sci. 2005, 36, 1400–1408. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, D. Differential Mobility Particle Sizers for Nanoparticle Characterization. J. Nanotechnol. Eng. Med. 2014, 5, 9. [Google Scholar] [CrossRef]
- Seifert, M.; Tiede, R.; Schnaiter, M.; Linke, C.; Möhler, O.; Schurath, U.; Ström, J. Operation and performance of a differential mobility particle sizer and a TSI 3010 condensation particle counter at stratospheric temperatures and pressures. J. Aerosol Sci. 2004, 35, 981–993. [Google Scholar] [CrossRef]
- North, G.R.; Pyle, J.A.; Zhang, F. (Eds.) Encyclopedia of Atmospheric Sciences; Elsevier: Amsterdam, The Netherlands, 2014; Volume 1. [Google Scholar]
- Loizidis, C.; Costi, M.; Lekaki, N.; Bezantakos, S.; Biskos, G. Improved performance of Differential Mobility Analyzers with 3D-printed flow straighteners. J. Aerosol Sci. 2020, 145, 105545. [Google Scholar] [CrossRef]
- Belosi, F.; Contini, D.; Donateo, A.; Santachiara, G.; Prodi, F. Aerosol size distribution at Nansen Ice Sheet Antarctica. Atmos. Res. 2012, 107, 42–50. [Google Scholar] [CrossRef]
- Lewis, A.; Edwards, P. Validate personal air-pol sensors lution. Nature 2016, 535, 29–31. [Google Scholar] [CrossRef]
- Schmale, J.; Baccarini, A.; Thurnherr, I.; Henning, S.; Efraim, A.; Regayre, L.; Bolas, C.; Hartmann, M.; Welti, A.; Lehtipalo, K.; et al. Overview of the Antarctic Circumnavigation Expedition: Study of Preindustrial-like Aerosols and Their Climate Effects (ACE-SPACE). Bull. Am. Meteorol. Soc. 2019, 100, 2260–2283. [Google Scholar] [CrossRef]
- Koponen, I.K.; Virkkula, A.; Hillamo, R.; Kerminen, V.-M.; Kulmala, M. Number size distributions and concentrations of the continental summer aerosols in Queen Maud Land, Antarctica. J. Geophys. Res. 2003, 108, D18. [Google Scholar] [CrossRef]
- Weller, R.; Legrand, M.; Preunkert, S. Size distribution and ionic composition of marine summer aerosol at the continental Antarctic site Kohnen. Atmos. Chem. Phys. 2018, 18, 2413–2430. [Google Scholar] [CrossRef]
- Järvinen, E.; Virkkula, A.; Nieminen, T.; Aalto, P.P.; Asmi, E.; Lanconelli, C.; Busetto, M.; Lupi, A.; Schioppo, R.; Vitale, V.; et al. Seasonal cycle and modal structure of particle number size distribution at Dome C, Antarctica. Atmos. Chem. Phys. 2013, 13, 7473–7487. [Google Scholar] [CrossRef]
- Hara, K.; Osada, K.; Nishita-Hara, C.; Yamanouchi, T. Seasonal variations and vertical features of aerosol particles in the Antarctic troposphere. Atmos. Chem. Phys. 2011, 11, 5471–5484. [Google Scholar] [CrossRef]
- Revell, L.E.; Kremser, S.; Hartery, S.; Harvey, M.; Mulcahy, J.P.; Williams, J.; Morgenstern, O.; McDonald, A.J.; Varma, V.; Bird, L.; et al. The sensitivity of Southern Ocean aerosols and cloud microphysics to sea spray and sulfate aerosol production in the HadGEM3-GA7.1 chemistry–climate model. Atmos. Chem. Phys. 2019, 19, 15447–15466. [Google Scholar] [CrossRef]
- Yu, F.; Luo, G. Oceanic Dimethyl Sulfide Emission and New Particle Formation around the Coast of Antarctica: A Modeling Study of Seasonal Variations and Comparison with Measurements. Atmosphere 2010, 1, 34–50. [Google Scholar] [CrossRef]
- Jokinen, T.; Sipilä, M.; Kontkanen, J.; Vakkari, V.; Tisler, P.; Duplissy, E.M.; Junninen, H.; Kangasluoma, J.; Manninen, H.E.; Petäjä, T.; et al. Ion-induced sulfuric acid-ammonia nucleation drives particle formation in coastal Antarctica. Sci. Adv. 2018, 4, eaat9744. [Google Scholar] [CrossRef]
- Burrell, E.; Kar, T.; Hansen, J.C. Computational Study of the Thermodynamics of New Particle Formation Initiated by Complexes of H2SO4–H2O–NHx, CH3SO3H–H2O–NHx, and HO2–H2O–NHx. ACS Earth Space Chem. 2019, 3, 1415–1425. [Google Scholar] [CrossRef]
- Lana, A.; Bell, T.G.; Simó, R.; Vallina, S.M.; Ballabrera-Poy, J.; Kettle, A.J.; Dachs, J.; Bopp, L.; Saltzman, E.S.; Stefels, J.; et al. An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean. Glob. Biogeochem. Cycles 2011, 25, GB1004. [Google Scholar] [CrossRef]
- Baccarini, A.; Dommen, J.; Lehtipalo, K.; Henning, S.; Modini, R.L.; Gysel-Beer, M.; Baltensperger, U.; Schmale, J. Low-Volatility Vapors and New Particle Formation Over the Southern Ocean During the Antarctic Circumnavigation Expedition. J. Geophys. Res. 2021, 126, e2021JD035126. [Google Scholar] [CrossRef]
- Hirsikko, A.; Nieminen, T.; Gagné, S.; Lehtipalo, K.; Manninen, H.E.; Ehn, M.; Hõrrak, U.; Kerminen, V.M.; Laakso, L.; McMurry, P.H.; et al. Atmospheric ions and nucleation: A review of observations. Atmos. Chem. Phys. 2011, 11, 767–798. [Google Scholar] [CrossRef]
- Stolzenburg, D.; Simon, M.; Ranjithkumar, A.; Kürten, A.; Lehtipalo, K.; Gordon, H.; Ehrhart, S.; Finkenzeller, H.; Pichelstorfer, L.; Nieminen, T.; et al. Enhanced growth rate of atmospheric particles from sulfuric acid. Atmos. Chem. Phys. 2020, 20, 7359–7372. [Google Scholar] [CrossRef]
- Zhang, R.; Khalizov, A.; Wang, L.; Hu, M.; Xu, W. Nucleation and growth of nanoparticles in the atmosphere. Chem. Rev. 2012, 112, 1957–2011. [Google Scholar] [CrossRef]
- Peltola, M.; Rose, C.; Trueblood, J.V.; Gray, S.; Harvey, M.; Sellegri, K. New particle formation in coastal New Zealand with a focus on open-ocean air masses. Atmos. Chem. Phys. 2022, 22, 6231–6254. [Google Scholar] [CrossRef]
- He, X.C.; Tham, Y.J.; Dada, L.; Wang, M.; Finkenzeller, H.; Stolzenburg, D.; Iyer, S.; Simon, M.; Kürten, A.; Shen, J.; et al. Role of iodine oxoacids in atmospheric aerosol nucleation. Science 2021, 371, 589–595. [Google Scholar] [CrossRef]
- Roscoe, H.K.; Jones, A.E.; Brough, N.; Weller, R.; Saiz-Lopez, A.; Mahajan, A.S.; Schoenhardt, A.; Burrows, J.P.; Fleming, Z.L. Particles and iodine compounds in coastal Antarctica. J. Geophys. Res. 2015, 120, 7144–7156. [Google Scholar] [CrossRef]
- Ning, A.; Liu, L.; Zhang, S.; Yu, F.; Du, L.; Ge, M.; Zhang, X. The critical role of dimethylamine in the rapid formation of iodic acid particles in marine areas. npj Clim. Atmos. Sci. 2022, 5, 92. [Google Scholar] [CrossRef]
- Sipilä, M.; Sarnela, N.; Jokinen, T.; Henschel, H.; Junninen, H.; Kontkanen, J.; Richters, S.; Kangasluoma, J.; Franchin, A.; Peräkylä, O.; et al. Molecular-scale evidence of aerosol particle formation via sequential addition of HIO3. Nature 2016, 537, 532–534. [Google Scholar] [CrossRef]
- Wang, S.; Yan, J.; Lin, Q.; Zhang, M.; Xu, S.; Zhao, S.; Ruan, M. Formation of marine secondary aerosols in the Southern Ocean, Antarctica. Environ. Chem. 2021, 18, 285–293. [Google Scholar] [CrossRef]
- Humphries, R.S.; Schofield, R.; Keywood, M.D.; Ward, J.; Pierce, J.R.; Gionfriddo, C.M.; Tate, M.T.; Krabbenhoft, D.P.; Galbally, I.E.; Molloy, S.B.; et al. Boundary layer new particle formation over East Antarctic sea ice—Possible Hg-driven nucleation? Atmos. Chem. Phys. 2015, 15, 13339–13364. [Google Scholar] [CrossRef]
- Yue, F.; Xie, Z.; Yan, J.; Zhang, Y.; Jiang, B. Spatial Distribution of Atmospheric Mercury Species in the Southern Ocean. J. Geophys. Res. 2021, 126, e2021JD034651. [Google Scholar] [CrossRef]
- Kerminen, V.-M.; Chen, X.; Vakkari, V.; Petäjä, T.; Kulmala, M.; Bianchi, F. Atmospheric new particle formation and growth: Review of field observations. Environ. Res. Lett. 2018, 13, 103003. [Google Scholar] [CrossRef]
- Chen, X.; Yu, F.; Yang, W.; Sun, Y.; Chen, H.; Du, W.; Zhao, J.; Wei, Y.; Wei, L.; Du, H.; et al. Global–regional nested simulation of particle number concentration by combing microphysical processes with an evolving organic aerosol module. Atmos. Chem. Phys. 2021, 21, 9343–9366. [Google Scholar] [CrossRef]
- Shirsat, S.V.; Graf, H.F. An emission inventory of sulfur from anthropogenic sources in Antarctica. Atmos. Chem. Phys. 2009, 9, 3397–3408. [Google Scholar] [CrossRef]
- Twohy, C.H.; DeMott, P.J.; Russell, L.M.; Toohey, D.W.; Rainwater, B.; Geiss, R.; Sanchez, K.J.; Lewis, S.; Roberts, G.C.; Humphries, R.S.; et al. Cloud-Nucleating Particles Over the Southern Ocean in a Changing Climate. Earth’s Future 2021, 9, e2020EF001673. [Google Scholar] [CrossRef]
- Pierce, J.R.; Adams, P.J. Efficiency of cloud condensation nuclei formation from ultrafine particles. Atmos. Chem. Phys. 2007, 7, 1367–1379. [Google Scholar] [CrossRef]
- Merikanto, J.; Spracklen, D.V.; Mann, G.W.; Pickering, S.J.; Carslaw, K.S. Impact of nucleation on global CCN. Atmos. Chem. Phys. 2009, 9, 8601–8616. [Google Scholar] [CrossRef]
- McCoy, I.L.; Bretherton, C.S.; Wood, R.; Twohy, C.H.; Gettelman, A.; Bardeen, C.G.; Toohey, D.W. Influences of Recent Particle Formation on Southern Ocean Aerosol Variability and Low Cloud Properties. J. Geophys. Res. 2021, 126, e2020JD033529. [Google Scholar] [CrossRef]
- Salim, S.N.; Adhikari, A.; Shaikh, A.A.; Menon, H.B.; Kumar, N.; Rajeev, K. Aerosol-boundary layer dynamics and its effect on aerosol radiative forcing and atmospheric heating rate in the Indian Ocean sector of Southern Ocean. Sci. Total Environ. 2023, 858, 159770. [Google Scholar] [CrossRef]
- Angeles Suazo, J.M.; Suarez Salas, L.; Huaman De La Cruz, A.R.; Angeles Vasquez, R.; Rosales Aylas, G.; Rocha Condor, A.; Requena Rojas, E.; Muñoz Ccuro, F.; Flores Rojas, J.L.; Abi Karam, H. Direct Radiative Forcing Due to Aerosol Properties at the Peruvian Antarctic Station and Metropolitan Huancayo Area. Anuário. do Instituto de Geociências 2020, 43, 404–412. [Google Scholar] [CrossRef]
- Yu, F.; Luo, G.; Turco, R.P.; Ogren, J.A.; Yantosca, R.M. Decreasing particle number concentrations in a warming atmosphere and implications. Atmos. Chem. Phys. 2012, 12, 2399–2408. [Google Scholar] [CrossRef]
- Bracegirdle, T.J.; Connolley, W.M.; Turner, J. Antarctic climate change over the twenty first century. J. Geophys. Res. 2008, 113, D03103. [Google Scholar] [CrossRef]
- Yan, J.; Jung, J.; Lin, Q.; Zhang, M.; Xu, S.; Zhao, S. Effect of sea ice retreat on marine aerosol emissions in the Southern Ocean, Antarctica. Sci. Total Environ. 2020, 745, 140773. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Instrument | Manufacturing Company | Principle | Size Range (nm) | Reference |
|---|---|---|---|---|
| AIS | Airel | electrical mobility size | 0.46–55 | [27] |
| CPC 3007 | TSI | aerodynamic size | 10–1000 | [42] |
| DMPS | GRIMM | electrical mobility size | 10–800 | [43] |
| PSM | Airmodus | aerodynamic size | <1 | [42] |
| APS | TSI | optical size | 500–20,000 | [44] |
| Location | Lat. And Long. | Study Period | Wind Speed (m s−1) | Relative Humidity (%) | Total PNC (cm−3) | FR (cm−3 s−1) | GR (nm h−1) | Reference |
|---|---|---|---|---|---|---|---|---|
| The Finnish Antarctic station Aboa (Antarctica) | 73°03′ S, 13°25′ W | January 2010 | ~ | ~ | ~ | 0.003–0.3 (J10) | ~ | [2] |
| The Marambio Station (Antarctic Peninsula) | 64°15′ S, 56°38′ W | 15 January 2018–25 February 2018 | 1–20 | <75 | ~ | 0.69 (J3) | 4.2 (3.8–12 nm) | [29] |
| Nansen Ice Sheet glacier (the Ross Sea) | 74°30′ S, 163°27′ E | 2006 | 1–5 | >80 | 169–1385 | ~ | 4 | [49] |
| The Finnish Antarctic station Aboa (Antarctica) | 73°03′ S, 13°25′ W | 5 January 2000–22 January 2000 and 1 January 2001–26 January 2001 | ~ | 40–100 | 200–2000 | ~ | 2 (3–15 nm) | [52] |
| The Concordia station (Dome C) | 75°06′ S, 123°23′ E | 14 December 2007–7 November 2009 | ~ | ~ | 109 (median) | ~ | 2.5 | [54] |
| The King Sejong Station (Antarctic Peninsula) | 62°12′ S, 58°48′ W | May 2009–December 2016 | ~ | ~ | 1707–83,120 | 2.79 ± 1.05 (J2.5–10) | 0.68 ± 0.27 | [3] |
| Nucleation Mechanism | Region | Location | Reference |
|---|---|---|---|
| H2SO4-DMA-H2O | Antarctic Peninsula | 62°36′ S, 60°30′ W | [28] |
| H2SO4-MSA-DMA | Antarctic Peninsula | 64°15′ S, 56°38′ W | [29] |
| H2SO4-DMA-H2O | Antarctic Peninsula | 64°15′ S, 56°38′ W | [29] |
| H2SO4-NH3-H2O | Coastal Antarctica | 73°03′ S, 13°25′ W | [58] |
| OxA-MA-MSA | Polluted area | ~ | [30] |
| Iodine-organics | Coastal areas | 53°20′ N, 9°54′ W | [12] |
| IA-MSA | Marine | ~ | [41] |
| IA-DMA | Marine | ~ | [41] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Xu, G.; Chen, L.; Chen, K. Atmospheric Particle Number Concentrations and New Particle Formation over the Southern Ocean and Antarctica: A Critical Review. Atmosphere 2023, 14, 402. https://doi.org/10.3390/atmos14020402
Wang J, Xu G, Chen L, Chen K. Atmospheric Particle Number Concentrations and New Particle Formation over the Southern Ocean and Antarctica: A Critical Review. Atmosphere. 2023; 14(2):402. https://doi.org/10.3390/atmos14020402
Chicago/Turabian StyleWang, Jiayu, Guojie Xu, Liqi Chen, and Kui Chen. 2023. "Atmospheric Particle Number Concentrations and New Particle Formation over the Southern Ocean and Antarctica: A Critical Review" Atmosphere 14, no. 2: 402. https://doi.org/10.3390/atmos14020402
APA StyleWang, J., Xu, G., Chen, L., & Chen, K. (2023). Atmospheric Particle Number Concentrations and New Particle Formation over the Southern Ocean and Antarctica: A Critical Review. Atmosphere, 14(2), 402. https://doi.org/10.3390/atmos14020402