
Nicotine Affects Multiple Biological Processes in EpiDermTM Organotypic Tissues and Keratinocyte Monolayers



Abstract
1. Introduction
2. Materials and Methods
2.1. EpiDermTM Treatment with Nicotine
2.2. Sample Preparation for Proteomics
2.2.1. Sample Digestion
2.2.2. TMT Labeling and CIF Fractionation
2.2.3. LC-MS Acquisition and Analysis
2.3. Keratinocyte Culturing
2.4. Cell Transfection
2.5. Mitochondrial Morphology Analysis
2.6. Peroxisome Morphology Analysis
2.7. Fluorescence Microscopy
2.8. Western Blot
3. Results
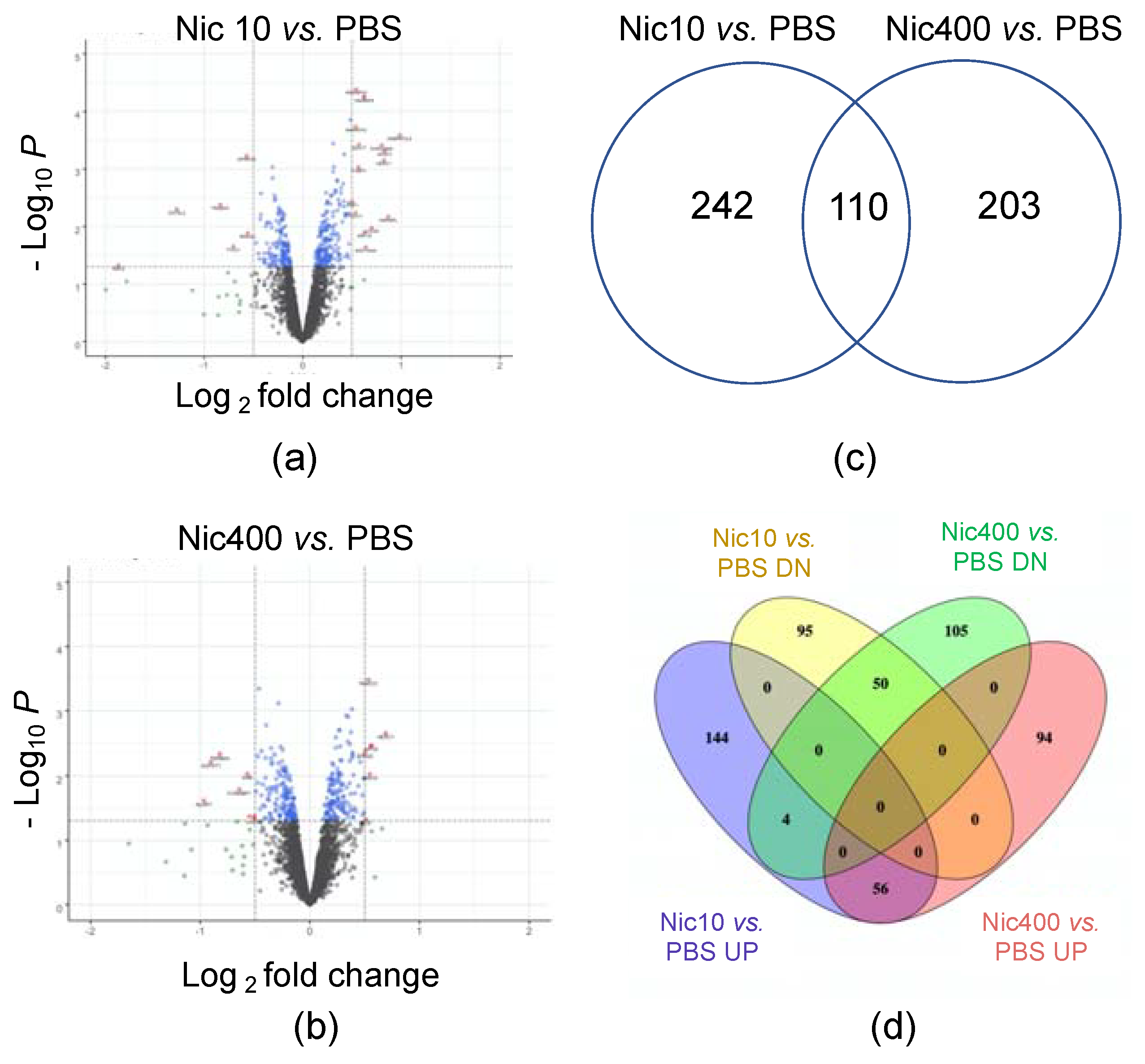
3.1. Proteomics Analysis of EpiDermTM Treated with Nic10 or Nic400
3.2. Canonical Pathways Affected in Nicotine-Treated EpiDermTM
3.3. IPA Toxicity Analysis
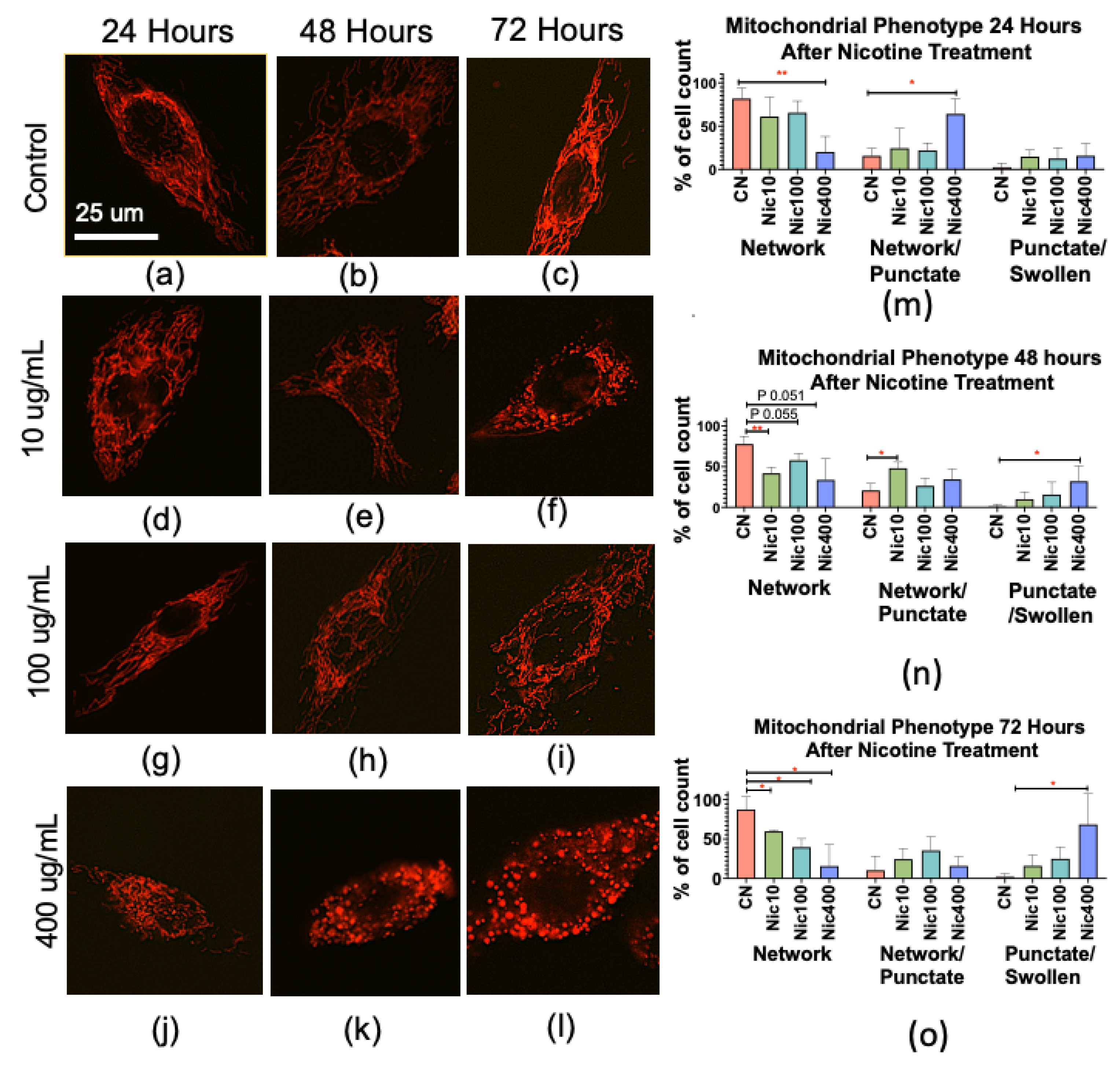
3.4. Nicotine Alters Mitochondrial Morphology in Human Keratinocytes
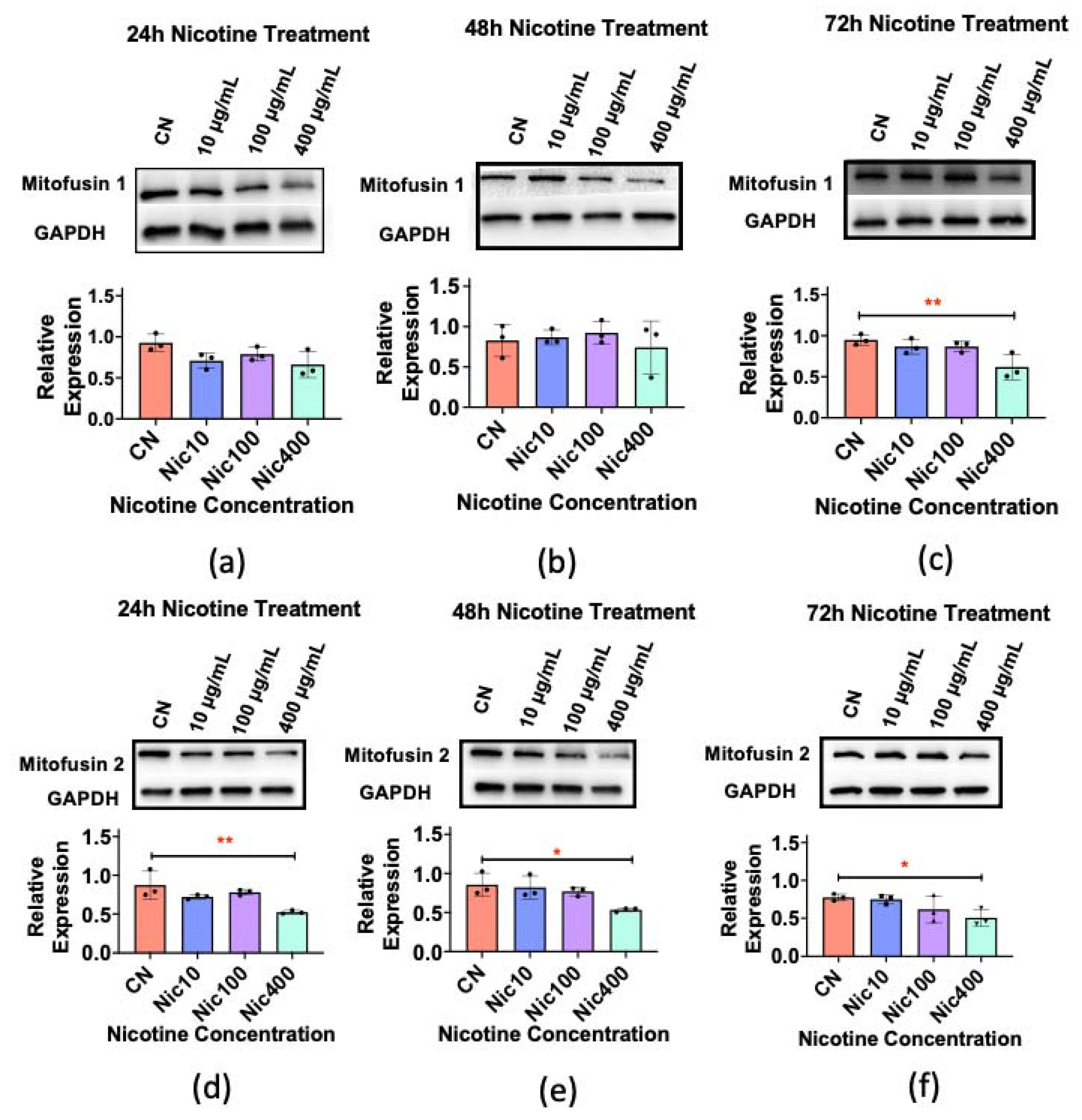
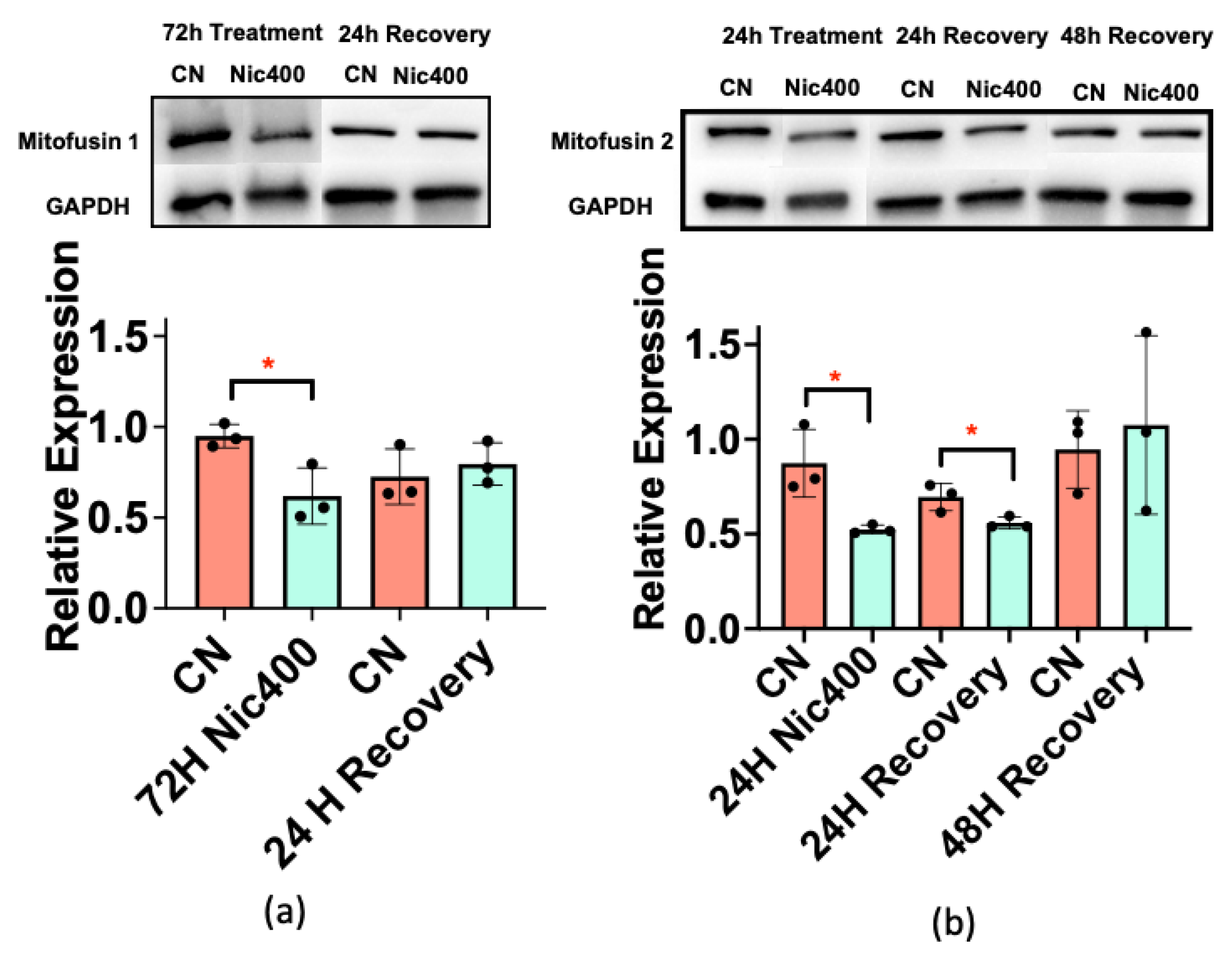
3.5. Mitofusin 1/2 Decreased in Keratinocytes Treated with Nic400
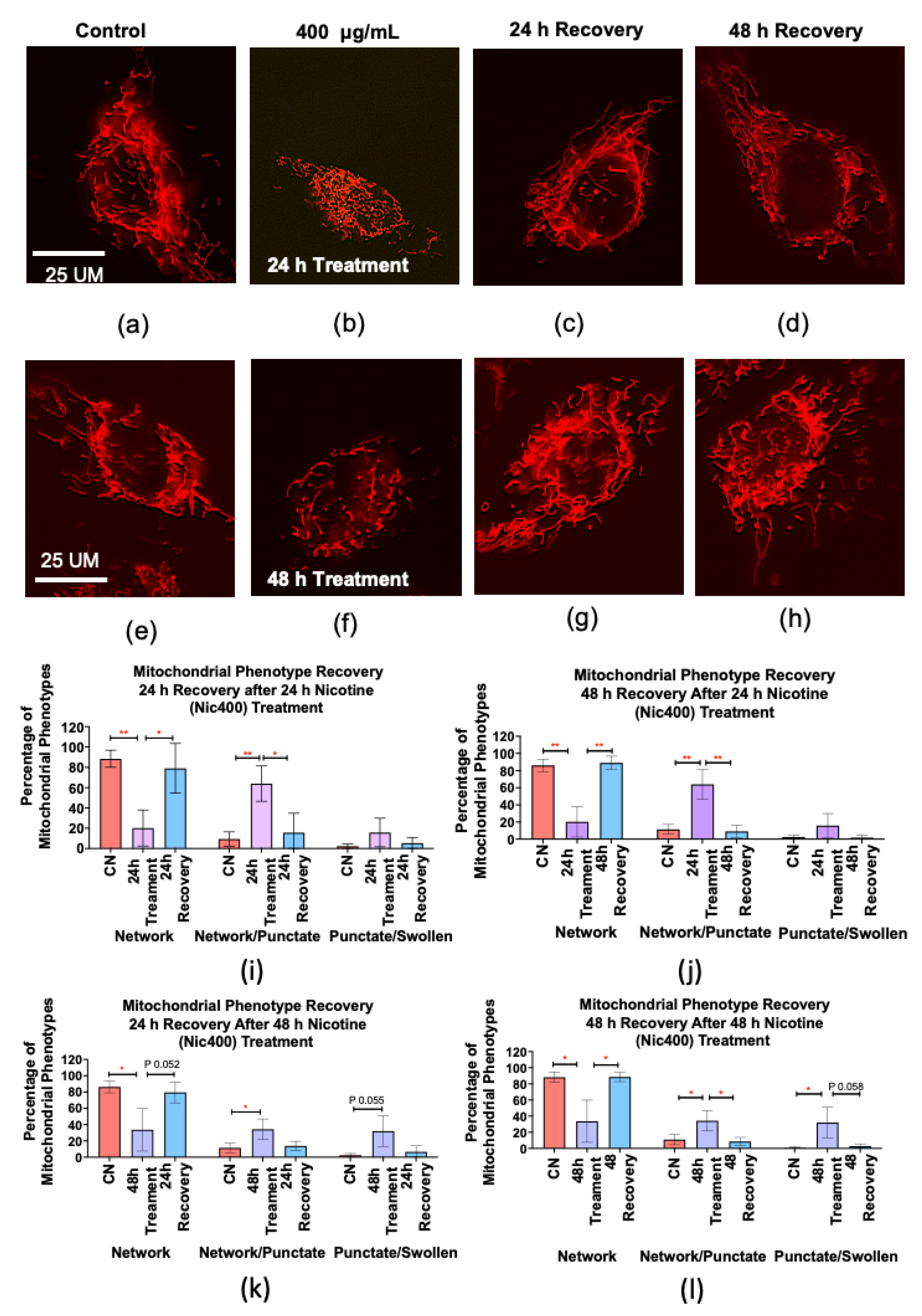
3.6. Recovery of Mitochondrial Morphology in Keratinocytes after Nicotine Treatment
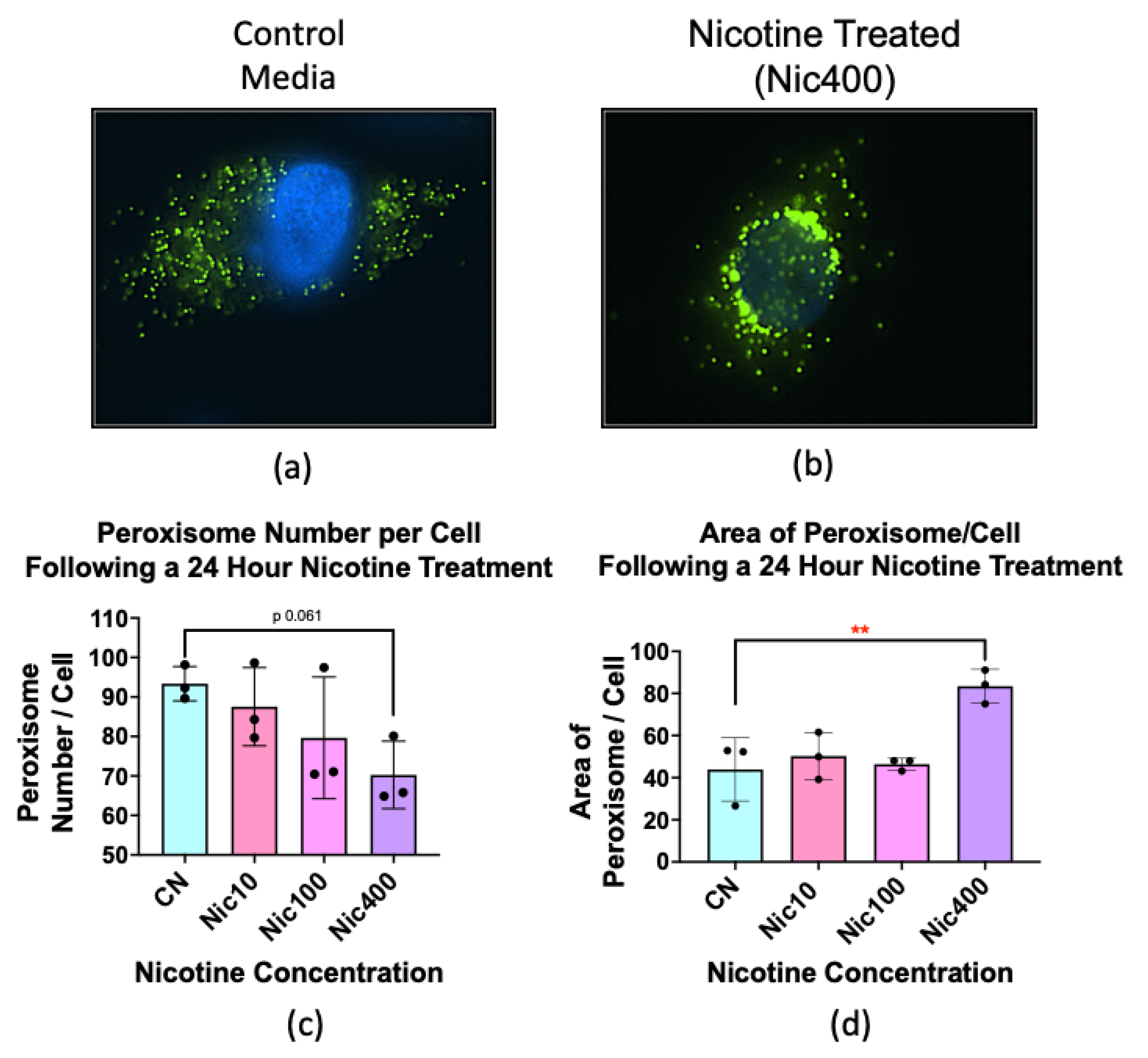
3.7. Nicotine Treatment Altered the Number and Size of Peroxisomes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mishra, A.; Chaturvedi, P.; Datta, S.; Sinukumar, S.; Joshi, P.; Garg, A. Harmful effects of nicotine. Indian J. Med. Paediatr. Oncol. 2015, 36, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Jacob, P., III; Benowitz, N.L.; Destaillats, H.; Gundel, L.; Hang, B.; Martins-Green, M.; Matt, G.E.; Quintana, P.J.E.; Samet, J.M.; Schick, S.F.; et al. Thirdhand Smoke: New Evidence, Challenges, and Future Directions. Chem. Res. Toxicol. 2017, 30, 270–294. [Google Scholar] [CrossRef] [PubMed]
- Solarino, B.; Rosenbaum, F.; Riesselmann, B.; Buschmann, C.T.; Tsokos, M. Death due to ingestion of nicotine-containing solution: Case report and review of the literature. Forensic Sci. Int. 2010, 195, e19–e22. [Google Scholar] [CrossRef] [PubMed]
- Tjoncke, J.A.; Goncalves, R.; Castaing, N.; Molimard, M.; Tovagliaro, F.; Titier, K. Death related to nicotine replacement therapy: A case report. Forensic Sci. Int. 2020, 309, 110223. [Google Scholar] [CrossRef]
- Mayer, B. How much nicotine kills a human? Tracing back the generally accepted lethal dose to dubious self-experiments in the nineteenth century. Arch. Toxicol. 2014, 88, 5–7. [Google Scholar] [CrossRef]
- McKnight, R.H.; Spiller, H.A. Green tobacco sickness in children and adolescents. Public. Health Rep. 2005, 120, 602–605. [Google Scholar] [CrossRef]
- Sørensen, L.T.; Zillmer, R.; Agren, M.; Ladelund, S.; Karlsmark, T.; Gottrup, F. Effect of smoking, abstention, and nicotine patch on epidermal healing and collagenase in skin transudate. Wound Repair Regen. 2009, 17, 347–353. [Google Scholar] [CrossRef]
- Matt, G.E.; Quintana, P.J.; Hovell, M.F.; Chatfield, D.; Ma, D.S.; Romero, R.; Uribe, A. Residual tobacco smoke pollution in used cars for sale: Air, dust, and surfaces. Nicotine Tob. Res. 2008, 10, 1467–1475. [Google Scholar] [CrossRef]
- Matt, G.E.; Quintana, P.J.; Destaillats, H.; Gundel, L.A.; Sleiman, M.; Singer, B.C.; Jacob, P.; Benowitz, N.; Winickoff, J.P.; Rehan, V.; et al. Thirdhand tobacco smoke: Emerging evidence and arguments for a multidisciplinary research agenda. Environ. Health Perspect. 2011, 119, 1218–1226. [Google Scholar] [CrossRef]
- Matt, G.E.; Quintan, P.J.; Zakarian, J.M.; Fortmann, A.L.; Chatfield, D.A.; Hoh, E.; Uribe, A.M.; Hovell, M.F. When smokers move out and non-smokers move in: Residential thirdhand smoke pollution and exposure. Tob. Control 2011, 20, e1. [Google Scholar] [CrossRef]
- Sleiman, M.; Logue, J.M.; Luo, W.; Pankow, J.F.; Gundel, L.A.; Destaillats, H. Inhalable Constituents of Thirdhand Tobacco Smoke: Chemical Characterization and Health Impact Considerations. J. Environ. Sci. Technol. 2014, 48, 13093–13101. [Google Scholar] [CrossRef] [PubMed]
- Bahl, V.; Jacob, P., III; Havel, C.; Schick, S.F.; Talbot, P. Thirdhand cigarette smoke: Factors affecting exposure and remediation. PLoS ONE 2014, 9, e108258. [Google Scholar] [CrossRef] [PubMed]
- Bahl, V.; Shim, H.J.; Payton, J., III; Dias, K.; Schick, S.F.; Talbot, P. Thirdhand Smoke: Chemical dynamics, cytotoxicity, and genotoxicity in outdoor and indoor environments. Toxicol. In Vitro 2016, 32, 220–231. [Google Scholar] [CrossRef]
- Pozuelos, G.L.; Jacob, P., III; Schick, S.F.; Omaiye, E.E.; Talbot, P. Adhesion and Removal of Thirdhand Smoke from Indoor Fabrics: A Method for Rapid Assessment and Identification of Chemical Repositories. Int. J. Environ. Res. Public Health 2021, 18, 3592. [Google Scholar] [CrossRef] [PubMed]
- Northrup, T.; Khan, A.M.; Jacob, P., III; Benowitz, N.L.; Hoh, E.; Hovell, M.F.; Matt, G.E.; Stotts, A.L. Thirdhand smoke contamination in hospital settings: Assessing exposure risk for vulnerable pediatric patients. Tob. Control 2016, 25, 619–623. [Google Scholar] [CrossRef]
- Matt, G.E.; Quintana, P.J.; Hovell, M.F.; Bernert, J.T.; Song, S.; Novianti, N.; Juarez, T.; Floro, J.; Gehrman, C.; Garcia, M.; et al. Households contaminated by environmental tobacco smoke: Sources of infant exposures. Tob. Control 2004, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Omaiye, E.E.; McWhirter, K.J.; Luo, W.; Pankow, J.F.; Talbot, P. High-Nicotine Electronic Cigarette Products: Toxicity of JUUL Fluids and Aerosols Correlates Strongly with Nicotine and Some Flavor Chemical Concentrations. Chem. Res. Toxicol. 2019, 32, 1058–1069. [Google Scholar] [CrossRef]
- Omaiye, E.E.; McWhirter, K.J.; Luo, W.; Tierney, P.A.; Pankow, J.F.; Talbot, P. High concentrations of flavor chemicals are present in electronic cigarette refill fluids. Sci. Rep. 2019, 9, 2468. [Google Scholar] [CrossRef]
- Davis, B.; Dang, M.; Kim, J.; Talbot, P. Nicotine concentrations in electronic cigarette refill and do-it-yourself fluids. Nicotine Tob. Res. 2015, 17, 134–141. [Google Scholar] [CrossRef]
- Trtchounian, A.; Talbot, P. Electronic nicotine delivery systems: Is there a need for regulation? Tob. Control 2010, 20, 47–52. [Google Scholar] [CrossRef]
- Garcia, R.; Allem, J.P.; Baezconde-Garbanati, L.; Unger, J.B.; Sussman, S. Employee and customer handling of nicotine-containing e-liquids in vape shops. Tob. Prev. Cessat 2016, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, F.L. Adverse effects of e-cigarette exposures. J. Community Health 2014, 39, 614–616. [Google Scholar] [CrossRef] [PubMed]
- Khachatoorian, C.; Jacob, P., III; Sen, A.; Zhu, Y.; Benowitz, N.L.; Talbot, P. Identification and quantification of electronic cigarette exhaled aerosol residue chemicals in field sites. Environ. Res. 2019, 170, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Maina, G.; Castagnoli, C.; Ghione, G.; Passini, V.; Adami, G.; Filon, F.L.; Crosera, M. Skin contamination as pathway for nicotine intoxication in vapers. Toxicol. In Vitr. 2017, 17, 30061–30069. [Google Scholar] [CrossRef]
- Casas, J.W.; Lewerenz, G.M.; Rankin, E.A.; Willoughby, J.A.; Blakeman, L.C.; McKim, J.M.; Coleman, K.P. In vitro human skin irritation test for evaluation of medical device extracts. Toxicol. In Vitr. 2013, 27, 2175–2183. [Google Scholar] [CrossRef]
- Kandárová, H.; Willoughby, J.A.; De Jong, W.H.; Letasiova, S.; Milasova, T.; Bachelor, M.A.; Breyfogle, B.; Handa, Y.; De La Fonteyne, L.; Coleman, K.P. Pre-validation of an in vitro skin irritation test for medical devices using the reconstructed human tissue model EpiDermTM. Toxicol. In Vitr. 2018, 50, 407–417. [Google Scholar] [CrossRef]
- Hughes, C.S.; Foehr, S.; Garfield, D.A.; Furlong, E.E.; Steinmetz, L.M.; Krijgsveld, J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 2014, 10, 757. [Google Scholar] [CrossRef]
- Deng, W.; Sha, J.; Plath, K.; Wohlschlegel, J.A. Carboxylate-Modified Magnetic Bead (CMMB)-Based Isopropanol Gradient Peptide Fractionation (CIF) Enables Rapid and Robust Off-Line Peptide Mixture Fractionation in Bottom-Up Proteomics. Mol. Cell. Proteom. 2021, 20, 100039. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Huang, T.; Choi, M.; Tzouros, M.; Golling, S.; Pandya, N.J.; Banfai, B.; Dunkley, T.; Vitek, O. MSstatsTMT: Statistical Detection of Differentially Abundant Proteins in Experiments with Isobaric Labeling and Multiple Mixtures. Mol. Cell. Proteom. 2020, 19, 1706–1723. [Google Scholar] [CrossRef]
- Abramoff, M.D.; Magalhaes, P.J.; Ram, S.J. Image Processing with Image. J. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Ganguli, G.; Mukherjee, U.; Sonawane, A. Peroxisomes and Oxidative Stress: Their Implications in the Modulation of Cellular Immunity During Mycobacterial Infection. Front. Microbiol. 2019, 10, 1121. [Google Scholar] [CrossRef] [PubMed]
- Tsoyi, K.; Jang, H.J.; Kim, J.W.; Chang, H.K.; Lee, Y.S.; Pae, H.O.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Chung, H.T.; et al. Stimulation of alpha7 nicotinic acetylcholine receptor by nicotine attenuates inflammatory response in macrophages and improves survival in experimental model of sepsis through heme oxygenase-1 induction. Antioxid. Redox Signal 2011, 14, 2057–2070. [Google Scholar] [CrossRef]
- Kalra, R.; Singh, S.P.; Pena-Philippides, J.C.; Langley, R.J.; Razani-Boroujerdi, S.; Sopori, M.L. Immunosuppressive and anti-inflammatory effects of nicotine administered by patch in an animal model. Clin. Diagn. Lab Immunol. 2004, 11, 563–568. [Google Scholar] [CrossRef]
- Staples, J.; Klein, D. Can nicotine use alleviate symptoms of psoriasis? Can. Fam. Physician 2012, 58, 404–408. [Google Scholar]
- Scott, D.A.; Martin, M. Exploitation of the nicotinic anti-inflammatory pathway for the treatment of epithelial inflammatory diseases. World J. Gastroenterol. 2006, 12, 7451–7459. [Google Scholar] [CrossRef]
- Misery, L. Nicotine effects on skin: Are they positive or negative? Exp. Dermatol. 2004, 13, 665–670. [Google Scholar] [CrossRef]
- Ingram, J.R. Nicotine: Does it have a role in the treatment of skin disease? Postgrad. Med. J. 2009, 85, 196–201. [Google Scholar] [CrossRef]
- Stegemann, A.; Böhm, M. Targeting the α7 nicotinic acetylcholine receptor-A novel road towards the future treatment of skin diseases. Exp. Dermatol. 2020, 29, 924–931. [Google Scholar] [CrossRef]
- Radek, K.A.; Elias, P.M.; Taupenot, L.; Mahata, S.K.; O’Connor, D.T.; Gallo, R.L. Neuroendocrine nicotinic receptor activation increases susceptibility to bacterial infections by suppressing antimicrobial peptide production. Cell Host Microbe 2010, 7, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Kishibe, M.; Griffin, T.M.; Radek, K.A. Keratinocyte nicotinic acetylcholine receptor activation modulates early TLR2-mediated wound healing responses. Int. Immunopharmacol. 2015, 29, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Cañe-Dorantes, L.; Cañedo-Ayala, M. Skin acute wound healing: A comprehensive review. Int. J. Inflamm. 2019, 2019, 3706315. [Google Scholar] [CrossRef]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef] [PubMed]
- Seeger, M.A.; Paller, A.S. The Roles of Growth Factors in Keratinocyte Migration. Adv. Wound Care 2015, 4, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Chernyavsky, A.I.; Arredondo, J.; Qian, J.; Galitovskiy, V.; Grando, S.A. Coupling of ionic events to protein kinase signaling cascades upon activation of alpha7 nicotinic receptor: Cooperative regulation of alpha2-integrin expression and Rho kinase activity. J. Biol. Chem. 2009, 284, 22140–22148. [Google Scholar] [CrossRef]
- Chernyavsky, A.I.; Arredondo, J.; Marubio, L.M.; Grando, S.A. Differential regulation of keratinocyte chemokinesis and chemotaxis through distinct nicotinic receptor subtypes. J. Cell Sci. 2004, 117, 5665–5679. [Google Scholar] [CrossRef]
- Martins-Green, M.; Adhami, N.; Frankos, M.; Valdez, M.; Goodwin, B.; Lyubovitsky, J.; Dhall, S.; Garcia, M.; Egiebor, I.; Martinez, B.; et al. Cigarette smoke toxins deposited on surfaces: Implications for human health. PLoS ONE 2014, 9, e86391. [Google Scholar] [CrossRef]
- Liu, B.; Qian, S.B. Translational reprogramming in cellular stress response. Wiley Interdiscip. Rev. RNA 2014, 5, 301–315. [Google Scholar] [CrossRef]
- Warnakulasuriyarachchi, D.; Cerquozzi, S.; Cheung, H.H.; Holcik, M. Translational induction of the inhibitor of apoptosis protein HIAP2 during endoplasmic reticulum stress attenuates cell death and is mediated via an inducible internal ribosome entry site element. J. Biol. Chem. 2004, 279, 17148–17157. [Google Scholar] [CrossRef]
- Huang, W.; Placzek, A.N.; Viana Di Prisco, G.; Khatiwada, S.; Sidrauski, C.; Krnjević, K.; Walter, P.; Dani, J.A.; Costa-Mattioli, M. Translational control by eIF2α phosphorylation regulates vulnerability to the synaptic and behavioral effects of cocaine. Elife 2016, 5, e12052. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.K.; Holloway, A.C.; Hardy, D.B. Nicotine directly induces endoplasmic reticulum stress response in rat placental trophoblast giant cells. Toxicol. Sci. 2016, 151, 23–34. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Placzek, A.N.; Molfese, D.L.; Khatiwada, S.; Di Prisco, G.V.; Huang, W.; Sidrauski, C.; Krnjević, K.; Amos, C.L.; Ray, R.; Dani, J.A.; et al. Translational control of nicotine-evoked synaptic potentiation in mice and neuronal responses in human smokers by eIF2α. Elife 2016, 5, e12056. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Kim, H.; Kim, I. Ribosomal protein L19 overexpression activates the unfolded protein response and sensitizes MCF7 breast cancer cells to endoplasmic reticulum stress-induced cell death. Biochem. Biophys. Res. Commun. 2014, 450, 673–678. [Google Scholar] [CrossRef]
- Meng, T.T.; Wang, W.; Meng, F.L.; Wang, S.Y.; Wu, H.H.; Chen, J.M.; Zheng, Y.; Wang, G.X.; Zhang, M.X.; Li, Y.; et al. Nicotine Causes Mitochondrial Dynamics Imbalance and Apoptosis Through ROS Mediated Mitophagy Impairment in Cardiomyocytes. Front Physiol. 2021, 12, 650055. [Google Scholar] [CrossRef]
- Zahedi, A.; Phandthong, R.; Chaili, A.; Leung, S.; Omaiye, E.; Talbot, P. Mitochondrial Stress Response in Neural Stem Cells Exposed to Electronic Cigarettes. iScience 2019, 16, 250–269. [Google Scholar] [CrossRef]
- Grando, S.A.; Horton, R.M.; Mauro, T.M.; Kist, D.A.; Lee, T.X.; Dahl, M.V. Activation of keratinocyte nicotinic cholinergic receptors stimulates calcium influx and enhances cell differentiation. J. Investig. Dermatol. 1996, 107, 412–418. [Google Scholar] [CrossRef]
- Hirata, N.; Yamada, S.; Asanagi, M.; Sekino, Y.; Kanda, Y. Nicotine induces mitochondrial fission through mitofusin degradation in human multipotent embryonic carcinoma cells. Biochem. Biophys. Res. Commun. 2016, 470, 300–305. [Google Scholar] [CrossRef]
- Bahl, V.; Johnson, K.; Phandthong, R.; Zahedi, A.; Schick, S.F.; Talbot, P. Thirdhand cigarette smoke causes stress-induced mitochondrial hyperfusion and alters the transcriptional profile of stem cells. Toxicol. Sci. 2016, 153, 55–69. [Google Scholar] [CrossRef]
- Pascual-Ahuir, A.; Manzanares-Estreder, S.; Proft, M. Pro- and Antioxidant Functions of the Peroxisome-Mitochondria Connection and Its Impact on Aging and Disease. Oxid. Med. Cell. Longev. 2017, 2017, 9860841. [Google Scholar] [CrossRef]
- Tripathi, D.N.; Walker, C.L. The peroxisome as a cell signaling organelle. Curr. Opin. Cell Biol. 2016, 39, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Till, A.; Lakhani, R.; Burnett, S.F.; Subramani, S. Pexophagy: The selective degradation of peroxisomes. Int. J. Cell Biol. 2012, 2012, 512721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Jean Beltran, P.M.; Cook, K.C.; Hashimoto, Y.; Galitzine, C.; Murray, L.A.; Vitek, O.; Cristea, I.M. Infection-Induced Peroxisome Biogenesis Is a Metabolic Strategy for Herpesvirus Replication. Cell. Host Microbe 2018, 24, 526–541.e7. [Google Scholar] [CrossRef]
- Baumgart, E.; Vanhorebeek, I.; Grabenbauer, M.; Borgers, M.; Declercq, P.E.; Fahimi, H.D.; Baes, M. Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model for Zellweger syndrome (PEX5 knockout mouse). Am. J. Pathol. 2001, 159, 1477–1494. [Google Scholar] [CrossRef]
- Huang, K.; Kim, J.; Vo, P.; Miao, T.; Bai, H. Peroxisome import stress impairs ribosome biogenesis and induces integrative stress response through eIF2α phosphorylation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kim, J.; Bai, H. Peroxisomal Stress Response and Inter-Organelle Communication in Cellular Homeostasis and Aging. Antioxidants 2022, 11, 192. [Google Scholar] [CrossRef]
- Kovacs, W.J.; Charles, K.N.; Walter, K.M.; Shackelford, J.E.; Wikander, T.M.; Richards, M.J.; Fliesler, S.J.; Krisans, S.K.; Faust, P.L. Peroxisome deficiency-induced ER stress and SREBP-2 pathway activation in the liver of newborn PEX2 knock-out mice. Biochim. Biophys. Acta 2012, 1821, 895–907. [Google Scholar] [CrossRef]
- Piao, L.; Dorotea, D.; Jiang, S.; Koh, E.H.; Oh, G.T.; Ha, H. Impaired Peroxisomal Fitness in Obese Mice, a Vicious Cycle Exacerbating Adipocyte Dysfunction via Oxidative Stress. Antioxid. Redox Signal 2019, 31, 1339–1351. [Google Scholar] [CrossRef]
- Ahlemeyer, B.; Gottwald, M.; Baumgart-Vogt, E. Deletion of a single allele of the Pex11β gene is sufficient to cause oxidative. stress, delayed differentiation and neuronal death in mouse brain. Dis. Model Mech. 2012, 5, 125–140. [Google Scholar] [CrossRef]
- López-Erauskin, J.; Galino, J.; Ruiz, M.; Cuezva, J.M.; Fabregat, I.; Cacabelos, D.; Boada, J.; Martínez, J.; Ferrer, I.; Pamplona, R.; et al. Impaired mitochondrial oxidative phosphorylation in the peroxisomal disease X-linked adrenoleukodystrophy. Hum. Mol. Genet. 2013, 22, 3296–3305. [Google Scholar] [CrossRef] [PubMed]
- Pozuelos, G.L.; Kagda, M.S.; Schick, S.; Girke, T.; Volz, D.C.; Talbot, P. Experimental Acute Exposure to Thirdhand Smoke and Changes in the Human Nasal Epithelial Transcriptome: A Randomized Clinical Trial. JAMA Netw. Open 2019, 2, e196362. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Canonical Pathways | −log(p-Value) | z-Score | Proteins |
|---|---|---|---|
| Protein-Synthesis-Related Pathway | |||
| EIF2 Signaling | 19.2 | 2.53 | EIF1, EIF3C, EIF3D, EIF3L, EIF4A2, MAP2K1, PTBP1, RPL15, RPL18A, RPL19, RPL24, RPL30, RPL38, RPL5, RPL6, RPL7A, RPS11, RPS13, RPS17, RPS2, RPS21, RPS24, RPS26, RPS3, RPS4X, RPS4Y2, RPS8, RPS9, RPSA |
| Oxidative-Stress-Related Pathway | |||
| Oxidative Phosphorylation | 4.39 | 1 | MT-ATP6, NDUFA4, NDUFB5, NDUFB8, NDUFS1, NDUFS6, UQCRC1, UQCRC2, UQCRQ |
| Canonical Pathways | −log(p-Value) | z-Score | Proteins |
|---|---|---|---|
| Apoptosis-Related Pathways | |||
| Apoptosis Signaling | 3.27 | −0.378 | BAX, CASP7, CASP8, LMNA, MAP2K1, NFKBIB, RELA |
| Induction of Apoptosis by HIV1 | 2.73 | −1.342 | BAX, CASP8, FADD, NFKBIB, RELA |
| TNFR1 Signaling | 3.17 | −2.236 | CASP7, CASP8, FADD, NFKBIB, RELA |
| Cell–Cell Adhesion-Related Pathways | |||
| ILK Signaling | 2.22 | −1.342 | DSP, GSK3A, ITGB4, KRT18, MTOR, RAC2, RELA, TMSB10/TMSB4X |
| Immune-Related Pathways | |||
| Role of MAPK Signaling in Promoting the Pathogenesis of Influenza | 1.68 | −0.447 | ATP6V1C1, BAX, MAP2K1, MAP2K3, NFKBIB |
| IL-6 Signaling | 2.1 | −0.816 | IL36B, MAP2K1, MAP2K3, NFKBIB, RELA, TAB1 |
| IL-1 Signaling | 1.34 | −1 | MAP2K3, NFKBIB, RELA, TAB1 |
| LPS-Stimulated MAPK Signaling | 1.55 | −1 | MAP2K1, MAP2K3, NFKBIB, RELA |
| IL-15 Production | 1.59 | −1.342 | CSK, MAP2K1, MAP2K3, PTK2B, RELA |
| IL-8 Signaling | 2.04 | −1.89 | BAX, EIF4EBP1, MAP2K1, MTOR, NFKBIB, PTK2B, RAC2, RELA |
| Canonical Pathways | −log(p- Value) | z-Score | Proteins |
|---|---|---|---|
| Protein-Synthesis-Related Pathway | |||
| EIF2 Signaling | 6.91 | 2 | EIF1, EIF2B2, EIF2S2, EIF3M, EIF4A2, PTBP1, RPL18A, RPL38, RPS12, RPS15, RPS24, RPS26, RPS3, RPS4X, RRAS2 |
| Canonical Pathways | −log(p-Value) | z-Score | Proteins |
|---|---|---|---|
| Immune-Related Pathways | |||
| IL-1 Signaling | 2.15 | −0.447 | MAP2K3, NFKBIB, PRKAR1A, RELA, TAB1 |
| ERK/MAPK Signaling | 2.29 | −0.707 | PAK1, PLA2G4A, PLA2G4D, PPP2R5A, PRKAR1A, PRKCI, PTK2B, RRAS2 |
| MIF-Mediated Glucocorticoid Regulation | 4.05 | −1.342 | MIF, NFKBIB, PLA2G4A, PLA2G4D, RELA |
| MIF Regulation of Innate Immunity | 3.6 | −1.342 | MIF, NFKBIB, PLA2G4A, PLA2G4D, RELA |
| Apoptosis-Related Pathway | |||
| Death Receptor Signaling | 1.54 | −1 | ARHGDIB, CASP8, NFKBIB, RELA |
| IPA Toxicity Lists | −log(p-Value) | Proteins |
|---|---|---|
| Mitochondrial Dysfunction | 5.41 | CASP8, CPT1A, MT-ATP6, NDUFA4, NDUFB5, NDUFB8, NDUFS1, NDUFS6, PARK7, UQCRC1, UQCRC2, UQCRQ |
| NRF2-Mediated Oxidative Stress Response | 2.27 | AKR1A1, BACH1, DNAJB1, DNAJB11, DNAJB2, DNAJC11, DNAJC5, MAP2K1, MAP2K3 |
| Decreases Respiration of Mitochondria | 1.83 | MFN1, PKM |
| Pro-Apoptosis | 1.72 | BAX, CASP7, CASP8 |
| Increases Transmembrane Potential of Mitochondria and Mitochondrial Membrane | 1.52 | BAX, CASP7, DNAJB1 |
| Mechanism of Gene Regulation by Peroxisome Proliferators via PPARα | 1.4 | MAP2K1, NFKBIB, RELA, TAB1 |
| IPA Toxicity Lists | −log(p-Value) | Proteins |
|---|---|---|
| Mitochondrial Dysfunction | 4.34 | CASP8, COX7A2, COX7C, NDUFA4, NDUFA8, NDUFA9, NDUFB5, NDUFS1, TXN2, UQCRQ |
| PXR/RXR Activation | 2.95 | GSTM2, PCK2, PRKAR1A, RELA, SCD |
| NRF2-Mediated Oxidative Stress Response | 2.59 | AKR1A1, BACH1, CDC34, DNAJC13, DNAJC5, GSTM2, MAP2K3, PRKCI, RRAS2 |
| Mechanism of Gene Regulation by Peroxisome Proliferators via PPARα | 2.23 | FAT1, NFKBI, PRKAR1A, RELA, TAB1 |
| Decreases Depolarization of Mitochondria and Mitochondrial Membrane | 2.07 | PAK1, PLA2G4A, PPIA |
| Hypoxia-Inducible Factor Signaling | 1.99 | EIF1, EIF2B2, EIF2S2, UBE2N |
| RAR Activation | 1.91 | PRKAR1A, PRKCI, PRMT1, RDH12, RELA, SDR16C5, SMARCC2 |
| Aryl Hydrocarbon Receptor Signaling | 1.87 | ALDH3A1, CDKN2A, CTSD, GSTM2,NEDD8, RELA |
| Cholesterol Biosynthesis | 1.81 | IDI1, SQLE |
| LXR/RXR Activation | 1.77 | ACACA, IL36B, PLTP, RELA, SCD |
| Oxidative Stress | 1.5 | GSTM2, RELA, S100A7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pozuelos, G.L.; Rubin, M.; Vargas, S.; Ramirez, E.; Bandaru, D.; Sha, J.; Wohlschlegel, J.; Talbot, P. Nicotine Affects Multiple Biological Processes in EpiDermTM Organotypic Tissues and Keratinocyte Monolayers. Atmosphere 2022, 13, 810. https://doi.org/10.3390/atmos13050810
Pozuelos GL, Rubin M, Vargas S, Ramirez E, Bandaru D, Sha J, Wohlschlegel J, Talbot P. Nicotine Affects Multiple Biological Processes in EpiDermTM Organotypic Tissues and Keratinocyte Monolayers. Atmosphere. 2022; 13(5):810. https://doi.org/10.3390/atmos13050810
Chicago/Turabian StylePozuelos, Giovanna L., Matine Rubin, Samantha Vargas, Erik Ramirez, Dhiresh Bandaru, Jihui Sha, James Wohlschlegel, and Prue Talbot. 2022. "Nicotine Affects Multiple Biological Processes in EpiDermTM Organotypic Tissues and Keratinocyte Monolayers" Atmosphere 13, no. 5: 810. https://doi.org/10.3390/atmos13050810
APA StylePozuelos, G. L., Rubin, M., Vargas, S., Ramirez, E., Bandaru, D., Sha, J., Wohlschlegel, J., & Talbot, P. (2022). Nicotine Affects Multiple Biological Processes in EpiDermTM Organotypic Tissues and Keratinocyte Monolayers. Atmosphere, 13(5), 810. https://doi.org/10.3390/atmos13050810