Abstract
The chemical characterization of aerosols, especially fine organic fraction, is a relevant atmospheric challenge because their composition highly depends on localization. Herein, we studied the concentration of multi-oxygenated organic compounds in the western Mediterranean area, focusing on sources and the effect of air patterns. The organic aerosol fraction ranged 3–22% of the total organic mass in particulate matter (PM)2.5. Seventy multi-oxygenated organic pollutants were identified by gas chromatography–mass spectrometry, including n-alkanones, n-alcohols, anhydrosugars, monocarboxylic acids, dicarboxylic acids, and keto-derivatives. The highest concentrations were found for carboxylic acids, such as linoleic acid, tetradecanoic acid and, palmitic acid. Biomarkers for vegetation sources, such as levoglucosan and some fatty acids were detected at most locations. In addition, carboxylic acids from anthropogenic sources—mainly traffic and cooking—have been identified. The results indicate that the organic PM fraction in this region is formed mainly from biogenic pollutants, emitted directly by vegetation, and from the degradation products of anthropogenic and biogenic volatile organic pollutants. Moreover, the chemical profile suggested that this area is interesting for aerosol studies because several processes such as local costal breezes, industrial emissions, and desert intrusions affect fine PM composition.
1. Introduction
Atmospheric aerosols affect climate and ecosystems, and they are related to human health problems [1]. The magnitude of these effects depends on the aerosol composition, which is temporally and spatially highly variable [2]. The main sources of particulate matter can be anthropogenic, including traffic, different industrial activities, building, biomass burning, and emissions from housing or farming activities, or natural, including vegetation, deserts, soil, volcanoes, wildfires, and seas and oceans [3]. Several studies have shown that a significant fraction of the particulate matter (PM) is attributed to the organic fraction. The reported percentages of total mass ranged from ~20% at continental mid-latitudes to ~90% at tropical forest areas [4,5]. One important challenge for atmospheric chemistry is to elucidate the sources, structure, chemistry, and fate of these organic atmospheric constituents, as this information is crucial for better understanding of local effects and global climate variations [6,7,8].
The organic PM can be divided into two fractions [3,9,10]. The primary organic aerosols (POAs) are associated with particles emitted directly by vegetation, fossil fuel combustion, biomass burning, coal-fired power plants emissions, motor vehicles exhaust, and cooking. The secondary organic aerosols (SOAs) are mostly produced by photochemical reactions of OH radical, NO3 radical, and ozone with biogenic and anthropogenic volatile and semi-volatile organic compounds (VOCs and SVOCs, respectively). In recent years, efforts have revealed an advance in the chemical characterization of SOA. Indeed, experiments performed in simulation chambers are a very interesting tool that reproduce atmospheric degradations of VOCs and SVOCs, and they have shown that the polar fraction of SOA is generally multi-oxygenated molecules with carboxylic, alcohol, ketone, aldehyde, and hydroperoxide moieties [11,12,13,14]. Data on the polar organic fraction are available for a roadway tunnel, biomass burning, forests, and others [15,16,17,18]. However, identifying functional groups of organic molecules within SOA remains a difficult analytical challenge, given that their inherent low volatility makes routine online analysis by mass spectrometry impossible without perturbation (thermal desorption, dissolution, derivatization, etc.). Several techniques including filter sampling coupled to gas-chromatography coupled to mass-spectrometry (GC-MS) plus different derivatization protocols—BF3/methanol, N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) pure with or without trimethylchlorosilane (TMCS) as catalyst, N-methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA) plus pyridine, and a combination with O-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine hydrochloride (PFBHA)—were applied to SOA characterization [12,19,20,21]. Recently, sophisticated in situ measurement methods have been developed such as thermal denuders coupled to aerosol mass spectrometers (AMS) and thermal desorption aerosol gas chromatograph (TAG) to measure the molecular composition of thermally desorbed aerosols. Other systems were an analogous collection of a thermal desorption inlet to a proton transfer reaction mass spectrometer (PTR-MS) to detect organic and inorganic compounds. Recently, the Filter Inlet for Gases and AEROsol (FIGAERO) instrument was developed, which allows both the separation of components in a volatility space via a temperature-programmed thermal desorption and the determination of the corresponding molecular composition on an hourly timescale [22,23]. Ion mobility spectrometry–mass spectrometry (IMS–MS) with the simultaneous characterization of the elemental composition and molecular structures of organic species in the gas and particulate phases [24] and high-resolution time-of-flight mass spectrometer have also been adapted to the AMS (HR-ToF-AMS) to distinguish the elemental composition of ions having the same nominal mass [25] and to improve the chemical aerosol characterization. With all these techniques and advances, progress has been made in the chemical characterization of the polar organic fraction of PM10 and PM2.5, measured in urban locations in North America [26], Asia [27], and Europe [28,29]. The latest research indicates that the PM organic carbon fraction comprises thousands of compounds which cover a wide range of concentrations, polarities, volatilities, and other physical and chemical properties. Nevertheless, their detailed characterization is still very challenging [6,25,30]. There are few studies about dicarboxylic acids and related compounds (oxocarboxylic acids), despite their significance as constituents in fine particulate matter and tracers of secondary processes in the Mediterranean area [17,25].
For all described above, the aim of this study was the characterization of the multi-oxygenated fine PM fraction in the western Mediterranean area, and the search for correlations to source assessment. The interest in this region lies in its climatic patterns and geographic characteristics, which produce air quality patterns with remarkable spatial and temporal variability if compared to other areas of the world [31]. PM properties are affected globally by the transport of polluted air masses from central-southern Europe, Azores anticyclone, and Sahara dust transportation. Locally, PM in coastal sites depends on sea thermal circulations and industrialization level. For instance, in the summer, the western Mediterranean basin is under the influence of the Azores anticyclone, which produces a generalized subsidence of the air masses located below 5 km. On the other hand, the nearby mountains favor vertical air injection in front of the combined breeze [32]. This specific regional circulation in the western Mediterranean basin leads to pollutants being emitted along the coastline. As a result of different emission sources and various wind regimes (direction, temperature of air masses, etc.), the outcomes of PM concentration prediction models, and for some species (NO3− and SO42−), differ from the rest of Europe [33]; this means that some models are not representative of the western Mediterranean area, since this geographical position favors photo-chemical reactions and accumulation of secondary aerosols [34]. For that, the goal of our research is also to discover general correlations associated with chemical fingerprint information in order to increase knowledge about the organic aerosols and their interaction between different sources and atmospheric processes.
2. Experimental Section
2.1. Location Description
PM samples (n = 59) were collected at industrial, sub-urban, and urban locations in eastern Spain (the western Mediterranean area, 25,000 km2). Figure S1 shows different sampling locations. The first site (39°59′45″ N, 0°03′54″ W) was an urban site, located near the city of Castellón (180,200 inhab.) in the surroundings of an industrial area and 15 km from the Mediterranean Sea. The second location (Valencia 1, 39°28′51.08″ N, 0°22′03.55″ W) was a sub-urban place in the coastal city of Valencia (800,000 inhab.) in the middle of a high urban park. The third site (Valencia 2, 39°28′56.53″ N, 0°0′ 36.88″ W) was an urban location in the same city, 3 km from the sea. The fourth site was a sub-urban location near the city of Alicante (335,000 inhab.) in a commercial and residential zone (Alicante 1, 38°20′25′′ N, 0°30′24″ W). The fifth site (Alicante 2, 39°28′51.08″ N, 0°22′03.55″ W) was an industrialized area near Alicante with a medium traffic flow, located 2 km from an industrial zone and 0.5 km from a cement plant. The number of samples was 12 per station (one per month) except for Alicante 2 because one sample was damaged during the campaign. Additionally, 24 filter aerosol samples were collected at two rural sites as remote sites, Villar del Arzobispo (Remote 1, 39°44′1″ N 0°49′39″ W) and Morella (Remote 2, 40°37′9″ N, 0°6′2″ W), at 60 km and 65 km to the coast, respectively.
The intensity of traffic close to the sampling sites was obtained from Spanish Ministry database [35]. Meteorological information about sampling periods was collected from a local meteorological database [36]. A specific database about Saharan intrusions was also consulted [37].
2.2. Sample Collection
The concentrations of some gaseous pollutants (NOx, O3, SO2, CO) and PM10 and PM2.5 mass concentrations were measured by monitoring air quality network systems (the data were previously validated). Moreover, the PM2.5 filter samples were collected for 24 h by high-volume samplers MCV (Barcelona, Spain), which worked at a flow rate of 30 m3 h−1. Before and after the sample collection, glass fiber filters (GF/A, 150 mm in diameter; Whatman, Brentford, UK) were conditioned to dryness for 24 h. Additionally, blank filters were prepared by purging in 99.995% pure nitrogen (Abelló-Linde, Barcelona, Spain) for 30 s and were then processed. Sampled filters and unexposed blanks were stored at −4 °C to prevent losses of SVOCs and analyzed before 72 h to avoid aerosol aging. Aerosol mass concentration was determined by direct gravimetric measurement.
2.3. Analysis of PM Composition
The multi-oxygenated organic PM analysis was similar to that previously described [30]. Briefly, the filter was extracted with CH2Cl2/CH3CN (1:1) by sonication and the derivatization with PFBHA, and MSTFA was carried out. The extract was injected into the gas chromatography–mass spectrometry (GC–MS) system. TRACE-DSQ II was used with an RTX-5MS column of 30 m × 0.25 mm I.D × 0.25 µm film thickness (Crosslinked 5% Ph Me Siloxane) supplied by Thermo Fisher Scientific (Waltham, MA, USA). Finally, the full scan mode (range m/z 50–650) was used. Standards and derivatization reagents (purity > 99%) were provided by Sigma Aldrich (Steinheim, Germany). Compounds were identified as derivatives by making a comparison with external standards and the quantification was performed from the selected extracted ions (m/z 73,181). The limits of detection (LODs) ranged from 0.02 ng m−3 to 0.1 ng m−3.
Cl−, NO3−, SO42−, and NH4+ inorganic ions were determined after extracting with 10 mL of distilled water in an ultrasonic bath for 30 min. Anions were analyzed by ionic chromatography (Dionex DX120; AS4A column; electrical autosuppression ASRS 300; and 1.8 mM Na2CO3/1.7 mM NaHNO3 eluent). Ammonium was analyzed by spectrophotometry using the indophenol blue method (Cary 100-Conc UV–visible Spectrophotometer, Varian). The LODs in mg L−1 were as follows: 0.05, 0.04, 0.09, and 0.002 in water and in 10, 8, 18, and 0.4 ng m−3 in air for Cl−, NO3−, SO42−, and NH4+, respectively.
2.4. Complementary Experiments
Quality assurance strategies were applied to guarantee sample stability and recoveries determination. Procedural blanks were analyzed, and no artifacts were detected. For confirming sampling efficiency and filter stability, an experiment to investigate the adsorption properties of the compounds in the glass fiber filters was performed. Twelve samples (PM2.5) were collected with a tandem of two filter stack systems at the Remote 1 location at the sampling flow rate of 30 m3 h−1. Organic and inorganic fractions were analyzed in both filters, including replicates. Since no compound was detected in the second tandem filter, complete sampling efficiency and stability was confirmed. The assay accuracy was checked from spiked filters, collected at Remote 2 location. Multi-oxygenated mixtures of 5 ng m−3 was recovered up to 95%.
3. Results
3.1. General Description
The present study was focused on the search of the common and specific atmospheric characteristics associated with the western Mediterranean zone. As the air composition is influenced by significant processes such as coastal breezes, industrial emissions, and desert intrusions, the chemical composition was determined in several locations of this area. Table 1 presents the mean concentrations of atmospheric gases and particulate matter in industrial, sub-urban, urban, and remote selected locations.

Table 1.
Average concentration and ranges in µg m−3 of main gaseous pollutants, particulate matter (PM) mass fractions, inorganic anions in fine fraction, and organic matter in the western Mediterranean area for twelve months.
The SO2, CO, NOx, and O3 values agreed with 24 h average concentration values obtained for similar locations [38]. The high concentrations of gaseous pollutants measured within the urban areas stress the importance of traffic emissions in urban air pollution. For PM10 and PM2.5 mass concentrations, the mean values were 22–37 µg m−3 and 10–27 µg m−3, respectively, and they were also comparable to other studies [39]. The highest PM2.5 mean value was observed at the urban location VLC2 (27 µg m−3), which is one of the most heavily traffic-exposed sites, followed by AL2 (21 µg m−3) with an important industrial emission contribution. Regarding PM10 mass levels, the average concentration of particulate matter was also significantly higher at urban location VLC2 site (37 µg m−3). The highest concentrations were reported in spring, coinciding with Saharan dust intrusions (three episodes). Correlations between PM10 to PM2.5 monthly in five locations were done (see Figure S2)
The mean mass concentration ratios of PM2.5/PM10 were calculated. The mean PM2.5/PM10 ratio was between 0.4 and 0.7. This can be attributed to larger contribution from photochemically produced organic aerosols, which are most likely enriched in fine PM fractions. This result is in accordance with our hypothesis that these geographical sampling locations are heavily influenced by photochemical reactions and accumulation of secondary aerosols. In fact, contributions of organic matter to the PM2.5 ranged from 4% to 35%, whereas those for PM10 ranged from 3% to 18%. Our values agreed with those previously reported in similar urban sites [39,40].
3.2. Identification and Quantification of Organic Tracers
The sampling campaign was focused on the characterization of multi-oxygenated polar organic fraction in fine PM. The average concentrations of the sum of all 70 compounds identified in this study varied in the range of 1.1–5 µg m−3, corresponding to 3–22% of OM, which corresponds to 0.03–4.4% of PM2.5. The mixture complexity reinforced the importance of obtaining fingerprint information. An example of a chromatogram is shown in Figure S3. Compounds were classified into different families: 8 n-alcohols; 1 anhydrosugar; 7 aldehydes; 47 carboxylic acids classified as 22 monocarboxylic acids (MCAs), 18 dicarboxylic acids (DCAs), 3 hydroxy-MCAs, 3 keto-MCAs, and 1 keto-DCA; and 7 hydroxy-aldehyde compounds (see Table S1 with a complete compound identification). Their concentrations (mean, range) in which these pollutants were detected are reported in Table 2.
Table 2.
Summary of 9 family compounds determined in fine PM. Range, average, and median concentrations in ng m−3.
The most frequently detected PM2.5 compounds were palmitic acid, myristic acid, pentadecanoic acid, pimelic acid, glyoxal, methylglyoxal and benzoic acid (present in >72% of the samples). The most abundant organic compounds were sebacic acid, 4-oxopimelic acid, heneicosanoic acid, pimelic acid, benzylalcohol, p-cresol, and levoglucosan (average concentration of >200 ng m−3). The concentrations are reported in Table S2. Figure S4 shows a box plot with detailed concentration values for each specie.
Relevant information was obtained analyzing the sampling sites factor. The average global concentrations ranged from 0.04 (Valencia 2) to 430 ng m−3 (Alicante 1). Chemical SOA composition varied between the studied locations. Figure 1 shows the relative contribution of different multi-oxygenated family species in the fine organic PM.
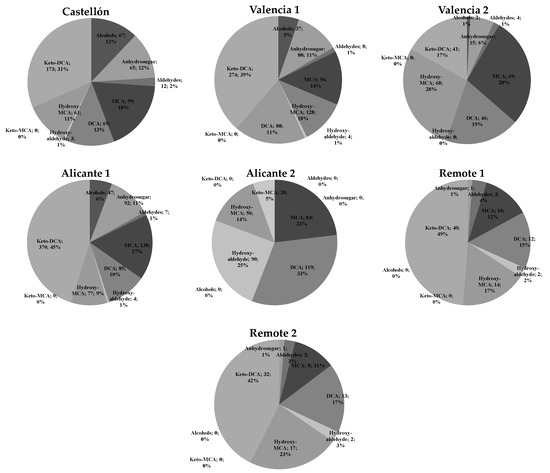
Figure 1.
Relative contribution of different multi-oxygenated family species in the fine PM organic fraction at the five sampling sites. Values include average mass concentration (µg m−3) of each family of oxygenated organic compound (OOCs) and its percentage contribution by sampling site. Castellón (urban site in the surroundings of an industrial area) ∑OOCs: 549 ng m−3; Valencia 1, (sub-urban place), ∑OOCs: 707 ng m−3; Valencia 2 (urban site) ∑OOCs: 245 ng m−3; location in the same city, 3 km from the sea. The fourth site was a near the city of Alicante 1 (sub-urban location in a commercial and residential zone) ∑OOCs: 821 ng m−3; and Alicante 2 (industrialized area) ∑OOCs: 363 ng m−3. Remote area 1 ∑OOCs: 85 ng m−3 and Remote area 2 ∑OOCs: 75 ng m−3 were selected as low-OOCs SOA.
The main family contribution was from keto-DCAs, about 32% of the total quantified fine organic mass. Anhydrosugar, whose main component is levoglucosan, was the second most important family and constituted 15% of the average total mass. Non-substituted MCAs and DCAs contributed with 18% and 15% of the average total quantified organic mass, respectively. The total amount of carboxylic acids classified as MCAs, DCAS, multi-oxygenated MCAs, and DCAs represented up to 80% of the determined oxygenated species of the fine fraction. Other multi-oxygenated compounds such as alcohols, alkenones, and aldehydes were minor compounds on PM2.5 samples. We want to point out that α-dicarbonyl compounds, glyoxal, and methylglyoxal were detected, which have recently attracted much attention as potential SOA precursors. These compounds are formed by photochemical oxidation of both biogenic and anthropogenic VOCs. Similar values of glyoxal were reported in Mexico City, which is a polluted city [41]. We also want to remark that alcohols were relevant at locations such as Castellón, Valencia 1, and Alicante 1; however, they were not detected at the other sampling sites, probably due to different SOA sub-urban emission sources influenced by biogenic and marine emissions.
3.3. Sources Assessment of Organic Tracers
In order to improve the insight on the origin and sources of fine organic aerosols in our samples, correlations and trends among the organic family compounds have been evaluated. Beyond the state of the art, there are some atmospheric phenomena affecting the aerosol composition in the western Mediterranean area [31,32]. In a first approach, an increment of more oxidized compounds was expected during the Azores anticyclone. It is responsible for dry, sunny, and hot weather during summer, even during autumn and spring, with low wind speeds and thus low dispersion of pollutants favoring the chemical reactivity of the air mass. The effect of regional air circulation, directed by frequent sea breezes and mountain barriers in the Mediterranean area, was studied. A high production of photochemical secondary aerosols was expected due to the accumulation of pollutants, both precursors and oxidants [8,33]. PM mass concentrations would also be suspected during the African dust intrusion, reducing the organic percentage. For confirming these hypotheses, the concentration of multi-oxygenated organic tracers (specific compounds and specific ratios between them) was evaluated and compared to other worldwide regions. In global terms, a high contribution of multi-oxygenated species due to photochemical processes was observed, and more details to reinforce this suggestion are developed and deepened in next sections.
3.3.1. Dicarboxylic Acids
The presence of DCA species in atmospheric particles has received much attention in the last years given the roles they play in affecting the global climate and human health. Dicarboxylic acids can be emitted in small quantities from several natural and anthropogenic primary sources such as vegetation and motor exhaust emissions, although the atmospheric photochemical transformation of volatile and semi-volatiles biogenic and anthropogenic compounds is considered the main source [16,34]. The average daily global value of DCAs was 81 ng m−3, ranged 46–119 ng m−3. These concentrations in the western Mediterranean area were in the same order as those determined in other regions [42]. However, they were lower than those reported in other European regions like Oporto, Copenhagen, Leipzig, and Zurich [43], and one order of magnitude lower than those reported in northern China [1].
It is widely studied that the major source of OH radicals during the day is the photodissociation of ozone. It is well known that ozonolysis of alkenes (mainly from biogenic sources) could be a significant OH radical source in zones with high biogenic emissions and high levels of ozone, but other anthropogenic sources such as HCHO photolysis and photolysis of nitrous acid (HONO) could [44] also be quite relevant. These DCA values can be explained by a highest photochemical production from the atmospheric degradation of VOCs expected in this Mediterranean area. It is well known that SOA particles are composed of low molecular weight compounds and formed from reactive organic gases (e.g., VOCs) emitted by the biosphere (mainly forests and phytoplankton) and from anthropogenic sources. The most relevant organic constituents in SOA are carboxylic acids, hydroxyl-carbonyl compounds, and nitro-compounds (Borras et al., 2012).
Concentration ranges for O3 and VOC in the summer (51–61 μg m−3, 21–29 μg m−3, respectively) were much higher than those in the winter (4.2–7.6 μg m−3, 8.2–13.9 μg m−3, respectively). These values showed high ozone values—coinciding with low VOC values—during summer, indicating a high photochemical activity. This photochemical activity can be corroborated by high intensity of solar radiation recorded. Average values obtained were 76 ± 4, 193 ± 16, 242.3 ± 0.4, and 139 ± 8 W m−2 for winter, spring, summer, and autumn, respectively, associated with long periods of anticyclonic time with poorly mixed air masses. This radiation comprises λ from 400 to 730 nm. Ultraviolet radiation (UVI factor) was also recorded with values of UVI 11 (0.275 W m−2) and UVI 2 (0.05 W m−2) for summer and winter, in all the studied region. OH radical production needs UV radiation (λ < 380 nm). In this sense, high global radiation perfectly correlates with high UV radiation, and our hypothesis can be confirmed: high values of DCAs can be assumed to come from photochemical degradation processes in summer periods during the day, possible reactions of O3 with unsaturated hydrocarbons, aromatic compounds in polluted air, and with terpenes emitted by vegetation. During summer, nighttime OH can exceed 1.2 × 105 molecules cm−3 in the western Mediterranean area [45] and could also contribute to chemical processes, in addition to daytime photochemistry. This value is ten times higher than in winter season in this area [45]. This affirmation was also supported by the detection of more oxidized species such as keto-DCAs (4-oxopimelic acid) and keto-MCAs (4-oxobutanoic acid, 5-oxopentanoic acid and C5H8O3 isomers.
At five locations, malonic acid, succinic acid, adipic acid, and azelaic acid were the most abundant dicarboxylic acids, and their maximum levels were reported in summer (July and September). August values were lower due to less anthropogenic VOCs emissions produced at summer holidays (general traffic decrease and several industrial activities are stopped or limited). Then, this behavior could be explained by a photochemical origin via OH oxidation of VOC compounds to form SOA compounds, favored by solar radiation and the slow air movement in the studied locations. Figure 2 shows seasonal trends of C2-C9 DCAs in the five areas studied. The specific contribution and origin of each compound were analyzed monthly.
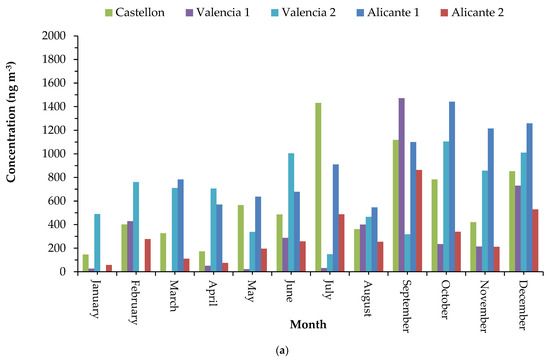
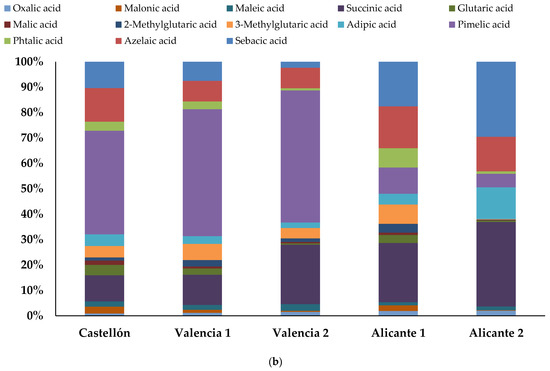
Figure 2.
(a) Monthly trends of main DCA species (C2 to C9); (b) average concentrations of DCAs (in percentage) in fine PM in 5 western Mediterranean areas studied.
The highest mean concentrations were found for pimelic acid (C7), which corresponds to 33% of the total oxygenated fine PM determined. It can be emitted directly from several biogenic and anthropogenic sources, such as fossil fuel burning, forest fires, meat cooking, and cigarette smoke, and it may also constitute a secondary aerosol product resulting from VOCs photochemistry. The annual average pimelic acid concentration was higher than those found in other areas [46] probably because all our locations presented a significant contribution of direct emissions by agriculture, traffic, and meat cooking. Succinic acid (C4), determined at a mean concentration of 122 ng m−3 (18%), has been identified in laboratory studies as a PM component from the atmospheric degradation of VOCs [11,12] and can be attributed to secondary formation. Azelaic acid (C9), measured at a mean concentration of 84 ng m−3 (12%), is generally produced by the atmospheric oxidation of unsaturated fatty acids from plants and from oleic acid degradation, whose major source in urban environment is food cooking. Then, an azelaic acid contribution can be associated with both anthropogenic sources in urban environments, since it was emitted from olive oil-based cooking (the main ingredient of Mediterranean cuisine), and with biogenic sources in agricultural or remote areas. Adipic acid (C6), measured at a mean concentration of 35 ng m−3 (4%), can be assumed from the photo-oxidation product of different cycloalkenes. Phthalic acid (C8) could be released from direct emission by incomplete combustion processes or formed by secondary oxidation of naphthalene and other polycyclic aromatic hydrocarbons (significant anthropogenic compounds) [42,46]. Finally, malic acid (C4:1), detected in 31 samples and measured at a mean concentration of 10 ng m−3 (1%), has also been identified and can be attributed of SOA from forest emissions—mainly fruit trees, pines groves, and holm oaks and from isoprene photo-oxidation, being the main probable origin in our locations [17].
Finally, short-chain DCAs were also determined. Different from other studies, oxalic acid has not been found to be the most abundant species. The average concentrations of oxalic acid (C2) and malonic acid (C3) were 10 ng m−3 (1.5%) and 14 ng m−3 (1.8%), respectively. Similar values were reported at Brno [47] and similar to an urban background and a road site in Barcelona [34]. Our levels were one order lower than those reported in North Carolina, Tokyo, and Hong Kong [27,46], and they were quite lower than those reported in several Chinese cities [1]. The lower contribution of C3-C5 DCA over C7-C9 DCA indicates the less aged SOA or new SOA generated. The SOA composition of DCAs, mainly C7-C9, suggests that both biogenic emissions and anthropogenic emissions such as traffic and cooking activities played an important role in this fine PM fraction.
3.3.2. Mono-Carboxylic Acids
The average value of MCAs was 97 ng m−3, ranged 69–138 ng m−3 with a mean average of 11.8% contribution to multi-oxygenated fine PM, and it was on the same order as those measured in Beijing [42] but higher than those reported in southeastern USA [27] or Jülich, Germany [25]. Short-chain MCAs (C < 20) derive mainly from direct emissions (primary pollutants) of meat cooking and traffic, while long-chain MCAs (C > 22) result from the uncontrolled combustion of organic materials in the open air, microbial activity on waste, and vascular plant wax [28,29,46].
At the sub-urban locations, the maximum levels of long-chain MCAs were reported in spring and autumn (2-fold higher). The observed guidelines can be explained by an increase in their primary emissions from biogenic emissions and heating. In our fine PM samples collected in the western Mediterranean area, the main MCAs were linoleic acid (C18:2), heneicosanoic acid (C21), palmitoleic acid (C16:1), and margaric acid (C17) (see Figure 3). We pointed out from an ANOVA test (p < 0.05) that compounds such as C14-carboxylic acid, myristic acid, tridecanoic acid, and octanoic acid were determined at a higher concentration in the more industrialized areas of Castellón and Alicante 2, showing industrial processes as the main sources of emission of those MCAs.
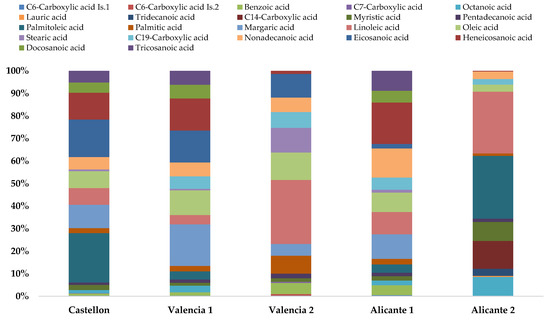
Figure 3.
The average concentrations of MCAs (in percentage) in fine PM at 5 western Mediterranean areas studied. Total average mass concentrations were 1688, 1637, 1038, 2354, and 1090 ng m−3 for Castellon, Valencia 1, Valencia 2, Alicante 1, and Alicante 2, respectively.
3.3.3. Levoglucosan
This anhydrosugar compound (1,6-anhydro-β-D-glucopyranose, C6) is considered a secondary pollutant formed by the pyrolysis of cellulose and is used as a tracer for biomass burning [25].
Levoglucosan was identified in many samples (33 of the 60 samples). The mean daily average concentration of levoglucosan in the western Mediterranean locations (431 ng m−3) was lower than that in cities such as Seattle, Fresno, and Bakersfiled (<7.6 µg m−3). It was also lower than a biomass smoke recorded in southeast Asia (1.4–40 µg m−3) [46]. Levoglucosan maximum values in autumn were between 2 and 5 times higher than those of the rest of the values obtained in the other seasons of the year. Principally, its high concentration values coincided with the legal permission of field burning of agriculture residue (rice crop straw) after the harvest in the western Mediterranean area to eliminate waste [33]. This contribution to atmospheric fine particles could also be due to typical western Mediterranean cooking and the use of new biomass burning-based home-heating technologies [46].
3.4. Ratios of Specific Tracers
The relative contributions of specific multi-oxygenated species on fine particulate matter have been demonstrated to be an interesting tool for source assessments [42,44]. For that, different ratios—previously established in the literature—have been calculated to understand the expected origin of fine PM collected in the present study (Table S3).
3.4.1. C3/C4 Ratio
The C3/C4 ratio—malonic acid/succinic acid—has been used as a tracer of the enhanced photochemical aging of organic aerosols. Because C4 is oxidized to C3, an increase in C3/C4 ratio indicates an increase in photochemical processing [48]. Our average quotient between malonic (C3) and succinic acid (C4) was 0.15. The dominance of succinic acid appeared to be associated with strong photochemical reactions from anthropogenic emission sources (mainly traffic). This suggests that other oxidation processes and/or other primary emission sources substantially contribute to the generation of succinic acid, which minimizes the draining that the conversion of succinic acid into malonic acid entails. The substantial contribution made by traffic can be explained by the C3/C4 ratios obtained because all the locations were close to or influenced by roadways. In fact, the C3/C4 ratio increased 2- or 3-fold during summer in the Castellón location, when secondary photochemical formation of diacids is higher. This increment can also be associated with traffic reduction during the holidays in this location, since the number of light vehicles and heavy vehicles was reduced to around 20% and 15%, respectively, according to the traffic database (http://www.dgt.es/es/el-trafico/). Nevertheless, more data would be needed to confirm this assumption.
3.4.2. C6/C9 and C8/C9 Ratios
C6/C9 and C8/C9 ratios were determined to evaluate the contribution of biogenic and anthropogenic sources to organic aerosol. An average quotient of 0.4 was observed between adipic acid (C6) and azelaic acid (C9) and also between phthalic acid (C8) and azelaic acid (C9). Adipic and phthalic acid have been attributed to anthropogenic precursors, while azelaic acid has been mainly referred to a photo-oxidation product of biogenic unsaturated fatty acids, i.e., oleic acid [1]. In fact, oleic acid and linoleic acid have been detected in almost all the analyzed PM samples. Their high concentration levels can explain high values of azelaic acid in the same samples. Hence, the results indicate that biogenic sources of SOA predominate in the western Mediterranean area as compared to the high anthropogenic contribution observed in Tokyo and Los Angeles with C6/C9 ratios and C8/C9 ratios of 0.4–7.4 and 0.3–8.0, respectively, and similar to that reported by [34]. Firstly, the traffic and industrial emissions at the study locations were obviously lower than in the two large cities. Secondly, an important agricultural area is present in the surroundings of the sampling sites, which produces an important input of biogenic compounds. Thirdly, the re-circulation of local sea breezes moved the precursors and/or the degradation products from the rural to the sub-urban or urban locations. Finally, we can conclude that both biogenic and anthropogenic sources have a significant contribution to fine PM in our western Mediterranean locations.
3.4.3. C18:0/C16 Ratio
The measured average ratio of stearic acid (C18:0) and palmitic acid (C16) was 0.7. Other studies obtained ratios lower than 0.25 resulting from foliar vegetation combustion, waxy leaf surface abrasions, and wood smoke; values between 0.25 and 0.5 have been recorded for vehicle exhaust fumes, and others of 0.5–1 have been obtained for paved and unpaved road dust [25]. Our results, although they should be corroborated over a longer time series, again indicate the contribution of anthropogenic sources associated with traffic as one of the two most relevant and significant factors that contribute to the formation of SOA in the western Mediterranean area.
3.4.4. C18:0/C18:1 Ratio
The ratio between stearic acid (C18:0) and oleic acid (C18:1) is used for aerosol aging [42] since unsaturated n-fatty acids are not stable in aerosols because they can be oxidized under atmospheric conditions. Values > 0.5 indicate that the air masses are more aged. These compounds are of the majority MCAs, with a C18:0/C18:1 ratio of 0.3. This indicates a predominance of recently formed fine PM, which can be explained by the usual sea breezes at these locations and by the subsequent air recirculation over the study locations. Moreover, the average C18:0/C18:1 ratios were 0.23, 0.33, 0.23, and 0.35 in winter, spring, summer, and autumn, respectively. These values were statistically similar at 95% confidence, indicating stable degradation rates of alkenoic acids in the western Mediterranean area promoting an early-aged PM over the year [46].
3.4.5. LG/OC
Levoglucosan is a product from the thermal degradation of cellulose and hemicellulose and is commonly used as tracer for wood smoke in the atmosphere, as described above. The ratio of levoglucosan to OC in aerosol is used to indicate biomass burning source contributions. The LG/OC ratio was 0.03, whereas the LG/PM2.5 was 0.005. These values are comparable to those reported for biomass combustion studies from various sites [49]. Our results demonstrate that biomass burning is an important input of levoglucosan to the atmosphere in regions (as ours) where brown coal is utilized as a domestic fuel, which also makes these values during winter higher. Its sources are also from typical western Mediterranean cooking, the use of new biomass burning-based home-heating technologies, and field burning of agriculture residue (rice crop straw) after the harvest in the western Mediterranean area to eliminate waste. The results obtained with both ratios confirm the allocation of sources previously carried out in previous sections of this study (Section 3.3.3).
3.5. Major Inorganic Ions
The objective of our research focused on the characterization of the organic fraction of PM2.5. Nevertheless, representative water-soluble inorganic ions were analyzed as a general approach to describe the PM composition and evaluate the effect of location and seasonal trends on the source assessments. Chloride, sulfate, nitrate, and ammonium corresponded to 7–76% of the total mass of PM2.5.
Sulfate is the most abundant anion in fine mode in all locations, ranging from 0.4 to 14 µg m−3. The concentrations mostly depended on the degree of industrialization (p value > 0.05). The highest values were monitored in Alicante 2 (mean 10 µg m−3), an industrial hotspot near a cement plant that suggests a significant contribution of anthropogenic sources. Nitrate levels in PM2.5 ranged from 0.1 to 10 µg m−3, and statistical differences were also associated with the hotspots (p value < 0.05). The highest mean concentration was measured at the highly industrialized hotspot (Alicante 2). In all the cases, NH4+ and Cl− values were not significant (p value > 0.05), indicating a low contribution of ocean-based aerosols.
The presence of seasonal trends on these inorganic ions has been previously described in regions with a high Atlantic influence [8]. Analysis of variance (ANOVA) showed that the concentration of inorganic ions in the western Mediterranean area varied as function of season (p value < 0.05), see Figure S5. Sulfate showed a summer maximum due to the greater SO2 oxidation velocity under high isolation conditions. Nitrate concentrations presented a slight seasonal trend. At these sites, nitrate particles are made up of two different species: NH4NO3 (unstable at high temperatures) and NaNO3. During winter pollution episodes (colder period), the most common compound is NH4NO3. During the rest of the year, the typical nitrate compound is NaNO3, given by the reaction of NaCl particles with nitric acid. Ammonium and chloride did not show any significant seasonal trend.
4. Conclusions
This study monitored 70 multi-oxygenated species, and different strategies have been applied to distinguish sources for the organic contribution to fine PM. The western Mediterranean Basin campaign in different seasons can be classified as a low-scale study, similar to others performed worldwide; however, due to the accurate quantification of a high number of species, we better understand specific pollution sources and reactivity of this atmospheric active zone. Indeed, sources and seasonal variations were assessed from the interpretation of individual makers—grouped into families—and ratios between specific tracers. It is important emphasize that, although this area shows particular characteristics, some correlations reported in this study can be extrapolated to other world areas.
In summary, from the results obtained in this study, there are three main sources of fine PM in the region studied. On the one hand, biogenic sources predominate in the western Mediterranean area, probably due to a huge agricultural area present in the surroundings of the sampling sites, which produced an important input of primary fine PM biogenic compounds and also contributed to SOA by photochemical chemistry of biogenic precursors emitted. On the other hand, results indicated a significant anthropogenic contribution associated to traffic and cooking processes related with strong photochemical reactions from anthropogenic emission sources. Finally, early-age PM has been explained by different assessments due to the local sea breezes at these locations, revealing the poor contribution of aged PM transported by air masses from central and northern Europe.
A comprehensive knowledge of the organic aerosol chemistry is of high importance for assessing biogenic and anthropogenic influences and evaluating the effect of radiative forcing. Further studies are needed to understand the main mechanisms by which the aerosols are lofted into the atmosphere and are transported to other regions.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4433/12/1/94/s1. Figure S1. Map showing the locations of the sampling sites of the western Mediterranean area, Spain. Figure S2 Correlations between PM10 and PM2.5 monthly in 5 locations (a) PM10 data; (b) PM2.5 data; (c) Correlations PM10 to PM2.5. Figure S3. GC-MS extracted ion chromatogram (m/z 73 + 181) obtained from sub-urban sample (Valencia 1). 1. Phenol, 2. Oxalic acid, 3. Benzoic acid, 4. Succinic acid, 5. Glutaric acid, 6. Malic acid, 7. Levoglucosan, 8. Phtalic acid, 9. Azelaic acid, 10. Myristic acid, 11. Glyoxal, 12. Pentadecanoic acid, 13. Palmitoleic acid, 14. Palmitic acid, 15. Margaric acid, 16. Tetradecanoic acid, 17. Oleic acid, 18. Linoelic acid, 19. Stearic acid, 20. Nonadecanoic acid, 21. Eicosanoic acid, 22. Heneicosanoic acid, and 23.Tricasanoic acid. Figure S4. Box plot data with a detailed concentration values (in ng m−3) for each specie. Figure S5. Seasonal trend of inorganic anions. Table S1. Classification of detected oxygenated organic compounds based on functional groups. Table S2. Concentration average and mean concentration (µg m−3) of oxygenated compounds of the fine PM. Table S3. Average ratios of concentrations of dicarboxylic acids collected at different locations and seasons.
Author Contributions
Investigation, E.B.; data curation, E.B., F.S.; writing—original draft prep-aration, E.B., L.A.T.-G.; writing—review and editing, E.B., A.M.; supervision, A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Spanish Ministry of Science and Innovation grant number [RTI2018-097768B-C21], and Generalitat Valenciana, grant number PROMETEO/2019/110.
Data Availability Statement
The data presented in this study are available in this article.
Acknowledgments
Fundación CEAM is supported by the Generalitat Valenciana, by the IMAGINA: IMpactos del cAmbio Global en la cuenca MediterráNeA occidental: Meteorología, contaminación atmosférica y ecosistemas forestales (PROMETEO/2019/110, Generalitat Valenciana) and by the CAPOX: Análisis de la modificación de la capacidad oxidativa de la atmósfera en Europa debido a cambios en emisión (RTI2018-097768B-C21, Spanish Ministry of Science, Innovation and Universities).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhu, Y.; Yang, L.; Chen, J.; Kawamura, K.; Sato, M.; Tilgner, A.; van Pinxteren, D.; Chen, Y.; Xue, L.; Wang, X.; et al. Molecular distributions of dicarboxylic acids, oxocarboxylic acids and α-dicarbonyls in PM2.5 collected at the top of Mt. Tai, North China, during the wheat burning season of 2014. Atmos. Chem. Phys. 2018, 18, 10741–10758. [Google Scholar] [CrossRef]
- Dongarrà, G.; Manno, E.; Varrica, D.; Lombardo, M.; Vultaggio, M. Study on ambient concentrations of PM10, PM10-2.5, PM2.5 and gaseous pollutants. Trace elements and chemical speciation of atmospheric particulates. Atmos. Environ. 2010, 44, 5244–5257. [Google Scholar] [CrossRef]
- Calvo, A.I.; Alves, C.; Castro, A.; Pont, V.; Vicente, A.M.; Fraile, R. Research on aerosol sources and chemical composition: Past, current and emerging issues. Atmos. Res. 2013, 120–121, 1–28. [Google Scholar] [CrossRef]
- Jimenez, J.L.; Canagaratna, M.R.; Donahue, N.M.; Prevot, A.S.; Zhang, Q.; Kroll, J.H.; DeCarlo, P.F.; Allan, J.D.; Coe, H.; Ng, N.L.; et al. Evolution of organic aerosols in the atmosphere. Science 2009, 326, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.J.; Zhang, Y.L.; Bozzetti, C.; Ho, K.F.; Cao, J.J.; Han, Y.M.; Dällenbach, K.R.; Slowik, J.G.; Platt, S.M.; Canonaco, F.; et al. High secondary aerosol contribution to particulate pollution during haze events in China. Nature 2014, 514, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Rabha, S.; Saikia, B.K. Advanced micro- and nanoscale characterization techniques for carbonaceous aerosols. Handbook of Nanomaterials in Analytical Chemistry. Mod. Trends Anal. 2020, 449–472. [Google Scholar] [CrossRef]
- Kanakidou, M.; Seinfeld, J.H.; Pandis, S.N.; Barnes, I.; Dentener, F.J.; Facchini, M.C.; Van Dingenen, R.; Ervens, B.; Nenes, A.; Nielsen, C.J.; et al. Organic aerosol and global climate modeling: A review. Atmos. Chem. Phys. 2005, 5, 1053–1123. [Google Scholar] [CrossRef]
- Querol, X.; Alastuey, A.; Pey, J.; Cusack, M.; Pérez, N.; Mihalopoulos, N.; Theodosi, C.; Gerasopoulos, E.; Kubilay, N.; Koçak, M. Variability in regional background aerosols within the Mediterranean. Atmos. Chem. Phys. 2009, 9, 4575–4591. [Google Scholar] [CrossRef]
- Atkinson, R.; Arey, J. Gas-phase tropospheric chemistry of biogenic volatile organic compounds: A review. Atmos. Environ. 2003, 37, 197–219. [Google Scholar] [CrossRef]
- Gieré, R.; Querol, X. Solid Particulate Matter in the Atmosphere. Elements 2010, 6, 215–222. [Google Scholar] [CrossRef]
- Tortajada-Genaro, L.A.; Borrás, E. Temperature effect of tapered element oscillating microbalance (TEOM) system measuring semi-volatile organic particulate matter. J. Environ. Monit 2011, 13, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Borrás, E.; Tortajada-Genaro, L.A. Secondary organic aerosol formation from the photo-oxidation of benzene. Atmos. Environ. 2012, 47, 154–163. [Google Scholar] [CrossRef]
- Yonghong, W.; Riva, M.; Xie, H.; Heikkinen, L.; Schallhart, S.; Zha, Q.; Yan, C.; He, X.-C.; Peräkylä, O.; Ehn, M. Formation of highly oxygenated organic molecules from chlorine-atom-initiated oxidation of alpha-pinene. Atmos. Chem. Phys. 2020, 20, 5145–5155. [Google Scholar] [CrossRef]
- Bianchi, F.; Kurtén, T.; Riva, M.; Mohr, C.; Rissanen, M.P.; Roldin, P.; Berndt, T.; Crounse, J.D.; Wennberg, P.O.; Mentel, T.F.; et al. Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol. Chem. Rev. 2019, 119, 3472–3509. [Google Scholar] [CrossRef] [PubMed]
- Falkovich, A.H.; Graber, E.R.; Schkolnik, G.; Rudich, Y.; Maenhaut, W.; Artaxo, P. Low molecular weight organic acids in aerosol particles from Rondonia, Brazil, during the biomass-burning, transition and wet periods. Atmos. Chem. Phys. 2005, 5, 781–797. [Google Scholar] [CrossRef]
- Kourtchev, I.; Warnke, J.; Maenhaut, W.; Hoffmann, T.; Claeys, M. Polar organic marker compounds in PM2.5 aerosol from a mixed forest site in western Germany. Chemosphere 2008, 73, 1308–1314. [Google Scholar] [CrossRef]
- Mirante, F.; Alves, C.; Pio, C.; Pindado, O.; Pérez, R.; Revuelta, M.A.; Artiñano, B. Organic composition of size segregated atmospheric particulate matter, during summer and winter sampling campaigns at representative sites in Madrid, Spain. Atmos. Res. 2013, 132–133, 345–361. [Google Scholar] [CrossRef]
- García, M.I.; van Drooge, B.L.; Rodríguez, S.; Alastuey, A. Speciation of organic aerosols in the Saharan Air Layer and in the free troposphere westerlies. Atmos. Chem. Phys. 2017, 17, 8939–8958. [Google Scholar] [CrossRef]
- Jaoui, M.; Kleindienst, T.E.; Lewandowski, M.; Offenberg, J.H.; Ednby, E.O. Identification and Quantification of Aerosol Polar Oxygenated Compounds Bearing Carboxylic or Hydroxyl Groups. 1. Method Development. Anal. Chem. 2004, 76, 4765–4778. [Google Scholar] [CrossRef]
- Wang, W.; Vas, G.; Dommisse, R.; Loones, K.; Claeys, M. Fragmentation study of diastereoisomeric 2-methyltetrols, oxidation products of isoprene, as their trimethylsilyl ethers, using gas chromatography/ion trap mass spectrometry. Rapid. Comm. Mass. Spectrom. 2004, 18, 1787–1797. [Google Scholar] [CrossRef]
- Lopez-Hilfiker, F.D.; Mohr, C.; Ehn, M.; Rubach, F.; Kleist, E.; Wildt, J.; Mentel, T.M.; Carrasquillo, A.J.; Daumit, K.E.; Hunter, J.F.; et al. Phase partitioning and volatility of secondary organic aerosol components formed from -pinene ozonolysis and OH oxidation: The importance of accretion products and other low volatility compounds. Atmos. Chem. Phys. 2015, 15, 7765–7776. [Google Scholar] [CrossRef]
- Chen, Y.; Takeuchi, M.; Nah, T.; Xu, L.; Canagaratna, M.R.; Stark, H.; Baumann, K.; Canonaco, F.; Prévôt, A.S.H.; Huey, L.G.; et al. Chemical Characterization of Secondary Organic Aerosol at a Rural Site in the Southeastern, U.S.: Insights from Simultaneous HR-ToF-AMS and FIGAERO-CIMS Measurements. Atmos. Chem. Phys. Discuss 2020. in review. [Google Scholar] [CrossRef]
- Krechmer, J.E.; Groessl, M.; Zhang, X.; Junninen, H.; Massoli, P.; Lambe, A.T.; Kimmel, J.R.; Cubison, M.J.; Graf, S.; Lin, Y.-H.; et al. Ion mobility spectrometry–mass spectrometry (IMS–MS) for on and offline analysis of atmospheric gas and aerosol species. Atmos. Meas. Tech. 2016, 9, 3245–3262. [Google Scholar] [CrossRef]
- DeCarlo, P.F.; Kimmel, J.R.; Trimborn, A.; Northway, M.J.; Jayne, J.T.; Aiken, A.C.; Gonin, M.; Fuhrer, K.; Horvath, T.; Docherty, K.; et al. Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer. Anal. Chem. 2006, 78, 8281–8289. [Google Scholar] [CrossRef]
- Lewandowski, M.; Jaoui, M.; Kleindienst, T.E.; Offenberg, J.H.; Edney, E.O. Composition of PM2.5 during the summer of 2003 in Research Triangle Park, North Carolina. Atmos. Environ. 2007, 41, 4073–4083. [Google Scholar] [CrossRef]
- Ren, G.; Yan, X.; Ma, Y.; Qiao, L.; Chen, Z.; Xin, Y.; Zhou, M.; Shi, Y.; Zheng, K.; Zhu, S.; et al. Characteristics and source apportionment of PM2.5-bound saccharides and carboxylic acids in Central Shanghai, China. Atmos. Res. 2020, 237, 104817. [Google Scholar] [CrossRef]
- Teich, M.; van Pinxteren, D.; Herrmann, H. A one year study of functionalised medium-chain carboxylic acids in atmospheric particles at a rural site in Germany revealing seasonal trends and possible sources. J. Atmos Chem. 2019, 76, 115–132. [Google Scholar] [CrossRef]
- Van Drooge, B.L.; Grimalt, J.O. Particle size-resolved source apportionment of primary and secondary organic tracer compounds at urban and rural locations in Spain. Atmos. Chem. Phys. 2015, 15, 7735–7752. [Google Scholar] [CrossRef]
- Borrás, E.; Tortajada-Genaro, L.A. Determination of oxygenated compounds in secondary organic aerosol from isoprene and toluene smog chamber experiments. Environ. Anal. Chem. 2012, 92, 110–124. [Google Scholar] [CrossRef]
- Millan, M.; Salvador, R.; Mantilla, E.; Artiñano, B. Meteorology and photochemical air pollution in southern Europe: Experimental results from EC research projects. Atmos. Environ. 1996, 30, 1909–1924. [Google Scholar] [CrossRef]
- Millan, M.; Artiñano, B.; Alonso, L.A.; Castro, M.; Fernández-Patier, R.; Goberna, J. Meso-meteorological cycles of air pollution in the Iberian Peninsula (MECAPIP); Air Pollution Research Report 44, EUR 14834; European Commission: Brussels, Belgium, 1992; pp. 1–219. [Google Scholar]
- Viana, M.; Kuhlbusch, T.A.J.; Querol, X.; Alastuey, A.; Harrison, R.M.; Hopke, P.K.; Winiwarter, W.; Vallius, M.; Szidat, S.; Prévôt, A.S.H.; et al. Source apportionment of particulate matter in Europe: A review of methods and results. Aero. Sci. 2008, 39, 827–849. [Google Scholar] [CrossRef]
- Alier, M.; van Drooge, B.L.; Dall’Osto, M.; Querol, X.; Grimalt, J.O.; Tauler, R. Source apportionment of submicron organic aerosol at an urban background and a road site in Barcelona (Spain) during SAPUSS. Atmos. Chem. Phys. 2013, 13, 10353–10371. [Google Scholar] [CrossRef]
- Ministerio de Transportes, Mobilidad y Agenda Urbana. Gobierno de España Home Page. Available online: www.mitma.es (accessed on 23 October 2019).
- Fundación Centro de Estudios Ambientales del Mediterráneo Home Page. Available online: www.ceam.es/ceamet (accessed on 23 October 2019).
- Ministerio para la Transición Ecológica y Reto Demográfico. Gobierno de España Home Page. Available online: www.miteco.gob.es/es (accessed on 23 October 2019).
- Salvador, P.; Artiñano, B.; Querol, X.; Alastuey, A.; Costoya, M. Characterisation of local and external contributions of atmospheric particulate matter at a background coastal site. Atmos. Environ. 2007, 41, 1–17. [Google Scholar] [CrossRef]
- Pey, J.; Querol, X.; Alastuey, A. Variations of levels and composition of PM10 and PM2.5 at an insular site in the Western Mediterranean. Atmos. Res. 2009, 94, 285–299. [Google Scholar] [CrossRef]
- Ram, K.; Sarin, M.M. Day and night variability of EC, OC, WSOC and inorganic ions in urban environment of Indo-Gangetic Plain: Implications to secondary aerosol formation. Atmos. Environ. 2011, 45, 460–468. [Google Scholar] [CrossRef]
- Curry, L.A.; Tsui, W.G.; McNeill, V.F. Technical note: Updated parameterization of the reactive uptake of glyoxal and methylglyoxal by atmospheric aerosols and cloud droplets. Atmos. Chem. Phys. 2018, 18, 9823–9830. [Google Scholar] [CrossRef]
- He, L.Y.; Hu, M.; Huang, X.-F.; Zhang, Y.-H.; Tang, X.-Y. Seasonal pollution characteristics of organic compounds in atmospheric fine particles in Beijing. Sci. Total Environ. 2006, 359, 167–176. [Google Scholar] [CrossRef]
- Van Pinxteren, D.; Neusüß, C.; Herrmann, H. On the abundance and source contributions of dicarboxylic acids in size-resolved aerosol particles at continental sites in central Europe. Atmos. Chem. Phys. 2014, 14, 3913–3928. [Google Scholar] [CrossRef]
- Ho, K.F.; Ho, S.S.H.; Lee, S.C.; Kawamura, K.; Zou, S.C.; Cao, J.J.; Xu, H.M. Summer and winter variations of dicarboxylic acids, fatty acids and benzoic acid in PM2.5 in Pearl Delta River Region, China. Atmos. Chem. Phys. 2011, 11, 2197–2208. [Google Scholar] [CrossRef]
- Elshorbany, Y.; Barnes, I.; Becker, K.; Kleffmann, J.; Wiesen, P. Sources and Cycling of Tropospheric Hydroxyl Radicals—An Overview. Z. Phys. Chem. Int. J. Res. Phys. Chem. Chem. Phys. 2010, 224, 967–987. [Google Scholar] [CrossRef]
- Lelieveld, J.; Gromov, S.; Pozzer, A.; Taraborrelli, D. Global tropospheric hydroxyl distribution, budget and reactivity. Atmos. Chem. Phys. 2016, 16, 12477–12493. [Google Scholar] [CrossRef]
- Capka, L.; Mikuška, P.; Kamil, K. Determination of dicarboxylic acids in atmospheric aerosols using continuous aerosol sampler with on-line connected ion chromatography system. Atmos. Environ. 2020, 222, 117178. [Google Scholar] [CrossRef]
- Oliveira, C.; Pio, C.; Alves, C.; Evtyugina, M.; Santosa, P.; Goncalves, V.; Nunes, T.; Silvestre, A.J.D.; Palmgren, F.; Wahlin, P.; et al. Seasonal distribution of polar organic compounds in the urban atmosphere of two large cities from the North and South of Europe. Atmos. Environ. 2007, 41, 5555–5570. [Google Scholar] [CrossRef]
- Deshmukh, D.K.; Kawamura, K.; De, M.K. Dicarboxylic acids, u-oxocarboxylic acids, a-dicarbonyls, WSOC, OC, EC, and inorganic ions in wintertime size-segregated aerosols from central India: Sources and formation processes. Chemosphere 2016, 161, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Reche, C.; Viana, M.; Amato, F.; Alastuey, A.; Moreno, T.; Hillamo, R.; Teinilä, K.; Saarnio, K.; Secoe, R.; Peñuelas, J.; et al. Biomass burning contributions to urban aerosols in a coastal Mediterranean City. Sci. Total Environ. 2012, 427–428, 175–190. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).