
Secondary Organic and Inorganic Aerosol Formation from a GDI Vehicle under Different Driving Conditions

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Vehicle and Emissions Testing
2.2. Driving Cycles
2.3. Photooxidation Chamber Experiments
3. Results
3.1. Tailpipe Gaseous and Particulate Emissions
3.2. Tailpipe Aerosol Composition
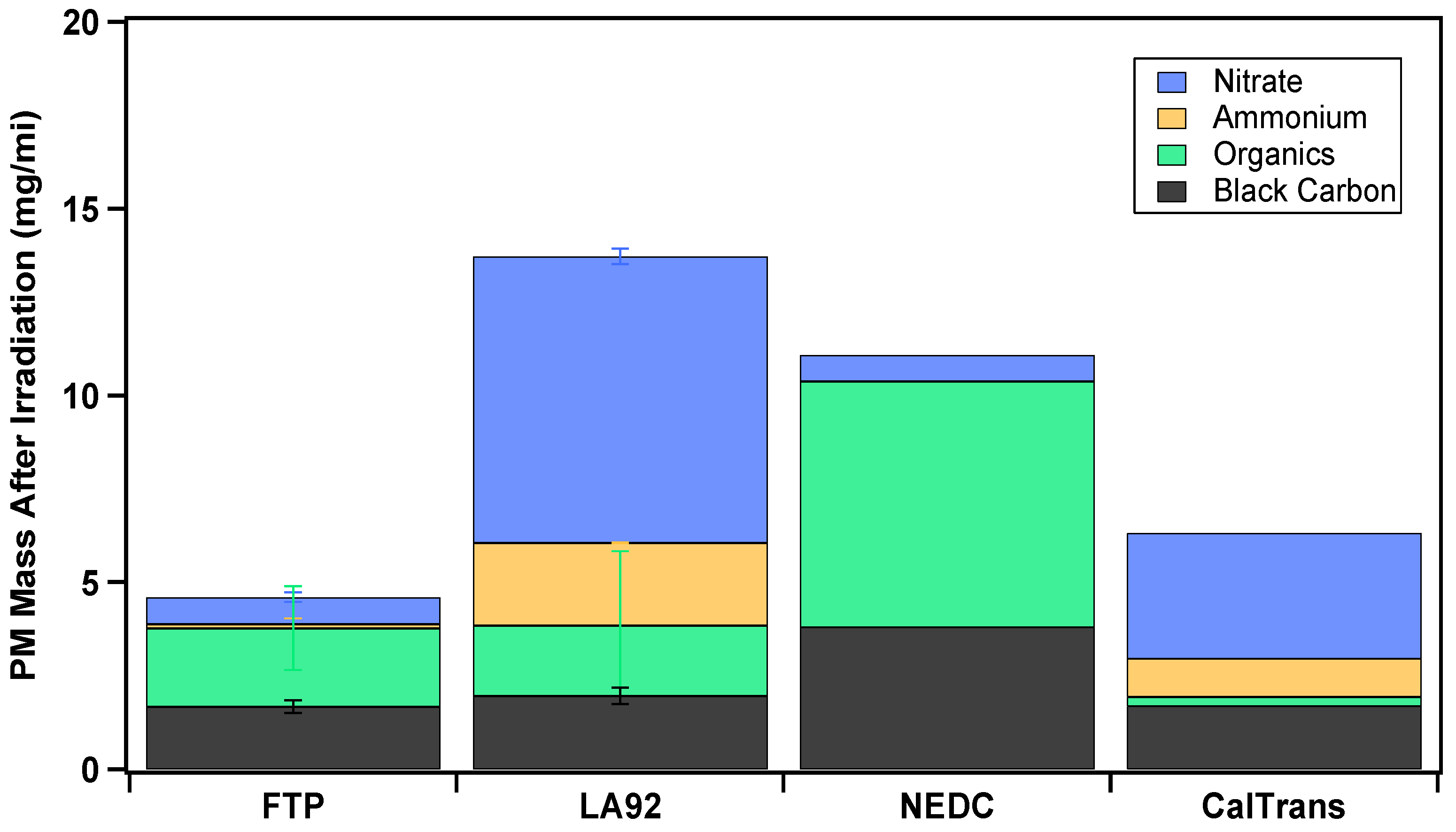
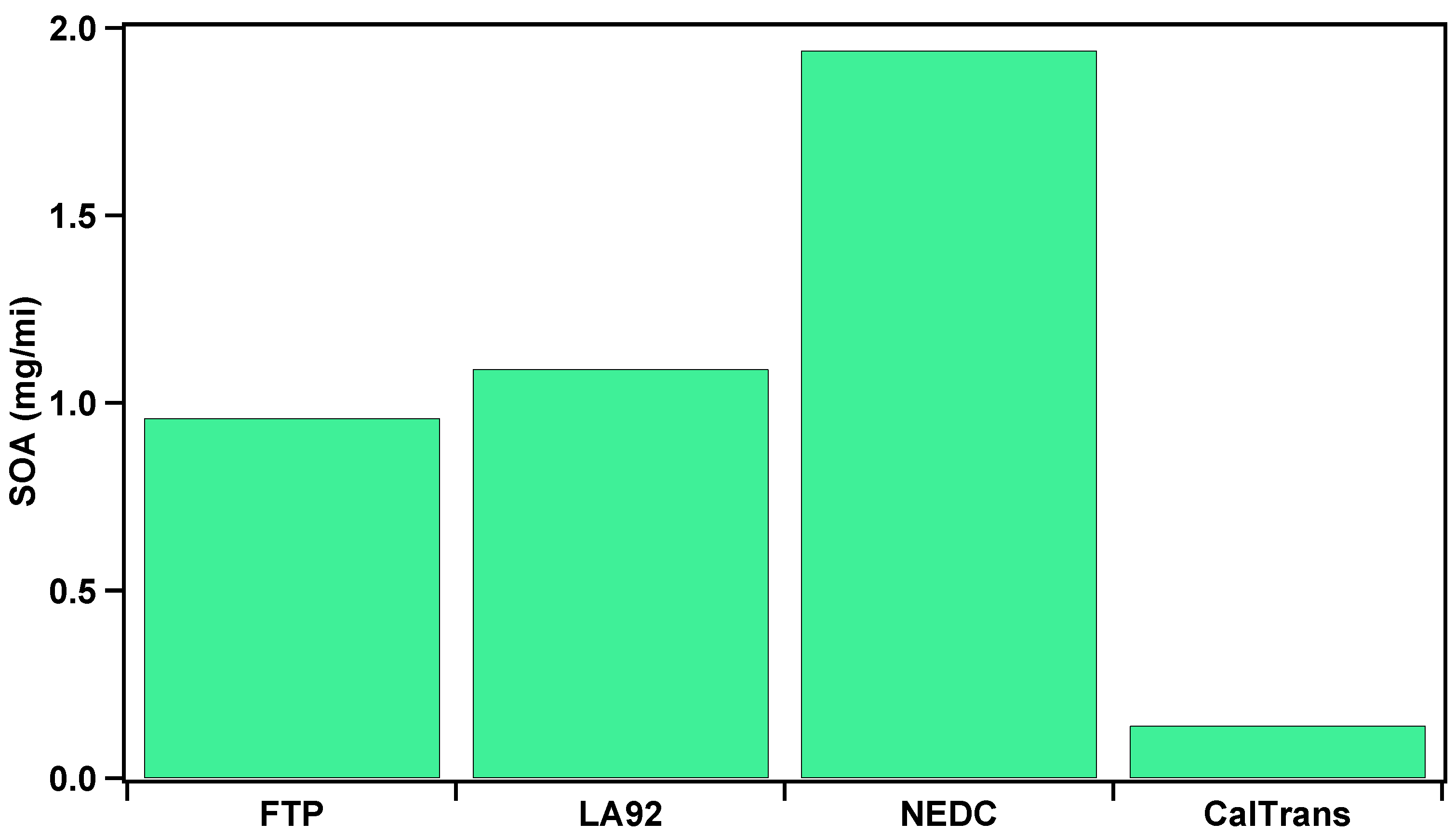
3.3. Total Secondary Aerosol Composition and SOA Formation
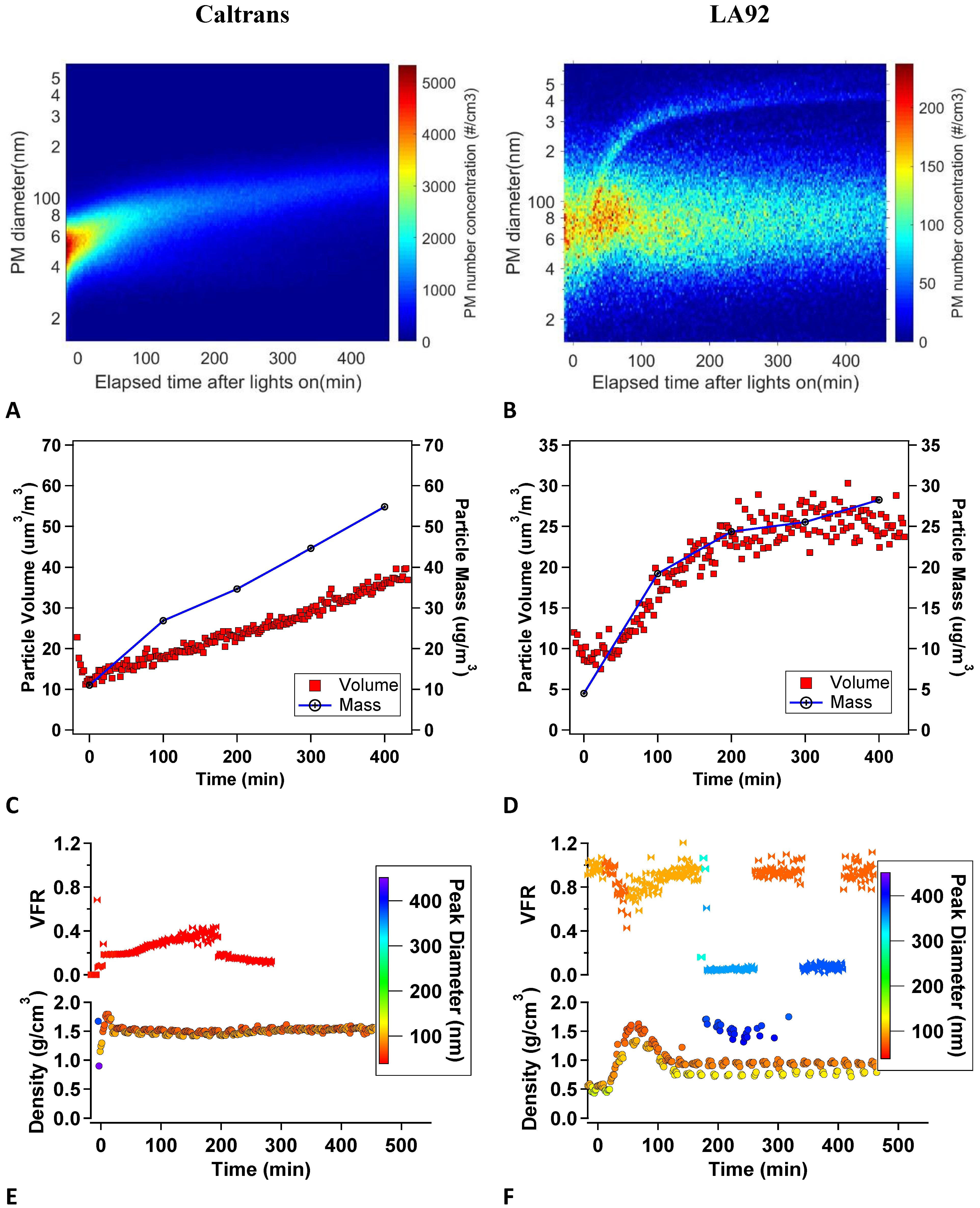
3.4. Particle Size Distributions and Composition
3.5. Ammonium Nitrate Condensation Experiments
3.6. Atmospheric Relevance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- May, A.A.; Nguyen, N.T.; Presto, A.A.; Gordon, T.; Lipsky, E.M.; Karve, M.; Gutierrez, A.; Robertson, W.H.; Zhang, M.; Brandow, C.; et al. Gas- and particle-phase primary emissions from in-use, on-road gasoline and diesel vehicles. Atmos. Environ. 2014, 88, 247–260. [Google Scholar] [CrossRef]
- Pang, Y.; Fuentes, M.; Rieger, P. Trends in the emissions of Volatile Organic Compounds (VOCs) from light-duty gasoline vehicles tested on chassis dynamometers in Southern California. Atmos. Environ. 2014, 83, 127–135. [Google Scholar] [CrossRef]
- Bahreini, R.; Middlebrook, A.; de Gouw, J.; Warneke, C.; Trainer, M.; Brock, C.A.; Stark, H.; Brown, S.S.; Dube, W.P.; Gilman, J.B.; et al. Gasoline emissions dominate over diesel in formation of secondary organic aerosol mass. Geophys. Res. Lett. 2012, 39, L06805. [Google Scholar] [CrossRef] [Green Version]
- Jathar, S.H.; Woody, M.; Pye, H.O.T.; Baker, K.R.; Robinson, A.L. Chemical transport model simulations of organic aerosol in southern California: Model evaluation and gasoline and diesel source contributions. Atmos. Chem. Phys. 2017, 17, 4305–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, S.M.; El Haddad, I.; Pieber, S.M.; Zardini, A.A.; Suarez-Bertoa, R.; Clairotte, M.; Daellenbach, K.R.; Slowik, J.G.; Baltensperger, U.; Temime-Roussel, B.; et al. Gasoline cars produce more carbonaceous particulate matter than modern filter-equipped diesel cars. Sci. Rep. 2017, 7, 4926. [Google Scholar] [CrossRef]
- Robinson, A.L.; Donahue, N.M.; Shrivastava, M.K.; Weitkamp, E.A.; Sage, A.M.; Grieshop, A.P.; Lane, T.E.; Pierce, J.R.; Pandis, S.N. Rethinking organic aerosols: Semivolatile emissions and photochemical aging. Science 2007, 315, 1259–1262. [Google Scholar] [CrossRef]
- Jimenez, J.L.; Canagaratna, M.R.; Donahue, N.M.; Prevot, A.S.H.; Zhang, Q.; Kroll, J.H.; Decarlo, P.F.; Allan, J.D.; Coe, H.; Ng, N.L.; et al. Evolution of organic aerosols in the atmosphere. Science 2009, 326, 1525–1529. [Google Scholar] [CrossRef]
- Drozd, G.T.; Zhao, Y.; Saliba, G.; Frodin, B.; Maddox, C.; Oliver Chang, M.-C.; Maldonado, H.; Sardar, S.; Weber, R.J.; Robinson, A.L.; et al. Detailed speciation of intermediate volatility and semivolatile organic compound emissions from gasoline vehicles: Effects of cold-starts and implications for secondary organic aerosol formation. Environ. Sci. Technol. 2018, 53, 1706–1714. [Google Scholar] [CrossRef]
- Gentner, D.R.; Jathar, S.H.; Gordon, T.D.; Bahreini, R.; Day, D.A.; Haddad, I.E.; Haynes, P.L.; Pieber, S.M.; Platt, S.M.; De Gouw, J.; et al. Review of urban secondary organic aerosol formation from gasoline and diesel motor vehicle emissions. Environ. Sci. Technol. 2017, 51, 1074–1093. [Google Scholar] [CrossRef]
- Peng, J.; Hu, M.; Du, Z.; Wang, Y.; Zheng, J.; Zhang, W.; Yang, Y.; Qin, Y.; Zheng, R.; Xiao, Y.; et al. Gasoline aromatics: A critical determinant of urban secondary organic aerosol formation. Atmos. Chem. Phys. 2017, 17, 10743–10752. [Google Scholar] [CrossRef] [Green Version]
- Roth, P.; Yang, J.; Stamatis, C.; Barsanti, K.C.; Cocker, D.R., III; Durbin, T.D.; Asa-Awuku, A.; Karavalakis, G. Evaluating the relationships between aromatic and ethanol levels in gasoline on secondary aerosol formation from a gasoline direct injection vehicle. Sci. Total Environ. 2020, 737, 140333. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.D.; Presto, A.A.; May, A.A.; Nguyen, N.T.; Lipsky, E.M.; Donahue, N.M.; Gutierrez, A.; Zhang, M.; Maddox, C.; Rieger, P.; et al. Secondary organic aerosol formation exceeds primary particulate matter emissions for light-duty gasoline vehicles. Atmos. Chem. Phys. 2014, 14, 4661–4678. [Google Scholar] [CrossRef] [Green Version]
- Vu, D.; Roth, P.; Berte, T.; Yang, J.; Cocker, D.; Durbin, T.D.; Karavalakis, G.; Asa-Awuku, A. Using a new Mobile Atmospheric Chamber (MACh) to investigate the formation of secondary aerosols from mobile sources: The case of gasoline direct injection vehicles. J. Aerosol Sci. 2019, 133, 1–11. [Google Scholar] [CrossRef]
- Herrington, J.S.; Hays, M.D.; George, B.J.; Baldauf, R.W. The effects of operating conditions on semivolatile organic compounds emitted from light-duty, gasoline-powered motor vehicles. Atmos. Environ. 2012, 54, 53–59. [Google Scholar] [CrossRef]
- Kuittinen, N.; McCaffery, C.; Peng, W.; Zimmerman, S.; Roth, P.; Simonen, P.; Karjalainen, P.; Keskinen, J.; Cocker, D.R.; Durbin, T.D.; et al. Effects of driving conditions on secondary aerosol formation from a GDI vehicle using an oxidation flow reactor. Environ. Pollut. 2021, 282, 117069. [Google Scholar] [CrossRef] [PubMed]
- Platt, S.M.; El Haddad, I.; Zardini, A.A.; Clairotte, M.; Astorga, C.; Wolf, R.; Slowik, J.G.; Temime-Roussel, B.; Marchand, N.; Ježek, I.; et al. Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber. Atmos. Chem. Phys. 2013, 13, 9141–9158. [Google Scholar] [CrossRef] [Green Version]
- Nordin, E.Z.; Eriksson, A.C.; Roldin, P.; Nilsson, P.T.; Carlsson, J.E.; Kajos, M.K.; Hellén, H.; Wittbom, C.; Rissler, J.; Löndahl, J.; et al. Secondary organic aerosol formation from idling gasoline passenger vehicle emissions investigated in a smog chamber. Atmos. Chem. Phys. 2013, 13, 6101–6116. [Google Scholar] [CrossRef] [Green Version]
- Simonen, P.; Kalliokoski, J.; Karjalainen, P.; Rönkkö, T.; Timonen, H.; Saarikoski, S.; Aurela, M.; Bloss, M.; Triantafyllopoulos, G.; Kontses, A.; et al. Characterization of laboratory and real driving emissions of individual Euro 6 light-duty vehicles—Fresh particles and secondary aerosol for-mation. Environ. Pollut. 2019, 255, 113175. [Google Scholar] [CrossRef]
- Zimmerman, N.; Rais, K.; Jeong, C.-H.; Pant, P.; Delgado-Saborit, J.M.; Wallace, J.S.; Evans, G.J.; Brook, J.R.; Pollitt, K.J.G. Carbonaceous aerosol sampling of gasoline direct injection engine exhaust with an integrated organic gas and particle sampler. Sci. Total Environ. 2019, 652, 1261–1269. [Google Scholar] [CrossRef]
- Miersch, T.; Czech, H.; Stengel, B.; Abbaszade, G.; Orasche, J.; Sklorz, M.; Streibel, T.; Zimmermann, R. Composition of car-bonaceous fine particulate emissions of a flexible fuel DISI engine under high velocity and municipal conditions. Fuel 2019, 236, 1465–1473. [Google Scholar] [CrossRef]
- Zhang, Y.; Deng, W.; Hu, Q.; Wu, Z.; Yang, W.; Zhang, H.; Wang, Z.; Fang, Z.; Zhu, M.; Li, S.; et al. Comparison between idling and cruising gasoline vehicles in primary emissions and secondary organic aerosol formation during photochemical ageing. Sci. Total Environ. 2020, 722, 137934. [Google Scholar] [CrossRef]
- Zhao, Y.; Lambe, A.T.; Saleh, R.; Saliba, G.; Robinson, A.L. Secondary organic aerosol production from gasoline vehicle exhaust: Effects of engine technology, cold start, and emission certification standard. Environ. Sci. Technol. 2018, 52, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, P.; Timonen, H.; Saukko, E.; Kuuluvainen, H.; Saarikoski, S.; Aakko-Saksa, P.; Murtonen, T.; Dal Maso, M.; Ahl-berg, E.; Svenningsson, B.; et al. Time-resolved characterization of primary and secondary particle emissions of a modern gasoline passenger car. Atmos. Chem. Phys. 2016, 16, 8559–8570. [Google Scholar] [CrossRef] [Green Version]
- Link, M.F.; Kim, J.; Park, G.; Lee, T.; Park, T.; Bin Babar, Z.; Sung, K.; Kim, P.; Kang, S.; Kim, J.S.; et al. Elevated production of NH 4 NO 3 from the photochemical processing of vehicle exhaust: Implications for air quality in the Seoul Metropolitan Region. Atmos. Environ. 2017, 156, 95–101. [Google Scholar] [CrossRef]
- Pusede, S.E.; Duffey, K.C.; Shusterman, A.A.; Saleh, A.; Laughner, J.L.; Wooldridge, P.J.; Zhang, Q.; Parworth, C.L.; Kim, H.; Capps, S.L.; et al. On the effectiveness of nitrogen oxide reductions as a control over ammonium nitrate aerosol. Atmos. Chem. Phys. 2016, 16, 2575–2596. [Google Scholar] [CrossRef] [Green Version]
- Nowak, J.B.; Neuman, J.; Bahreini, R.; Middlebrook, A.; Holloway, J.S.; McKeen, S.A.; Parrish, D.D.; Ryerson, T.B.; Trainer, M. Ammonia sources in the California South Coast Air Basin and their impact on ammonium nitrate formation: South coast air basin ammonia sources. Geophys. Res. Lett. 2012, 39, L07804. [Google Scholar] [CrossRef]
- Kim, M.J. Sensitivity of nitrate aerosol production to vehicular emissions in an urban street. Atmosphere 2019, 10, 212. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Tao, L.; Miller, D.J.; Pan, D.; Golston, L.; Zondlo, M.A.; Griffin, R.J.; Wallace, H.W.; Leong, Y.J.; Yang, M.M.; et al. Vehicle emissions as an important urban ammonia source in the United States and China. Environ. Sci. Technol. 2017, 51, 2472–2481. [Google Scholar] [CrossRef]
- Suarez-Bertoa, R.; Zardini, A.; Astorga, C. Ammonia exhaust emissions from spark ignition vehicles over the new European driving cycle. Atmos. Environ. 2014, 97, 43–53. [Google Scholar] [CrossRef]
- Roth, P.; Yang, J.; Peng, W.; Cocker III, D.R.; Durbin, T.D.; Asa-Awuku, A.; Karavalakis, G. Intermediate and high ethanol blends reduce secondary organic aerosol formation from gasoline direct injection vehicles. Atmos. Environ. 2020, 220, 117064. [Google Scholar] [CrossRef]
- Tkacik, D.S.; Lambe, A.T.; Jathar, S.; Li, X.; Presto, A.; Zhao, Y.; Blake, D.; Meinardi, S.; Jayne, J.T.; Croteau, P.L.; et al. Secondary organic aerosol formation from in-use motor vehicle emissions using a potential aerosol mass reactor. Environ. Sci. Technol. 2014, 48, 11235–11242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, P.; Yang, J.; Fofie, E.; Cocker, D.R.; Durbin, T.D.; Brezny, R.; Geller, M.; Asa-Awuku, A.; Karavalakis, G. Catalyzed gasoline particulate filters reduce secondary organic aerosol production from gasoline direct injection vehicles. Environ. Sci. Technol. 2019, 53, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- Malloy, Q.G.J.; Nakao, S.; Qi, L.; Austin, R.; Stothers, C.; Hagino, H.; Cocker, D.R. Real-time aerosol density determination utilizing a modified scanning mobility particle sizer—Aerosol particle mass analyzer system. Aerosol Sci. Technol. 2009, 43, 673–678. [Google Scholar] [CrossRef] [Green Version]
- Cocker, D.R.; Flagan, R.C.; Seinfeld, J.H. State-of-the-art chamber facility for studying atmospheric aerosol chemistry. Environ. Sci. Technol. 2001, 35, 2594–2601. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Roth, P.; Ruehl, C.R.; Shafer, M.M.; Antkiewicz, D.S.; Durbin, T.D.; Cocker, D.; Asa-Awuku, A.; Karavalakis, G. Physical, chemical, and toxicological characteristics of particulate emissions from current technology gasoline direct injection vehicles. Sci. Total Environ. 2019, 650, 1182–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saliba, G.; Saleh, R.; Zhao, Y.; Presto, A.A.; Lambe, A.T.; Frodin, B.; Sardar, S.; Maldonado, H.; Maddox, C.; May, A.; et al. Comparison of Gasoline Direct-Injection (GDI) and Port Fuel Injection (PFI) vehicle emissions: Emission certification standards, cold-start, secondary organic aerosol formation potential, and potential climate impacts. Environ. Sci. Technol. 2017, 51, 6542–6552. [Google Scholar] [CrossRef]
- Pirjola, L.; Karjalainen, P.; Heikkilä, J.; Saari, S.; Tzamkiozis, T.; Ntziachristos, L.; Kulmala, K.; Keskinen, J.; Rönkkö, T. Effects of fresh lubricant oils on particle emissions emitted by a modern gasoline direct injection passenger car. Environ. Sci. Technol. 2015, 49, 3644–3652. [Google Scholar] [CrossRef]
- Chen, L.; Liang, Z.; Zhang, X.; Shuai, S. Characterizing particulate matter emissions from GDI and PFI vehicles under transient and cold start conditions. Fuel 2017, 189, 131–140. [Google Scholar] [CrossRef]
- Duronio, F.; De Vita, A.; Montanaro, A.; Villante, C. Gasoline direct injection engines—A review of latest technologies and trends. Part 2. Fuel 2020, 265, 116947. [Google Scholar] [CrossRef]
- Bahreini, R.; Xue, J.; Johnson, K.; Durbin, T.; Quiros, D.; Hu, S.; Huai, T.; Ayala, A.; Jung, H. Characterizing emissions and optical properties of particulate matter from PFI and GDI light-duty gasoline vehicles. J. Aerosol Sci. 2015, 90, 144–153. [Google Scholar] [CrossRef]
- Liu, T.; Wang, X.; Deng, W.; Zhang, Y.; Chu, B.; Ding, X.; Hu, Q.; He, H.; Hao, J. Role of ammonia in forming secondary aerosols from gasoline vehicle exhaust. Sci. China Ser. B Chem. 2015, 58, 1377–1384. [Google Scholar] [CrossRef]
- Nakao, S.; Shrivastava, M.; Nguyen, A.; Cocker, D., III. Interpretation of secondary organic aerosol formation from diesel exhaust photooxidation in an environmental chamber. Aerosol Sci. Technol. 2011, 45, 964–972. [Google Scholar] [CrossRef] [Green Version]
- Bond, T.C.; Doherty, S.J.; Fahey, D.W.; Forster, P.M.; Berntsen, T.; DeAngelo, B.J.; Flanner, M.G.; Ghan, S.J.; Kärcher, B.; Koch, D.; et al. Bounding the role of black carbon in the climate system: A scientific assessment. J. Geophys. Res. Atmos. 2013, 118, 5380–5552. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LA92 | FTP | NEDC | Caltrans | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cold-Start | Hot-Running | Hot-Start | Weighted | Cold-Start | Hot-Running | Hot-Start | Weighted | UDC | EUDC | Weighted | ||
| THC (g/mile) | 0.091 | 0.002 | 0.010 | 0.008 | 0.090 | 0.002 | 0.003 | 0.020 | 0.151 | 0.003 | 0.058 | 0.004 |
| NMHC (g/mile) | 0.070 | 0.002 | −0.004 | 0.005 | 0.072 | 0.000 | 0.000 | 0.015 | 0.123 | 0.003 | 0.047 | 0.003 |
| CO (g/mile) | 1.227 | 0.276 | 1.037 | 0.379 | 0.900 | 0.124 | 0.202 | 0.306 | 1.524 | 0.160 | 0.660 | 0.859 |
| NOx (g/mile) | 0.034 | 0.011 | 0.003 | 0.012 | 0.013 | 0.003 | 0.005 | 0.006 | 0.015 | 0.002 | 0.007 | 0.004 |
| PM Mass (mg/mile) | - | - | - | 2.85 | - | - | - | 5.53 | - | - | 8.76 | 2.19 |
| Black Carbon (mg/mile) | 21.55 | 0.92 | 0.26 | 1.95 | 20.63 | 0.44 | 0.83 | 1.29 | 15.98 | 0.30 | 3.79 | 1.69 |
| Total Particle Number (particles/mile) | 1.39 × 1013 | 1.37 × 1012 | 6.02 × 1011 | 2.09 × 1012 | 1.43 × 1013 | 1.23 × 1012 | 1.67 × 1012 | 4.34 × 1012 | 1.13 × 1013 | 6.78 × 1011 | 4.57 × 1012 | 3.93 × 1012 |
| Solid Particle Number (particles/mile) | 1.42 × 1013 | 1.23 × 1012 | 4.80 × 1011 | 1.97 × 1012 | 1.09 × 1013 | 1.01 × 1012 | 1.44 × 1012 | 3.41 × 1012 | 1.07 × 1013 | 5.38 × 1011 | 4.26 × 1012 | 7.38 × 1011 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, W.; McCaffery, C.; Kuittinen, N.; Rönkkö, T.; Cocker, D.R.; Karavalakis, G. Secondary Organic and Inorganic Aerosol Formation from a GDI Vehicle under Different Driving Conditions. Atmosphere 2022, 13, 433. https://doi.org/10.3390/atmos13030433
Peng W, McCaffery C, Kuittinen N, Rönkkö T, Cocker DR, Karavalakis G. Secondary Organic and Inorganic Aerosol Formation from a GDI Vehicle under Different Driving Conditions. Atmosphere. 2022; 13(3):433. https://doi.org/10.3390/atmos13030433
Chicago/Turabian StylePeng, Weihan, Cavan McCaffery, Niina Kuittinen, Topi Rönkkö, David R. Cocker, and Georgios Karavalakis. 2022. "Secondary Organic and Inorganic Aerosol Formation from a GDI Vehicle under Different Driving Conditions" Atmosphere 13, no. 3: 433. https://doi.org/10.3390/atmos13030433
APA StylePeng, W., McCaffery, C., Kuittinen, N., Rönkkö, T., Cocker, D. R., & Karavalakis, G. (2022). Secondary Organic and Inorganic Aerosol Formation from a GDI Vehicle under Different Driving Conditions. Atmosphere, 13(3), 433. https://doi.org/10.3390/atmos13030433