Abstract
High-level coupled-cluster calculations in combination with two-dimensional master equation simulations were used to study the HO2 + CH3O2 reaction, which plays an important role in the oxidation of methane and hydrocarbons in the Earth’s atmosphere and low-temperature combustion. The main reaction pathways taking place on the lowest-lying triplet and singlet potential energy surfaces (PES) were characterized. Interestingly, methyl hydroperoxide (CH3OOH), the sole product, could be produced from both the triplet and singlet PESs, with a ratio of roughly 9:1. Formaldehyde is not made as a primary product, but can be formed via secondary chemistry. The formation of methyl tetraoxide (MTO) from the singlet PES is unimportant. The calculated reaction rate coefficients were found to be practically pressure-independent for p ≤ 760 Torr and can be given by (in cm3/s), an expression useful for kinetics modeling over the range T = 200–800 K. The rate constant has a slight negative Arrhenius energy dependence of about −1.75 kcal mol–1, falling about a factor of 30 from 200 K to 800 K.
1. Introduction
Methane is the simplest stable hydrocarbon and is released in tremendous quantities into the Earth’s atmosphere from both natural (biogenic) and anthropogenic processes [1,2,3]. In the atmosphere, it is mainly oxidized by highly reactive hydroxyl radical (OH) via Equation (1) to yield methyl radical (CH3), followed by the association of CH3 with oxygen molecule (O2) to make methyl peroxy radical (CH3O2) via Equation (2) [4,5,6,7,8]. In polluted environments (i.e., urban or industrial areas) where NOx concentrations are high, CH3O2 radicals primarily react with NO via Equation (3) to yield CH3O and NO2. Subsequent photolysis of NO2 will then lead to the formation of tropospheric ground-level ozone, which is a contributor to smog, harmful to both animal and plant life. However, in clean environments (such as rural or forest areas), CH3O2 is mainly consumed by reacting with HO2 radicals (Equations (4) and (5)) [4,5,6,7,8]. The same sequential reaction steps are expected to occur in the flames of CH4 (or hydrocarbons) at low temperatures.
CH4 + OH → CH3 + H2O
CH3 + O2 + M → CH3O2 + M
CH3O2 + NO + M → CH3O + NO2 + M
CH3O2 + HO2 → CH3OOH + O2
CH3O2 + HO2 → CH2O + H2O + O2
It is well established that the title reaction mainly (if not exclusively) produces methyl hydroperoxide (CH3OOH) via Equation (4) [9,10,11,12,13,14,15,16,17,18,19]. Conversely, the formation of formaldehyde (CH2O) via Equation (5) remains debated. Some experiments [12,19] have found CH2O while others [13,14,15] did not. It is widely believed that the production of CH2O is at most minor, less than 10% [3,13]. It should be mentioned that the CH2O observed in some experiments could be produced (as a secondary product) through the (photo-) oxidation processes of the primary product CH3OOH, Equations (6) and (7).
CH3OOH + X → CH2OOH (unstable) + HX → CH2O + OH + HX
Earlier, the title reaction was theoretically studied using CCSD(T)//B3LYP [20,21] and CASPT2//CASSCF [22] levels of theory. Both the lowest-lying triplet and singlet electronic state potential energy surfaces (PES) were characterized [20,22]. The reaction pathway taking place on the triplet PES to yield CH3OOH (Equation (4)) was reported to be dominant while the contribution of the singlet PES was postulated to be negligibly small [20,22]. The potential catalytic role of one water molecule was also investigated, but found to be insignificant [23,24]. The reaction rate coefficients were computed by assuming the thermal equilibrium condition [22], which can only be achieved at extremely high pressures. Such conditions cannot be fulfilled in the Earth’s atmosphere and in experiments for this reaction system, which features a shallow well (i.e., van der Waals complex) and a submerged TS (see Figure 1). The title reaction as displayed in Figure 1 is expected to be (slightly) pressure-dependent above 1 atm. In such a case, solving a master equation is required to obtain the reaction rate constants.

Figure 1.
Schematic reaction energy profiles for the reaction of HO2 and CH3O2, constructed using the mHEAT method (see text). The left-hand side shows the important reaction pathways on the singlet PES while the right-hand side displays the reaction pathway on the triplet PES. Benchmark ATcT values (in parentheses) are also included for comparison.
In this work, the title reaction is restudied using high-level accuracy coupled-cluster calculations. We reexamine the potential roles of the singlet PES; particularly, we address questions of the potential formation of methyl tetraoxide (MTO, CH3OOOOH), methyl peroxide (CH3OOH), and formaldehyde from the singlet PES. We then solve an E,J-resolved master equation for the temperature range 200–800 K and a pressure range of 1–1000 Torr (i.e., conditions applicable to the Earth’s atmosphere and low-temperature combustion processes) to quantify product branching ratios and to provide reliable phenomenological rate constants for kinetics modeling.
2. Theoretical Methodologies
2.1. High-Level Coupled-Cluster Calculations
The radical–radical association of HO2 and CH3O2 can, in principle, take place on both the lowest-lying triplet and singlet PESs. The geometries of all key stationary points were fully optimized using the frozen-core (fc) CCSD(T) method [25,26,27] in combination with a triple-zeta atomic natural orbital (ANO1) [28,29] basis set. As usual, unrestricted and restricted Hartree–Fock reference wave functions in the CCSD(T) calculations were used for triplet and singlet stationary points, respectively. However, for singlet open-shell structures (such as TS2, TS3, and 1PRC, see Figure 1), broken-symmetry UHF reference wave functions were used. Harmonic vibrational frequency analyses were then carried out to verify the located stationary points as well as to compute harmonic force fields. To obtain anharmonic zero-point vibrational energy (ZPE) corrections and anharmonic constants for subsequent kinetics simulations, cubic and quartic force constants were also computed using fc-CCSD(T)/aug-cc-pVDZ [30] level of theory.
Total electronic energies (including anharmonic ZPE corrections) of all stationary points were then obtained with the composite mHEAT-345(Q) method. As detailed elsewhere, the mHEAT-345(Q) protocol [31] generally comprises a series of high-level (single-point energy) coupled-cluster calculations; it recovers a large part of electron correlation with the perturbative triple excitations method (CCSD(T)) and a smaller part of electron correlation with the fully iterative triple (CCSDT) and non-iterative (perturbative) quadruple (CCSDT(Q)) methods [31]. In addition, other smaller corrections include the diagonal Born–Oppenheimer correction (DBOC), scalar relativity, and spin-orbit [31]. As seen in Figure 1, mHEAT calculations provide a high accuracy (relative) energy of about ±0.3 kcal mol−1 as compared to benchmark ATcT [32] for the reaction enthalpies. Although experimental activation energies are not available to be compared with the theory in this circumstance, a (conservative) accuracy energy of ± 1.0 kcal mol−1 may be expected for other stationary points.
The calculations made for the a1Δg state of 1O2 (an RHF-based treatment starting from the state) are clearly not adequate (as evidenced by the fairly large discrepancy with ATcT), but it should be emphasized that the thermodynamic energy of this species is not important for the kinetics calculations presented in this work. Nevertheless, the (relative) energy of the a1Δg state of 1O2 is estimated using the energy of triplet ground state of O2 obtained with the mHEAT method and a singlet-triplet energy gap of 22.57 ± 0.24 kcal mol−1 taken from ATcT [32].
A comparison of relative energies of various stationary points calculated with the mHEAT method in this work and values obtained with lower levels of theory reported in the literature are given in Table 1. A difference of a few kcal mol–1 can be seen there. To the best of our knowledge, the mHEAT method used in this work is the highest level of theory that has ever been applied to the title reaction. It is well known that kinetic results are sensitively dependent on the accuracy of the calculated barrier heights. Therefore, high-accuracy relative energies are required for the following kinetic analysis.

Table 1.
A comparison of relative energies (in kcal mol−1) of various stationary points for the title reaction occurring on the triplet and singlet PESs calculated with mHEAT method and values in the literature.
2.2. Two-Dimensional Master Equation Calculations
As shown in Figure 1, the title reaction proceeds via energized adducts (pre-reactive complex (PRC) and methyl tetraoxide (MTO)) before leading to products; it is therefore expected to depend on pressure. As a result, a master equation approach must be used to compute phenomenological rate constants as functions of temperature and pressure. An E,J-resolved master equation [33,34,35,36,37,38,39] for a chemically activated reaction (see Figure 1), which describes the competition of unimolecular dissociation reactions and energy transfer processes through collisions between a bath gas and vibrationally excited intermediates as a function of time, can be expressed by Equation (8) for the reaction pathway on the triplet PES and in Equation (9) for the reaction pathways on the singlet PES.
On the triplet PES
On the singlet PES
Here 1, 2, and 3 designate for singlet PRC, MTO, and triplet PRC, respectively. In Equations (8) and (9), is the maximum angular momentum; is the maximum internal energy; represents the (time-dependent) mole fractions of triplet PRC in the state and time t; (in s−1) is the Lennard–Jones collisional frequency [40,41,42]; and (in ) is the -resolved microcanonical rate coefficient for the isomerization step of singlet PRC to MTO. is the E,J-resolved collisional transfer probability distribution function of triplet PRC from the state to state . OST stands for the original source term, and is given by [43,44,45,46]:
where is the capture rate constant—that can be calculated using micro-variational transition state theory (μνTST) [47,48,49,50] (see Equation (13) below)—for the barrier-less association step of HO2 and CH3O2 leading to triplet PRC. is the E,J-resolved initial distribution function for the nascent energized triplet PRC and given by [43,46]:
In Equation (12), is the density of ro-vibrational states for triplet PRC, and is the microcanonical rate constant for the triplet PRC → HO2 + CH3O2 step, which is calculated using micro-variational TST [50,51].
Here, h is Planck’s constant, kB is Boltzmann’s constant, and σ is the reaction path degeneracy, which is equal to 3 × 2/2 ± 2 = 3/2 for the triplet PES in this case, 2 on the nominator is because TS1 is a chiral structure. Note that electronic partition functions for all stationary points are set to 1, 2, and 3 for singlet, doublet and triplet electronic states, respectively. T is the reaction temperature and E is the total internal energy. stands for minimizing the chemical reaction flux at the given E and J. and are the complete partition functions for HO2 and CH3O2, respectively. Qtr is the translational partition function, and Qe is the electronic partition function of the TS (the superscripts “re” and “≠” designate reactants and transition state (TS), respectively). is the sum of ro-vibrational quantum states of the TS for the given E and J, which can be obtained from its vibrational counterpart using the J-shifting approximation [52,53,54], Equations (14) and (15):
In Equation (14), is the anharmonic (coupled) vibrational sum of states of TS that is calculated using Miller’s semiclassical TST (SCTST) theory [55,56,57,58,59] based on the Wang–Landau algorithm [60,61,62,63]. SCTST theory [55,56,57,58,59] automatically includes coupled anharmonic vibrations and multidimensional quantum mechanical tunneling. is the (external) rotational energy level of TS, which is approximated by a symmetric top, [64] Equation (16):
In this work, hindered internal rotations with low vibrational frequencies in stationary points are separated and approximately treated as separable one-dimensional hindered internal rotors (1DHR). We then solve 1D Schrodinger equation independently to obtain a spectrum of eigenvalues for each 1DHR mode, whose sums of quantum states are then counted directly. Finally, we convolve these 1DHR motions with the remaining vibrations to obtain the overall density of vibrational states.
The phenomenological rate coefficients can be determined in two ways. First, they can be associated with the lowest (negative) eigenvalues (i.e., chemically significant eigenvalues, CSE) that are obtained by solving Equations (8) and (9). Second, the rate coefficients can also be calculated at a pseudo steady-state condition using Equations (17) and (18):
In Equations (17) and (18), is the mole fraction of HO2, which is produced by unimolecular re-dissociation of the triplet (or singlet) energized PRC back to initial reactants (HO2 and CH3O2), obtained at a pseudo steady state condition at a given T and p.
3. Results and Discussions
3.1. Reaction Mechanisms
The association of HO2 and CH3O2 can proceed on both the triplet and singlet electronic state PESs. The reaction pathways on the triplet PES are displayed on the right-hand side of Figure 1 while those on the singlet PES are presented on the left-hand side.
On the triplet PES: the barrier-less association of HO2 and CH3O2 leads to the formation of the vibrationally excited triplet pre-reactive complex (3PRC), which is a van der Waals complex stabilized by two H-bonded with a binding energy of 5.92 kcal mol−1. Starting at 3PRC, there are two feasible dissociating pathways: it can re-dissociate back to the initial reactants (HO2 + CH3O2) or it can carry out the internal H-abstraction from HO2 to yield CH3OOH and triplet O2. The latter pathway has a submerged barrier of −2.77 kcal mol−1 (relative to reactants), and thus formation of products is facile. This is the major (if not sole) product pathway, which was previously confirmed by both experimental [9,10,11,12,13,14,15,16,17,18,19] and theoretical [20,22] studies. It should be noted that HO2 could abstract an H atom from the CH3 group of CH3O2 leading to H2O2 and triplet CH2OO, but this H-abstraction pathway (not shown in Figure 1) has a very high barrier of ca. 28.6 kcal mol−1 at mHEAT method, and can be safely disregarded.
On the singlet PES: the barrier-less combination of HO2 and CH3O2 can also lead to an energized singlet adduct, 1PRC. Because the two unpaired electrons in PRC are far apart, their interaction is extremely weak. As a result, the binding energy of 1PRC (5.92 kcal mol−1) is nearly identical to that of 3PRC. When formed, 1PRC can carry out three plausible dissociative pathways. First, it can return without a potential barrier to the initial reactants. Second, it can undergo an O–O association via TS3 to lead to vibrationally excited methyl tetraoxide (MTO), overcoming a barrier of 5.77 kcal mol−1. When produced, MTO mostly isomerizes back to 1PRC because further decomposition of MTO yielding various products must overcome much higher barriers (not shown in Figure 1), which are therefore irrelevant under the conditions studied here. Third, it can do an internal H abstraction via TS2 to make CH3OOH and singlet O2, surmounting a barrier of 5.79 kcal mol−1. It is worthy of mention that, in addition to the triplet channel, this singlet channel provides an additional contribution (about 10%) to the overall formation of CH3OOH observed in experiments. To the best of our knowledge, this new finding has not previously been reported in the literature.
3.2. Statistical Kinetics Analysis
To solve a master equation, one must have the collisional parameters of energized adduct (CH4O4) and bath gas (both N2 and He used here) as well as the energy/angular momentum transfer probability distribution function. These parameters were empirically selected based on similar (known) systems [65] and are tabulated in Table S2. It is worth mentioning that the calculated rate constants in this work depend only slightly on pressure (see Figure 2), at pressures typical of atmospheric conditions (p ≤ 760 Torr), they are practically constant, as seen in Figure 2.
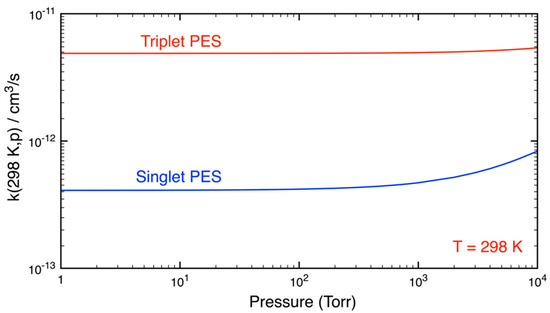
Figure 2.
Two falloff curves for the reaction of HO2 and CH3O2 calculated at T = 298 K and a function of pressure.
Figure 2 shows two falloff curves calculated at T = 298 K. The reaction on the triplet PES appeared to be pressure-independent (for p = 1–10,000 Torr) while that on the singlet PES depended slightly on pressure when P was greater than 1000 Torr. These results are completely consistent with experimental results, which show that the reaction does not depend on pressure when p ≤ 760 Torr.
To examine the formation of methyl tetraoxide (MTO), mole fractions of various species from the reaction on the singlet PES were computed at 298 K and a function of pressure. As revealed in Figure 3, the yield of the thermalized MTO—which is formed through collisional stabilization—was negligible (<0.1%) even at p = 10,000 Torr. There are two reasons for this observation: first, the chemical flux via the tighter TS3 is about two orders of magnitude smaller than that proceeding via the looser TS2; second, the energized intermediate MTO, when produced, prefers to isomerize back to 1PRC.
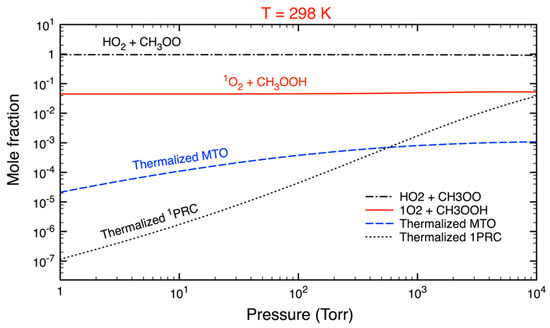
Figure 3.
Mole fractions of various species in the reaction of HO2 and CH3O2 occurring on the singlet electronic state PES, calculated at T = 298 K and a function of pressure.
Figure 3 also indicates that most of vibrationally excited 1PRC, when formed, returned to initial reactants (HO2 + CH3O2) while the rest further reacted to yield CH3OOH and singlet O2. Furthermore, the yield of collisional stabilization of 1PRC was found to be minor, especially at atmospheric pressures. It can be predicted that the thermalized 1PRC decreases significantly at higher temperatures in combustion environments. In practical applications, the reaction pathway on the singlet PES must be considered at the low-P limit as well. This finding differs from a previous theoretical study [22] where the high-P limit model (i.e., the thermal equilibrium was assumed) was used to compute k(T), which would appear to be inappropriate.
The results from the master equation analysis show that CH3OOH is the sole product under the conditions considered in this work. Importantly, CH3OOH can be produced from both the triplet and singlet PESs with a relative ratio of about 9:1, which is found to be very marginally dependent on temperature (see Figure 4). The yield of CH3OOH from the triplet PES is dominant for two reasons: first, the formation rate of 3PRC is three times as fast as that of 1PRC, due to the electronic degeneracy; and second, TS1 lies lower in energy than TS2.
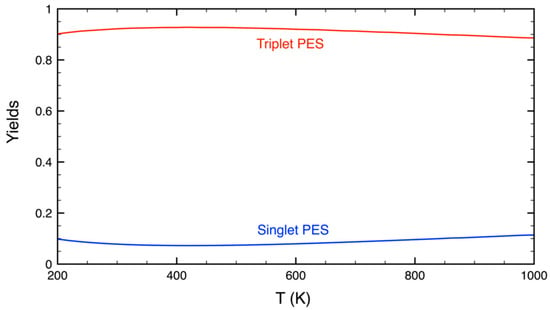
Figure 4.
A comparison of the yield of CH3OOH produced from the triplet and singlet PESs as a function of temperature.
Figure 5 shows the reaction rate coefficients calculated as a function of temperature, experimental data in symbols are also included for comparison. An inspection of Figure 5 reveals that the ab initio k(T) results (the dashed blue curve) were in good agreement with experiments at low temperatures (T < 260 K), but slightly too low at higher temperatures. By lowering the calculated barriers by 0.5 kcal mol−1, which is within the range of error expected with the mHEAT method, we were able to reproduce most experimental data within 20% (see the red solid curve). Combining the high-level theoretical results obtained in this work and available experimental data, we carried out curve fitting and obtained an Arrhenius equation:
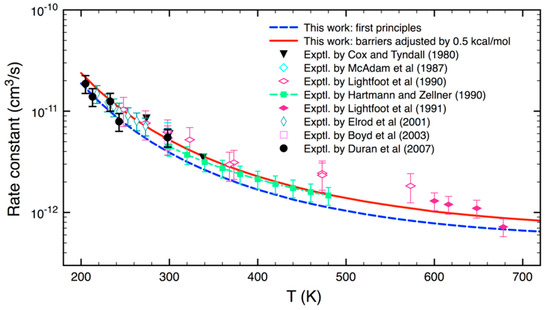
Figure 5.
Calculated thermal rate coefficients for the reaction of HO2 and CH3O2: ab initio k (T): dashed blue curve; the barriers lessened by 0.5 kcal mol−1: solid red curve. Experimental data (symbols) are also included for comparison.
As seen in Figure 6, Equation (20) presents a new set of predicted reaction rate constants, which agree well (within 10%) with most experimental data where they are available. In addition, it provides reliable rate constants where experimental data are absent. Therefore, we believe that Equation (20) can be useful for kinetics modeling.
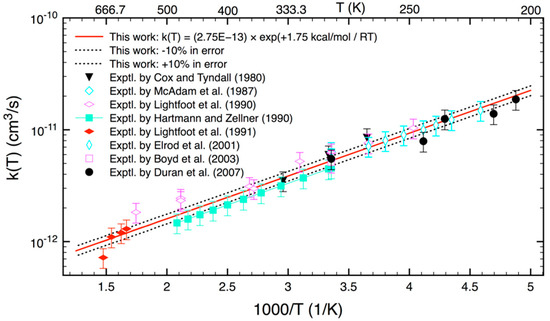
Figure 6.
The reaction rate coefficients (the solid red curve) for the reaction of HO2 and CH3O2 are recommended in this work. Experimental data (symbols) are also included for comparison.
4. Conclusions
The reaction of HO2 and CH3O2 was reinvestigated using high-accuracy coupled-cluster calculations, followed by computing phenomenological rate coefficients with an E,J-resolved master equation technique. Methyl hydroperoxide (CH3OOH) was found to be the sole product and can be produced from both triplet and singlet PESs with a ratio of about 9:1, a factor which is nearly independent on temperature and pressure. The formation of CH3OOH on the singlet PES is a new finding in this work. The yield of methyl tetraoxide (CH3OOOOH) from the singlet PES formed through collisional stabilization was found to be negligibly small. Formaldehyde (CH2O) was not found to be a primary product; if formed, it is most likely via succeeding photo-oxidation processes of CH3OOH in some experiments. The reaction was found to proceed through hydrogen-bonded pre-reactive complexes, followed by internal H-abstraction steps via submerged barriers leading to products, thus the reaction rate constant had a slight negative temperature dependence and did not depend on pressure (when p ≤ 1 atm). The rate coefficients fitted to the expression of (in cm3/s) for a temperature range of 200–800 K are recommended for kinetics modeling. These findings are completely consistent with experimental knowledge on this system.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/atmos13091397/s1.
Author Contributions
The original manuscript was prepared by T.L.N. It was reviewed and edited by J.F.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the U.S. Department of Energy, Office of Basic Energy Sciences under Award DE-SC0018164.
Acknowledgments
We would like to thank two anonymous reviewers who provided helpful comments that helped to improve the presentation of this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Atkinson, R. Gas-Phase Tropospheric Chemistry of Volatile Organic Compounds: 1. Alkanes and Alkenes. J. Phys. Chem. Ref. Data 1997, 26, 215–290. [Google Scholar] [CrossRef]
- Miller, J.A.; Pilling, M.J.; Troe, E. Unravelling combustion mechanisms through a quantitative understanding of elementary reactions. Proc. Combust. Inst. 2005, 30, 43–88. [Google Scholar]
- Baulch, D.L.; Bowman, C.T.; Cobos, C.J.; Cox, R.A.; Just, T.; Kerr, J.A.; Pilling, M.J.; Stocker, D.; Troe, J.; Tsang, W.; et al. Evaluated kinetic data for combustion modeling: Supplement II. J. Phys. Chem. Ref. Data 2005, 34, 757–1397. [Google Scholar]
- Orlando, J.J.; Tyndall, G.S. Laboratory studies of organic peroxy radical chemistry: An overview with emphasis on recent issues of atmospheric significance. Chem. Soc. Rev. 2012, 41, 6294–6317. [Google Scholar]
- Wallington, T.J.; Dagaut, P.; Kurylo, M.J. Ultraviolet-Absorption Cross-Sections and Reaction-Kinetics and Mechanisms for Peroxy-Radicals in the Gas-Phase. Chem. Rev. 1992, 92, 667–710. [Google Scholar]
- Tyndall, G.S.; Cox, R.A.; Granier, C.; Lesclaux, R.; Moortgat, G.K.; Pilling, M.J.; Ravishankara, A.R.; Wallington, T.J. Atmospheric chemistry of small organic peroxy radicals. J. Geophys Res.-Atmos 2001, 106, 12157–12182. [Google Scholar]
- Lightfoot, P.D.; Cox, R.A.; Crowley, J.N.; Destriau, M.; Hayman, G.D.; Jenkin, M.E.; Moortgat, G.K.; Zabel, F. Organic Peroxy-Radicals-Kinetics, Spectroscopy and Tropospheric Chemistry. Atmos. Environ. Part A Gen. Top. 1992, 26, 1805–1961. [Google Scholar]
- Onel, L.; Brennan, A.; Seakins, P.W.; Whalley, L.; Heard, D.E. A new method for atmospheric detection of the CH3O2 radical. Atmos. Meas. Tech. 2017, 10, 3985–4000. [Google Scholar]
- Cox, R.A.; Tyndall, G.S. Rate Constants for the Reactions of CH3O2 with HO2 NO and NO2 Using Molecular Modulation Spectrometry. J. Chem. Soc. Faraday Trans. 1980, 76, 153–163. [Google Scholar]
- Kurylo, M.J.; Dagaut, P.; Wallington, T.J.; Neuman, D.M. Kinetic Measurements of the Gas-Phase HO2 + CH3O2 Cross-Disproportionation Reaction at 298-K. Chem. Phys. Lett. 1987, 139, 513–518. [Google Scholar]
- Mcadam, K.; Veyret, B.; Lesclaux, R. Uv Absorption-Spectra of HO2 and CH3O2 Radicals and the Kinetics of Their Mutual Reactions at 298-K. Chem. Phys. Lett. 1987, 133, 39–44. [Google Scholar]
- Jenkin, M.E.; Cox, R.A.; Hayman, G.D.; Whyte, L.J. Kinetic-Study of the Reactions CH3O2 + CH3O2 and CH3O2 + HO2 Using Molecular Modulation Spectroscopy. J. Chem. Soc. Faraday Trans. 1988, 84, 913–930. [Google Scholar]
- Wallington, T.J. Fourier-Transform Infrared Product Study of the Reaction of CH3O2 + HO2 over the Pressure Range 15-700 Torr at 295-K. J. Chem. Soc. Faraday Trans. 1991, 87, 2379–2382. [Google Scholar]
- Lightfoot, P.D.; Roussel, P.; Caralp, F.; Lesclaux, R. Flash-Photolysis Study of the CH3O2 + CH3O2 and CH3O2 + HO2 Reactions between 60-K and 719-K-Unimolecular Decomposition of Methylhydroperoxide. J. Chem. Soc. Faraday Trans. 1991, 87, 3213–3220. [Google Scholar]
- Lightfoot, P.D.; Veyret, B.; Lesclaux, R. Flash-Photolysis Study of the CH3O2 + HO2 Reaction between 248 and 573-K. J. Phys. Chem. 1990, 94, 708–714. [Google Scholar] [CrossRef]
- Boyd, A.A.; Flaud, P.M.; Daugey, N.; Lesclaux, R. Rate constants for RO2 + HO2 reactions measured under a large excess of HO2. J. Phys. Chem. A 2003, 107, 818–821. [Google Scholar]
- Raventos-Duran, M.T.; Mcgillen, M.; Percival, C.J.; Hamer, P.D.; Shallcross, D.E. Kinetics of the CH3O2 + HO2 reaction: A temperature and pressure dependence study using chemical ionization mass Spectrometry. Int. J. Chem. Kinet. 2007, 39, 571–579. [Google Scholar]
- Moortgat, G.K.; Cox, R.A.; Schuster, G.; Burrows, J.P.; Tyndall, G.S. Peroxy Radical Reactions in the Photo-Oxidation of CH3CHO. J. Chem. Soc. Faraday Trans. 1989, 85, 809–829. [Google Scholar]
- Elrod, M.J.; Ranschaert, D.L.; Schneider, N.J. Direct kinetics study of the temperature dependence of the CH2O branching channel for the CH3O2+HO2 reaction. Int. J. Chem. Kinet. 2001, 33, 363–376. [Google Scholar]
- Hou, H.; Wang, B.S. A systematic computational study on the reactions of HO2 with RO2: The HO2+CH3O2(CD3O2) and HO2+CH2FO2 reactions. J. Phys. Chem. A 2005, 109, 451–460. [Google Scholar]
- Drougas, E. Quantum mechanical studies of the CH3O2 + HO2 reaction. Comput. Theor. Chem. 2016, 1093, 98–103. [Google Scholar]
- Anglada, J.M.; Olivella, S.; Sole, A. Mechanistic study of the CH3O2 + HO2 -> CH3O2H + O2 reaction in the gas phase. Computational evidence for the formation of a hydrogen-bonded diradical complex. J. Phys. Chem. A 2006, 110, 6073–6082. [Google Scholar] [PubMed]
- Zhang, T.L.; Wang, W.L.; Zhang, P.; Lu, J.; Zhang, Y. Water-catalyzed gas-phase hydrogen abstraction reactions of CH3O2 and HO2 with HO2: A computational investigation. Phys. Chem. Chem. Phys. 2011, 13, 20794–20805. [Google Scholar] [PubMed]
- English, A.M.; Hansen, J.C.; Szente, J.J.; Maricq, A.M. The effects of water vapor on the CH3O2 self-reaction and reaction with HO2. J. Phys. Chem. A 2008, 112, 9220–9228. [Google Scholar] [PubMed]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Headgordon, M. A 5th-Order Perturbation Comparison of Electron Correlation Theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar]
- Bartlett, R.J.; Watts, J.D.; Kucharski, S.A.; Noga, J. Noniterative 5th-Order Triple and Quadruple Excitation-Energy Corrections in Correlated Methods. Chem. Phys. Lett. 1990, 165, 513–522. [Google Scholar]
- Stanton, J.F. Why CCSD(T) works: A different perspective. Chem. Phys. Lett. 1997, 281, 130–134. [Google Scholar]
- Almlof, J.; Taylor, P.R. General Contraction of Gaussian-Basis Sets. 1. Atomic Natural Orbitals for 1st-Row and 2nd-Row Atoms. J. Chem. Phys. 1987, 86, 4070–4077. [Google Scholar]
- Almlof, J.; Taylor, P.R. General Contraction of Gaussian-Basis Sets. 2. Atomic Natural Orbitals and the Calculation of Atomic and Molecular-Properties. J. Chem. Phys. 1990, 92, 551–560. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian-Basis Sets for Use in Correlated Molecular Calculations. 1. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar]
- Thorpe, J.H.; Lopez, C.A.; Nguyen, T.L.; Baraban, J.H.; Bross, D.H.; Ruscic, B.; Stanton, J.F. High-accuracy extrapolated ab initio thermochemistry. IV. A modified recipe for computational efficiency. J. Chem. Phys. 2019, 150, 224102. [Google Scholar] [CrossRef] [PubMed]
- Ruscic, B.; Bross, D.H. Active Thermochemical Tables (ATcT) Values Based on Ver. 1.122r of the Thermochemical Network 2022. Available online: ATcT.anl.gov (accessed on 4 July 2022).
- Jeffery, S.J.; Gates, K.E.; Smith, S.C. Full Iterative Solution of the Two-Dimensional Master Equation for Thermal Unimolecular Reactions. J. Phys. Chem. 1996, 100, 7090–7096. [Google Scholar] [CrossRef]
- Robertson, S.H.; Pilling, M.J.; Green, N.J.B. Diffusion approximations of the two-dimensional master equation. Mol. Phys. 1996, 89, 1531–1551. [Google Scholar] [CrossRef]
- Miller, J.A.; Klippenstein, S.J.; Raffy, C. Solution of Some One- and Two-Dimensional Master Equation Models for Thermal Dissociation: The Dissociation of Methane in the Low-Pressure Limit. J. Phys. Chem. 2002, 106, 4904–4913. [Google Scholar] [CrossRef]
- Jasper, A.W.; Pelzer, K.M.; Miller, J.A.; Kamarchik, E.; Harding, L.B.; Klippenstein, S.J. Predictive a priori pressure-dependent kinetics. Science 2014, 346, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.; Stanton, J.F. A Steady-State Approximation to the Two-Dimensional Master Equation for Chemical Kinetics Calculations. J. Phys. Chem. A 2015, 119, 7627–7636. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Lee, H.; Matthews, D.A.; McCarthy, M.C.; Stanton, J.F. Stabilization of the Simplest Criegee Intermediate from the Reaction between Ozone and Ethylene: A High-Level Quantum Chemical and Kinetic Analysis of Ozonolysis. J. Phys. Chem. A 2015, 119, 5524–5533. [Google Scholar]
- Nguyen, T.L.; Stanton, J.F. Pragmatic Solution for a Fully E,J-Resolved Master Equation. J. Phys. Chem. A 2020, 124, 2907–2918. [Google Scholar] [CrossRef]
- Troe, J. Theory of Thermal Unimolecular Reactions at Low-Pressures.1. Solutions of Master Equation. J. Chem. Phys. 1977, 66, 4745–4757. [Google Scholar] [CrossRef]
- Reid, R.C.; Prausnitz, J.M.; Sherwood, T.K. The Properties of Gases and Liquids; McGraw-Hill: New York, NY, USA, 1977. [Google Scholar]
- Neufeld, P.D.; Aziz, R.A.; Janzen, A.R. Empirical Equations to Calculate 16 of Transport Collision Integrals-Omega(L, S)’ for Lennard-Jones (12-6) Potential. J. Chem. Phys. 1972, 57, 1100. [Google Scholar] [CrossRef]
- Holbrook, K.A.; Pilling, M.J.; Robertson, S.H.; Robinson, P.J. Unimolecular Reactions, 2nd ed.; Wiley: Chichester, UK; New York, NY, USA, 1996; 417p. [Google Scholar]
- Gilbert, R.G.; Smith, S.C. Theory of Unimolecular and Recombination Reactions; Blackwell Science Publications; Publishers’ Business Services Distributor: Oxford, UK; Boston, MA, USA, 1990; 356p. [Google Scholar]
- Forst, W. Adiabatic Rotations in Unimolecular Rate Theory. J. Chem. Phys. 1968, 48, 3665. [Google Scholar] [CrossRef]
- Forst, W. Unimolecular Reactions: A Concise Introduction; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2003; 319p. [Google Scholar]
- Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Some applications of the transition state method to the calculation of reaction velocities, especially in solution. Trans. Faraday Soc. 1935, 31, 0875–0893. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Garrett, B.C.; Klippenstein, S.J. Current status of transition-state theory. J. Phys. Chem. 1996, 100, 12771–12800. [Google Scholar] [CrossRef]
- Hase, W.L. Variational Unimolecular Rate Theory. Acc. Chem. Res. 1983, 16, 258–264. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Garrett, B.C. Variational Transition-State Theory. Annu. Rev. Phys. Chem. 1984, 35, 159–189. [Google Scholar] [CrossRef]
- Miller, W.H. Tunneling Corrections to Unimolecular Rate Constants, with Application to Formaldehyde. J. Am. Chem. Soc. 1979, 101, 6810–6814. [Google Scholar] [CrossRef]
- Bowman, J.M. Reduced Dimensionality Theory of Quantum Reactive Scattering. J. Phys. Chem. 1991, 95, 4960–4968. [Google Scholar] [CrossRef]
- Balakrishnan, N. Quantum mechanical investigation of the O + H2 -> OH + H reaction. J. Chem. Phys. 2003, 119, 195–199. [Google Scholar] [CrossRef]
- Miller, W.H. Semiclassical Theory for Non-Separable Systems-Construction of Good Action-Angle Variables for Reaction-Rate Constants. Faraday Discuss. 1977, 62, 40–46. [Google Scholar] [CrossRef]
- Miller, W.H.; Hernandez, R.; Handy, N.C.; Jayatilaka, D.; Willetts, A. Abinitio Calculation of Anharmonic Constants for a Transition-State, with Application to Semiclassical Transition-State Tunneling Probabilities. Chem. Phys. Lett. 1990, 172, 62–68. [Google Scholar] [CrossRef]
- Hernandez, R.; Miller, W.H. Semiclassical Transition-State Theory-a New Perspective. Chem. Phys. Lett. 1993, 214, 129–136. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Stanton, J.F.; Barker, J.R. A practical implementation of semi-classical transition state theory for polyatomics. Chem. Phys. Lett. 2010, 499, 9–15. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Stanton, J.F.; Barker, J.R. Ab Initio Reaction Rate Constants Computed Using Semiclassical Transition-State Theory: HO + H2 -> H2O + H and Isotopologues. J. Phys. Chem. A 2011, 115, 5118–5126. [Google Scholar] [CrossRef]
- Wang, F.G.; Landau, D.P. Efficient, multiple-range random walk algorithm to calculate the density of states. Phys. Rev. Lett. 2001, 86, 2050–2053. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.G.; Landau, D.P. Determining the density of states for classical statistical models: A random walk algorithm to produce a flat histogram. Phys. Rev. E 2001, 64, 056101. [Google Scholar] [CrossRef]
- Basire, M.; Parneix, P.; Calvo, F. Quantum anharmonic densities of states using the Wang-Landau method. J. Chem. Phys. 2008, 129, 081101. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Barker, J.R. Sums and Densities of Fully Coupled Anharmonic Vibrational States: A Comparison of Three Practical Methods. J. Phys. Chem. A 2010, 114, 3718–3730. [Google Scholar] [CrossRef]
- Baer, T.; Hase, W.L. Unimolecular Reaction Dynamics: Theory and Experiments; Oxford University Press: New York, NY, USA, 1996; 438p. [Google Scholar]
- Barker, J.R.; Nguyen, T.L.; Stanton, J.F.; Aieta, C.; Ceotto, M.; Gabas, F.; Kumar, T.J.D.; Li, C.G.L.; Lohr, L.L.; Maranzana, A.; et al. MULTIWELL Program Suite, Climate and Space Sciences and Engineering, University of Michigan. Ann. Arbor. MI 2021. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).