Long-Term Observations of Atmospheric Speciated Mercury at a Coastal Site in the Northern Gulf of Mexico during 2007–2018

 , ,
, ,
Abstract
1. Introduction
2. Site and Measurements
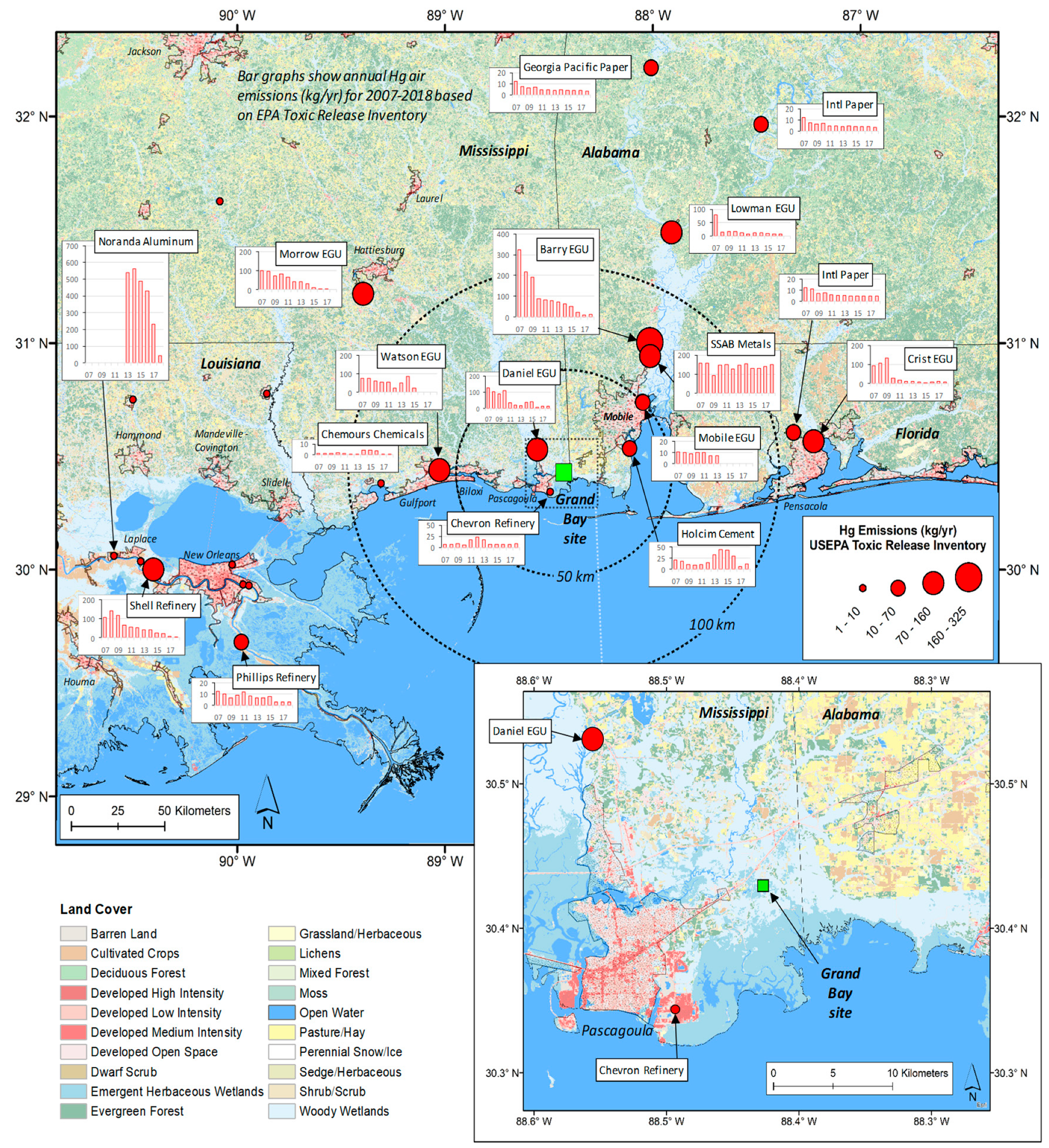
2.1. Site Description
2.2. Experimental Description
2.2.1. Measurements of Speciated Mercury
2.2.2. Ancillary Measurements
3. Results
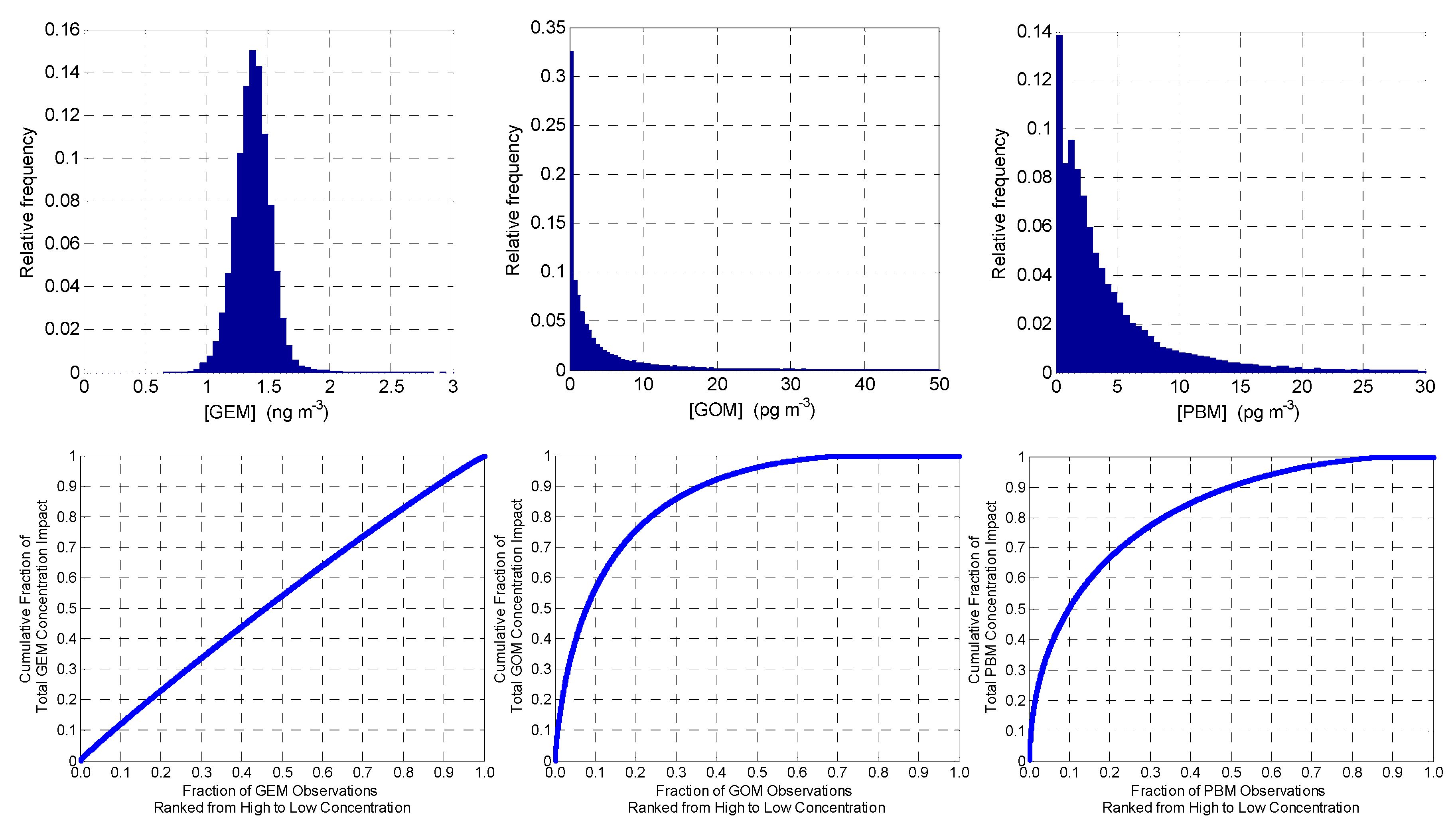
3.1. Overall Measurement Statistics
Inter-Annual Variation
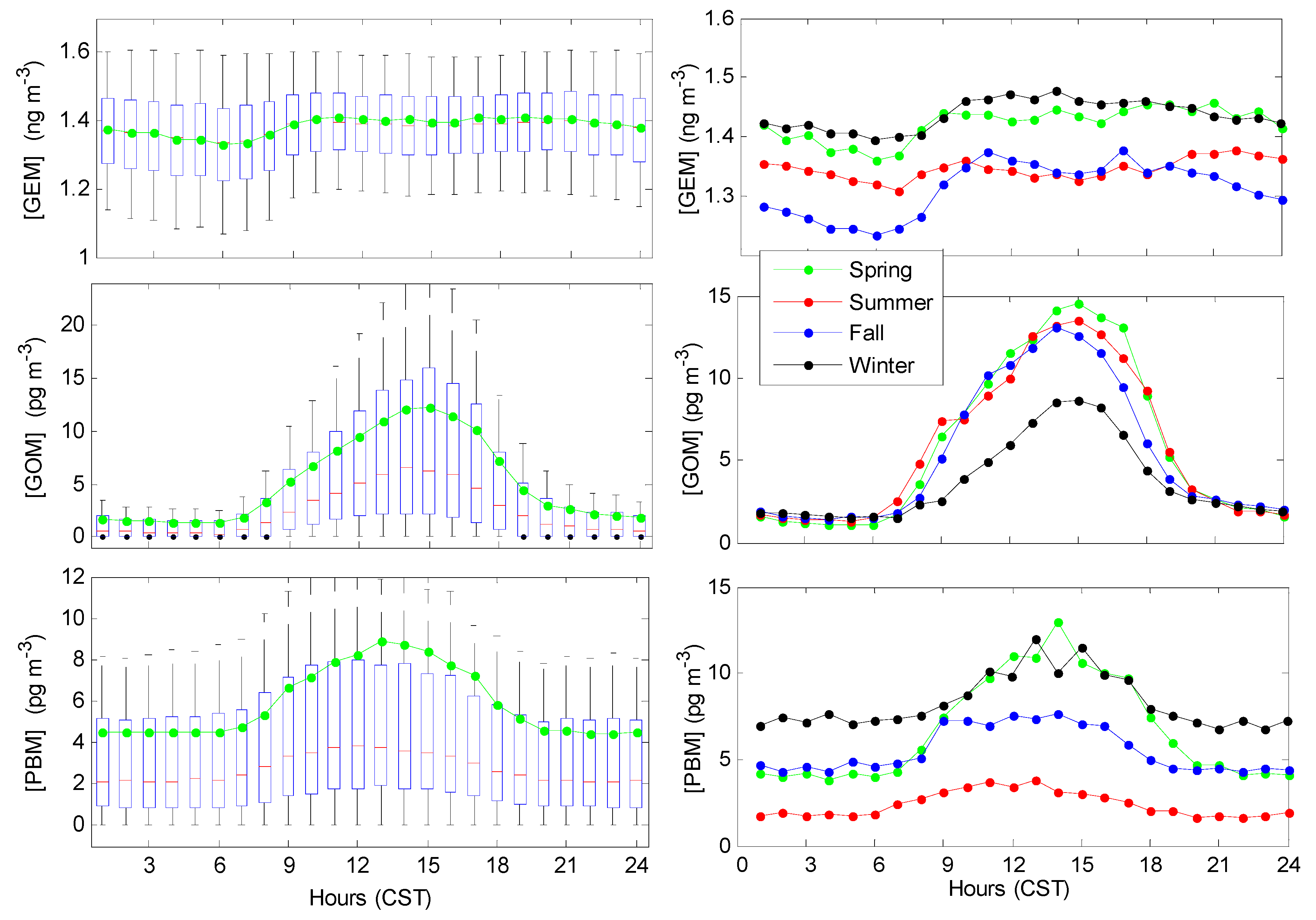
3.2. Seasonal Trends
3.3. Diurnal Trends of Hg Speciation
4. Discussion
4.1. Correlation between Observed Mercury Species and Other Chemical Species
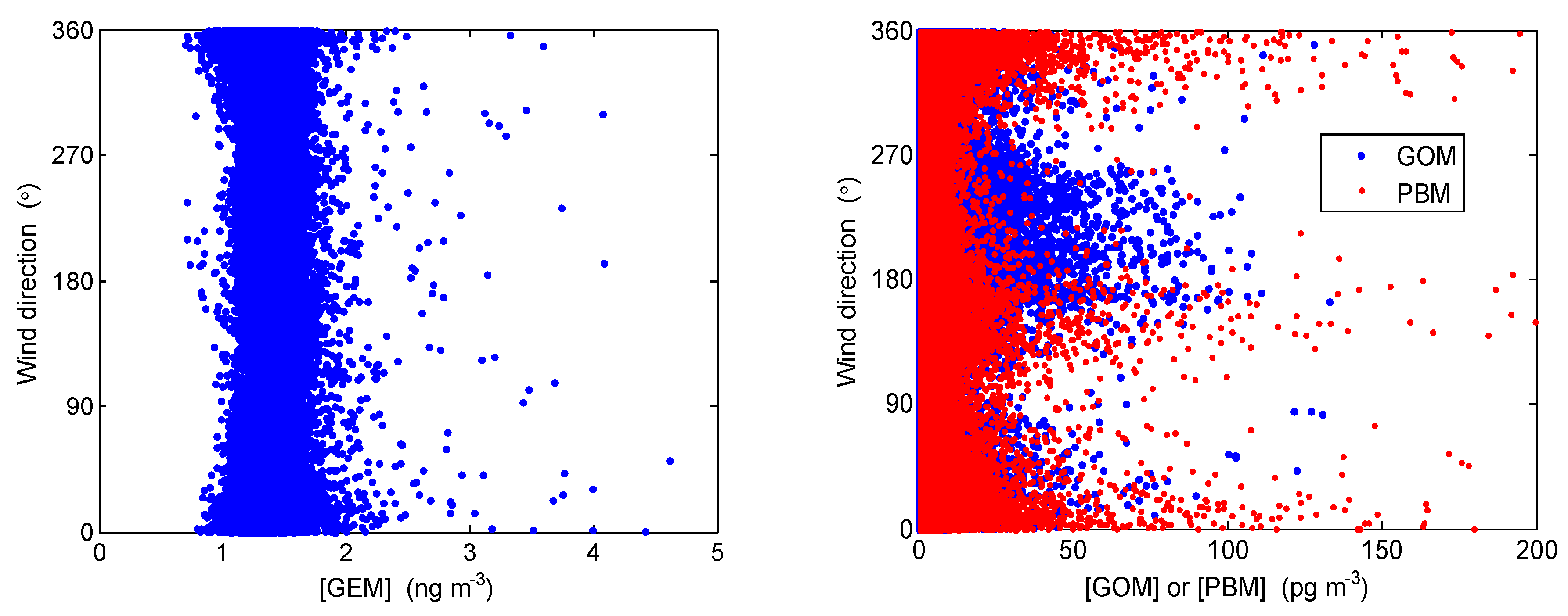
4.2. Correlation with Meteorological Parameters
4.3. GEM Depletion Events
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Choi, A.L.; Grandjean, P. Methylmercury exposure and health effects in humans. Environ. Chem. 2008, 5, 112–120. [Google Scholar] [CrossRef]
- Ruelas-Inzunza, J.; Delgado-Alvarez, C.; Frias-Espericueta, M.; Paez-Osuna, F. Mercury in the Atmospheric and Coastal Environments of Mexico. Rev. Environ. Contam. Toxicol. 2013, 226, 65–99. [Google Scholar]
- Gworek, B.; Dmuchowski, W.; Baczewska, A.H.; Bragoszewska, P.; Bemowska-Kalabun, O.; Wrzosek-Jakubowska, J. Air Contamination by Mercury, Emissions and Transformations-a Review. Water Air Soil Pollut. 2017, 228, 123. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.D.; Draxler, R.R.; Artz, R.S.; Blanchard, P.; Gustin, M.S.; Han, Y.J.; Holsen, T.M.; Jaffe, D.A.; Kelley, P.; Lei, H.; et al. Modeling the global atmospheric transport and deposition of mercury to the Great Lakes. Elem. Sci. Anthr. 2016, 4, 000118. [Google Scholar] [CrossRef]
- Zhang, L.M.; Lyman, S.; Mao, H.T.; Lin, C.J.; Gay, D.A.; Wang, S.X.; Gustin, M.S.; Feng, X.B.; Wania, F. A synthesis of research needs for improving the understanding of atmospheric mercury cycling. Atmos. Chem. Phys. 2017, 17, 9133–9144. [Google Scholar] [CrossRef]
- NPS. Fish Consumption Advisories. 2018. Available online: https://www.nps.gov/subjects/fishing/fish-consumption-advisories.htm (accessed on 5 December 2019).
- Evers, D.C.; DiGangi, J.; Petrlík, J.; Buck, D.G.; Šamánek, J.; Beeler, B.; Turnquist, M.A.; Hatch, S.K.; Regan, K. Global Mercury Hotspots: New Evidence Reveals Mercury Contamination Regularly Exceeds Health Advisory Levels in Humans and Fish Worldwide. BRI-IPEN Science Communications Series 2014-34. 2014. Available online: http://www.briloon.org/uploads/BRI_Documents/Mercury_Center/BRI-IPEN-report-update-102214%20for%20web.pdf (accessed on 5 December 2019).
- Fitzgerald, W.F.; Engstrom, D.R.; Mason, R.P.; Nater, E.A. The case for atmospheric mercury contamination in remote areas. Environ. Sci. Technol. 1998, 32, 1–7. [Google Scholar] [CrossRef]
- Schroeder, W.H.; Munthe, J. Atmospheric mercury—An overview. Atmos. Environ. 1998, 32, 809–822. [Google Scholar] [CrossRef]
- Lin, C.J.; Pehkonen, S.O. The chemistry of atmospheric mercury: A review. Atmos. Environ. 1999, 33, 2067–2079. [Google Scholar] [CrossRef]
- Harris, R.; Pollman, C.; Landing, W.; Evans, D.; Axelrad, D.; Hutchinson, D.; Morey, S.L.; Rumbold, D.; Dukhovskoy, D.; Adams, D.H.; et al. Mercury in the Gulf of Mexico: Sources to receptors. Environ. Res. 2012, 119, 42–52. [Google Scholar] [CrossRef]
- Butler, T.J.; Cohen, M.D.; Vermeylen, F.M.; Likens, G.E.; Schmeltz, D.; Artz, R.S. Regional precipitation mercury trends in the eastern USA, 1998–2005: Declines in the Northeast and Midwest, no trend in the Southeast. Atmos. Environ. 2008, 42, 1582–1592. [Google Scholar] [CrossRef]
- Coburn, S.; Dix, B.; Edgerton, E.; Holmes, C.D.; Kinnison, D.; Liang, Q.; ter Schure, A.; Wang, S.Y.; Volkamer, R. Mercury oxidation from bromine chemistry in the free troposphere over the southeastern US. Atmos. Chem. Phys. 2016, 16, 3743–3760. [Google Scholar] [CrossRef]
- Engle, M.A.; Tate, M.T.; Krabbenhoft, D.P.; Kolker, A.; Olson, M.L.; Edgerton, E.S.; DeWild, J.F.; McPherson, A.K. Characterization and cycling of atmospheric mercury along the central US Gulf Coast. Appl. Geochem. 2008, 23, 419–437. [Google Scholar] [CrossRef]
- Nair, U.S.; Wu, Y.L.; Walters, J.; Jansen, J.; Edgerton, E.S. Diurnal and seasonal variation of mercury species at coastal-suburban, urban, and rural sites in the southeastern United States. Atmos. Environ. 2012, 47, 499–508. [Google Scholar] [CrossRef]
- Castro, M.S.; Sherwell, J. Effectiveness of Emission Controls to Reduce the Atmospheric Concentrations of Mercury. Environ. Sci. Technol. 2015, 49, 14000–14007. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, H.M.; Jacob, D.J.; Zhang, Y.X.; Dibble, T.S.; Slemr, F.; Amos, H.M.; Schmidt, J.A.; Corbitt, E.S.; Marais, E.A.; Sunderland, E.M. A new mechanism for atmospheric mercury redox chemistry: Implications for the global mercury budget. Atmos. Chem. Phys. 2017, 17, 6353–6371. [Google Scholar] [CrossRef]
- Ren, X.R.; Luke, W.T.; Kelley, P.; Cohen, M.; Ngan, F.; Artz, R.; Walker, J.; Brooks, S.; Moore, C.; Swartzendruber, P.; et al. Mercury Speciation at a Coastal Site in the Northern Gulf of Mexico: Results from the Grand Bay Intensive Studies in Summer 2010 and Spring 2011. Atmosphere 2014, 5, 230–251. [Google Scholar] [CrossRef]
- USEPA. Toxic Release Inventory. 2020. Available online: https://www.epa.gov/toxics-release-inventory-tri-program/tri-basic-data-files-calendar-years-1987-2018 (accessed on 13 January 2020).
- ESRI. USA National Land Cover Database 2011, Based on Data from the Multi-Resolution Land Characteristics Consortium; ESRI ArcGIS Data Server: Redlands, CA, USA, 2019. [Google Scholar]
- Ren, X.R.; Luke, W.T.; Kelley, P.; Cohen, M.D.; Artz, R.; Olson, M.L.; Schmeltz, D.; Puchalski, M.; Goldberg, D.L.; Ring, A.; et al. Atmospheric mercury measurements at a suburban site in the Mid-Atlantic United States: Inter-annual, seasonal and diurnal variations and source-receptor relationships. Atmos. Environ. 2016, 146, 141–152. [Google Scholar] [CrossRef]
- Gay, D.A.; Schmeltz, D.; Prestbo, E.; Olson, M.; Sharac, T.; Tordon, R. The Atmospheric Mercury Network: Measurement and initial examination of an ongoing atmospheric mercury record across North America. Atmos. Chem. Phys. 2013, 13, 11339–11349. [Google Scholar] [CrossRef]
- Gustin, M.S.; Huang, J.Y.; Miller, M.B.; Peterson, C.; Jaffe, D.A.; Ambrose, J.; Finley, B.D.; Lyman, S.N.; Call, K.; Talbot, R.; et al. Do We Understand What the Mercury Speciation Instruments Are Actually Measuring? Results of RAMIX. Environ. Sci. Technol. 2013, 47, 7295–7306. [Google Scholar] [CrossRef]
- Lyman, S.N.; Cheng, I.; Gratz, L.E.; Weiss-Penzias, P.; Zhang, L. An updated review of atmospheric mercury. Sci. Total Environ. 2020, 707, 135575. [Google Scholar] [CrossRef]
- Lyman, S.N.; Jaffe, D.A.; Gustin, M.S. Release of mercury halides from KCl denuders in the presence of ozone. Atmos. Chem. Phys. 2010, 10, 8197–8204. [Google Scholar] [CrossRef]
- McClure, C.D.; Jaffe, D.A.; Edgerton, E.S. Evaluation of the KCI Denuder Method for Gaseous Oxidized Mercury using HgBr2 at an In-Service AMNet Site. Environ. Sci. Technol. 2014, 48, 11437–11444. [Google Scholar] [CrossRef]
- Holland, S.P.; Mansur, E.T.; Muller, N.; Yates, A.J. Decompositions and Policy Consequences of an Extraordinary Decline in Air Pollution from Electricity Generation; National Bureau of Economic Research: Cambridge, MA, USA, 2018. [Google Scholar]
- Mann, H.B. Nonparametric tests against trend. Econometrica 1945, 13, 245–259. [Google Scholar] [CrossRef]
- Kendall, M.G. Rank Correlation Methods; Griffin: London, UK, 1975. [Google Scholar]
- Weiss-Penzias, P.S.; Gay, D.A.; Brigham, M.E.; Parsons, M.T.; Gustin, M.S.; ter Schure, A. Trends in mercury wet deposition and mercury air concentrations across the US and Canada. Sci. Total Environ. 2016, 568, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhou, C.L.; Lynam, M.M.; Dvonch, J.T.; Barres, J.A.; Hopke, P.K.; Cohen, M.; Holsen, T.M. Atmospheric Mercury Temporal Trends in the Northeastern United States from 1992 to 2014: Are Measured Concentrations Responding to Decreasing Regional Emissions? Environ. Sci. Technol. Lett. 2017, 4, 91–97. [Google Scholar] [CrossRef]
- UNEP. Global Mercury Assessment 2018; UN Environment Programme, Chemicals and Health Branch: Geneva, Switzerland, 2019. [Google Scholar]
- Cheng, I.; Zhang, L.M.; Castro, M.; Mao, H.T. Identifying Changes in Source Regions Impacting Speciated Atmospheric Mercury at a Rural Site in the Eastern United States. J. Atmos. Sci. 2017, 74, 2937–2947. [Google Scholar] [CrossRef]
- Gustin, M.S.; Weiss-Penzias, P.S.; Peterson, C. Investigating sources of gaseous oxidized mercury in dry deposition at three sites across Florida, USA. Atmos. Chem. Phys. 2012, 12, 9201–9219. [Google Scholar] [CrossRef]
- Poissant, L.; Pilote, M.; Beauvais, C.; Constant, P.; Zhang, H.H. A year of continuous measurements of three atmospheric mercury species (GEM, RGM and Hg-p) in southern Quebec, Canada. Atmos. Environ. 2005, 39, 1275–1287. [Google Scholar] [CrossRef]
- Schleicher, N.J.; Schafer, J.; Blanc, G.; Chen, Y.; Chai, F.; Cen, K.; Norra, S. Atmospheric particulate mercury in the megacity Beijing: Spatiotemporal variations and source apportionment. Atmos. Environ. 2015, 109, 251–261. [Google Scholar] [CrossRef]
- Xu, L.L.; Chen, J.S.; Niu, Z.C.; Yin, L.Q.; Chen, Y.T. Characterization of mercury in atmospheric particulate matter in the southeast coastal cities of China. Atmos. Pollut. Res. 2013, 4, 454–461. [Google Scholar] [CrossRef]
- Brooks, S.; Luke, W.; Cohen, M.; Kelly, P.; Lefer, B.; Rappengluck, B. Mercury species measured atop the Moody Tower TRAMP site, Houston, Texas. Atmos. Environ. 2010, 44, 4045–4055. [Google Scholar] [CrossRef]
- Poissant, L. Potential sources of atmospheric total gaseous mercury in the St. Lawrence River valley. Atmos. Environ. 1999, 33, 2537–2547. [Google Scholar] [CrossRef]
- Ariya, P.A.; Amyot, M.; Dastoor, A.; Deeds, D.; Feinberg, A.; Kos, G.; Poulain, A.; Ryjkov, A.; Semeniuk, K.; Subir, M.; et al. Mercury Physicochemical and Biogeochemical Transformation in the Atmosphere and at Atmospheric Interfaces: A Review and Future Directions. Chem. Rev. 2015, 115, 3760–3802. [Google Scholar] [CrossRef] [PubMed]
- Kalinchuk, V.; Lopatnikov, E.; Astakhov, A. Gradient measurements of gaseous elemental mercury (Hg-0) in the marine boundary layer of the northwest Sea of Japan (East Sea). Environ. Pollut. 2018, 237, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Saiz-Lopez, A.; Mahajan, A.S.; Martin, J.C.G.; Armstrong, D.; Lemes, M.; Hay, T.; Prados-Roman, C. Enhanced production of oxidised mercury over the tropical Pacific Ocean: A key missing oxidation pathway. Atmos. Chem. Phys. 2014, 14, 1323–1335. [Google Scholar] [CrossRef]
- Ye, Z.Y.; Mao, H.T.; Lin, C.J.; Kim, S.Y. Investigation of processes controlling summertime gaseous elemental mercury oxidation at midlatitudinal marine, coastal, and inland sites. Atmos. Chem. Phys. 2016, 16, 8461–8478. [Google Scholar] [CrossRef]
- Brunke, E.G.; Labuschagne, C.; Ebinghaus, R.; Kock, H.H.; Slemr, F. Gaseous elemental mercury depletion events observed at Cape Point during 2007–2008. Atmos. Chem. Phys. 2010, 10, 1121–1131. [Google Scholar] [CrossRef]
- Mao, H.; Talbot, R.W.; Sigler, J.M.; Sive, B.C.; Hegarty, J.D. Seasonal and diurnal variations of Hg degrees over New England. Atmos. Chem. Phys. 2008, 8, 1403–1421. [Google Scholar] [CrossRef]
- Fu, X.W.; Zhu, W.; Zhang, H.; Sommar, J.; Yu, B.; Yang, X.; Wang, X.; Lin, C.J.; Feng, X.B. Depletion of atmospheric gaseous elemental mercury by plant uptake at Mt. Changbai, Northeast China. Atmos. Chem. Phys. 2016, 16, 12861–12873. [Google Scholar] [CrossRef]
- Lindberg, S.E.; Brooks, S.; Lin, C.J.; Scott, K.J.; Landis, M.S.; Stevens, R.K.; Goodsite, M.; Richter, A. Dynamic oxidation of gaseous mercury in the Arctic troposphere at polar sunrise. Environ. Sci. Technol. 2002, 36, 1245–1256. [Google Scholar] [CrossRef]
- Obrist, D.; Tas, E.; Peleg, M.; Matveev, V.; Fain, X.; Asaf, D.; Luria, M. Bromine-induced oxidation of mercury in the mid-latitude atmosphere. Nat. Geosci. 2011, 4, 22–26. [Google Scholar] [CrossRef]
- Steffen, A.; Lehnherr, I.; Cole, A.; Ariya, P.; Dastoor, A.; Durnford, D.; Kirk, J.; Pilote, M. Atmospheric mercury in the Canadian Arctic. Part I: A review of recent field measurements. Sci. Total Environ. 2015, 509, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; McNamara, S.M.; Moore, C.W.; Obrist, D.; Steffen, A.; Shepson, P.B.; Staebler, R.M.; Raso, A.R.W.; Pratt, K.A. Direct detection of atmospheric atomic bromine leading to mercury and ozone depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 14479–14484. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | [GEM] (ng m−3) | [GOM] (pg m−3) | [PBM] (pg m−3) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Min | Max | Median | Mean ± σ | Max | Median | Mean ± σ | Max | Median | Mean ± σ | |
| 2007 | 0.85 | 8.0 | 1.40 | 1.41 ± 0.29 | 140 | 1.2 | 4.0 ± 8.7 | 112 | 1.7 | 2.8 ± 4.3 |
| 2008 | 0.79 | 6.1 | 1.40 | 1.40 ± 0.16 | 256 | 2.1 | 7.5 ± 15.2 | 52 | 2.6 | 3.8 ± 4.1 |
| 2009 | 0.71 | 8.1 | 1.37 | 1.37 ± 0.17 | 133 | 1.7 | 5.2 ± 9.4 | 46 | 2.1 | 3.6 ± 4.4 |
| 2010 | 0.92 | 2.8 | 1.40 | 1.39 ± 0.12 | 130 | 1.7 | 6.1 ± 12.1 | 364 | 4.6 | 9.4 ± 17.5 |
| 2011 | 0.79 | 16.0 | 1.45 | 1.44 ± 0.23 | 127 | 1.7 | 6.4 ± 11.7 | 470 | 2.9 | 4.9 ± 8.7 |
| 2012 | 0.83 | 13.8 | 1.37 | 1.38 ± 0.25 | 98 | 1.5 | 4.8 ± 8.9 | 464 | 2.3 | 4.4 ± 10.4 |
| 2013 | 0.86 | 7.8 | 1.41 | 1.43 ± 0.32 | 104 | 2.2 | 5.5 ± 9.7 | 100 | 2.4 | 4.4 ± 7.5 |
| 2014 | 0.70 | 9.7 | 1.39 | 1.41 ± 0.34 | 94 | 1.8 | 4.5 ± 7.6 | 970 | 5.3 | 12.3 ± 28 |
| 2015 | 0.72 | 9.3 | 1.32 | 1.34 ± 0.23 | 71 | 1.9 | 3.5 ± 4.8 | 192 | 7.1 | 14.1 ± 19.4 |
| 2016 | 0.82 | 3.7 | 1.33 | 1.33 ± 0.14 | 42 | 1.4 | 2.9 ± 4.5 | 61 | 2.3 | 3.9 ± 5.2 |
| 2017 | 0.83 | 2.24 | 1.31 | 1.31 ± 0.14 | 46 | 0.89 | 2.7 ± 4.8 | 34 | 0.6 | 1.2 ± 2.2 |
| 2018 | 0.74 | 2.20 | 1.31 | 1.31 ± 0.13 | 27 | 0 | 1.5 ± 3.0 | 174 | 2.3 | 5.0 ± 12.2 |
| All | 0.70 | 16.0 | 1.38 | 1.39 ± 0.22 | 256 | 1.6 | 5.1 ± 10.2 | 970 | 2.7 | 5.9 ± 13.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, X.; Luke, W.T.; Kelley, P.; Cohen, M.D.; Olson, M.L.; Walker, J.; Cole, R.; Archer, M.; Artz, R.; Stein, A.A. Long-Term Observations of Atmospheric Speciated Mercury at a Coastal Site in the Northern Gulf of Mexico during 2007–2018. Atmosphere 2020, 11, 268. https://doi.org/10.3390/atmos11030268
Ren X, Luke WT, Kelley P, Cohen MD, Olson ML, Walker J, Cole R, Archer M, Artz R, Stein AA. Long-Term Observations of Atmospheric Speciated Mercury at a Coastal Site in the Northern Gulf of Mexico during 2007–2018. Atmosphere. 2020; 11(3):268. https://doi.org/10.3390/atmos11030268
Chicago/Turabian StyleRen, Xinrong, Winston T. Luke, Paul Kelley, Mark D. Cohen, Mark L. Olson, Jake Walker, Ronald Cole, Michael Archer, Richard Artz, and Ariel A. Stein. 2020. "Long-Term Observations of Atmospheric Speciated Mercury at a Coastal Site in the Northern Gulf of Mexico during 2007–2018" Atmosphere 11, no. 3: 268. https://doi.org/10.3390/atmos11030268
APA StyleRen, X., Luke, W. T., Kelley, P., Cohen, M. D., Olson, M. L., Walker, J., Cole, R., Archer, M., Artz, R., & Stein, A. A. (2020). Long-Term Observations of Atmospheric Speciated Mercury at a Coastal Site in the Northern Gulf of Mexico during 2007–2018. Atmosphere, 11(3), 268. https://doi.org/10.3390/atmos11030268