Electrochemical Evidence of non-Volatile Reduced Sulfur Species in Water-Soluble Fraction of Fine Marine Aerosols


Abstract
1. Introduction
2. Experiments
2.1. Materials and Methods
2.2. Aerosol Sampling and Preparation
2.3. Electrochemical Instrumentation and Procedure
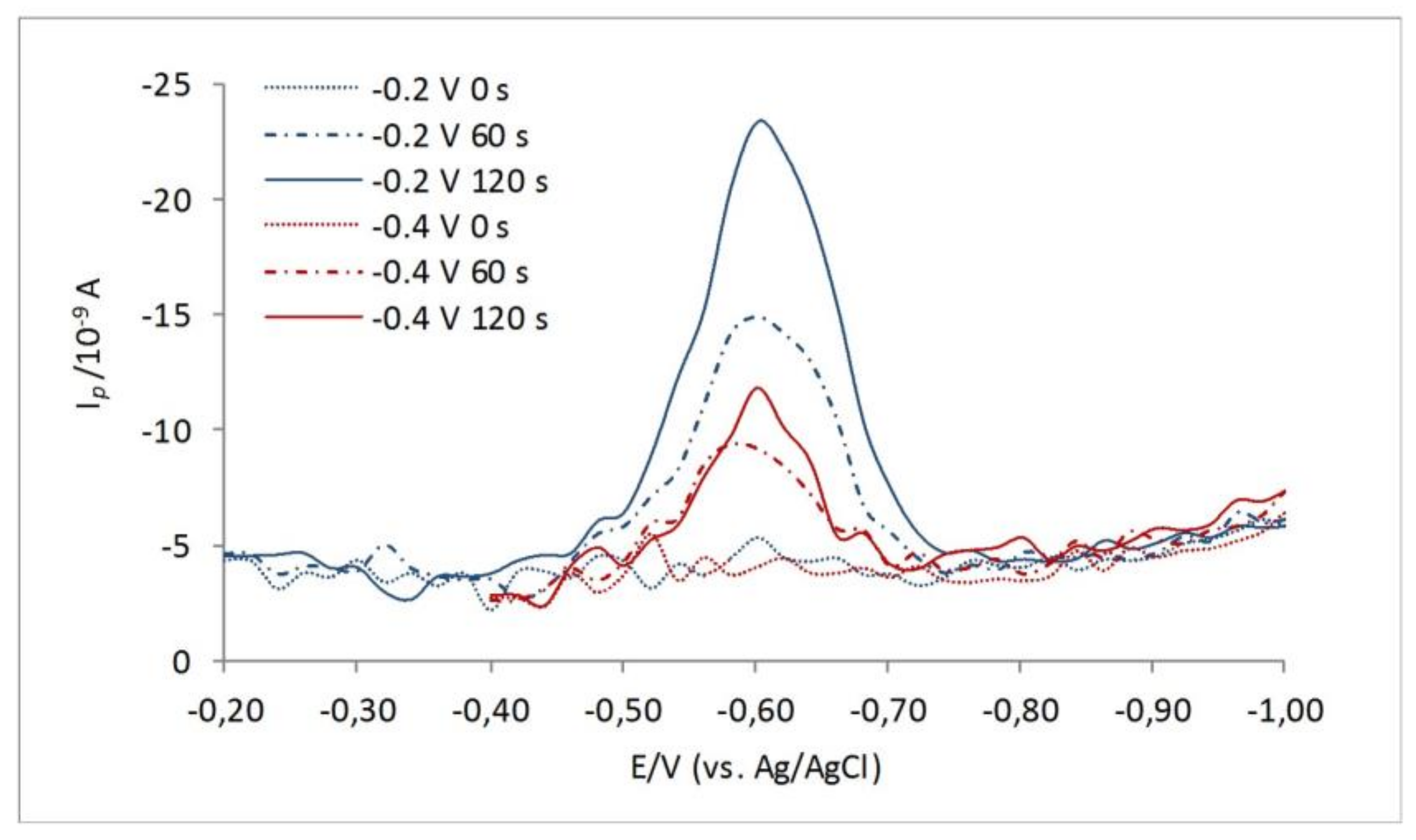
3. Results and Discussion
3.1. Electrochemical Behavior of 3-MPA
3.2. Mixture of Model RSS: 3-MPA and Na2S as Representatives for Organic and Inorganic RSS
3.3. Electrochemical RSS Responses in WS Fraction of Fine Marine Aerosols
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Luther, G.W.; Tsamakis, E. Concentration and form of dissolved sulfide in the oxic water column of the ocean. Mar. Chem. 1989, 27, 165–177. [Google Scholar] [CrossRef]
- Ciglenečki, I.; Ćosović, B. Electrochemical Study of Sulfur Species in Seawater and Marine Phytoplankton Cultures. Mar. Chem. 1996, 52, 87–97. [Google Scholar] [CrossRef]
- Ciglenečki, I.; Ćosović, B. Electrochemical Determination of Thiosulfate in Seawater in the Presence of Elemental Sulfur and Sulfide. Electroanalysis 1997, 9, 775–780. [Google Scholar] [CrossRef]
- Al-Farawati, R.K.; Van den Berg, C.M.G. The Determination of Sulfide in Seawater by Flow-Analysis with Voltammetric Detection. Mar. Chem. 1997, 57, 277–286. [Google Scholar] [CrossRef]
- Rozan, T.F.; Theberge, S.M.; Luther, G. Quantifying elemental sulfur (S0), bisulfide (HS-) and polysulfides (Sx2-) using a voltametric method. Anal. Chim. Acta 2000, 415, 175–184. [Google Scholar] [CrossRef]
- Laglera, L.M.; Van den Berg, C.M.G. Copper complexation by thiol compounds in estuarine waters. Mar. Chem. 2003, 82, 71–89. [Google Scholar] [CrossRef]
- Bura-Nakić, E.; Helz, G.R.; Ciglenečki, I.; Ćosović, B. Seasonal variations in reduced sulfur species in a stratified seawaterlake (Rogoznica Lake, Croatia): Evidence for organic carriers of reactive sulfur. Geochim. Cosmochim. Acta 2009, 73, 3738–3751. [Google Scholar] [CrossRef]
- Bura-Nakić, E.; Viollier, B.; Ciglenečki, I. Electrochemical and colorimetric measurements show the dominant role of FeS in a permanently anoxic lake. Environ. Sci. Technol. 2013, 47, 741–749. [Google Scholar] [CrossRef]
- Superville, P.-J.; Pižeta, I.; Omanović, D.; Billon, G. Identification and on-line monitoring of reduced sulphur species (RSS) by voltammetry in oxic waters. Talanta 2013, 112, 55–62. [Google Scholar] [CrossRef]
- Laglera, L.M.; Downes, J.; Tovar-Sánchez, A.; Monticelli, D. Cathodic pseudopolarography: A new tool for the identification and quantification of cysteine, cystine and other low molecular weight thiols in seawater. Anal. Chim. Acta 2014, 836, 24–33. [Google Scholar] [CrossRef]
- Marie, L.; Pernet-Coudrier, B.; Waeles, M.; Gabon, M.; Riso, R. Dynamics and sources of reduced sulfur, humic substances and dissolved organic carbon in a temperate river system affected by agricultural practices. Sci. Total Environ. 2015, 537, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Guéguen, C.; Smith, D.S. Assessing effects of pH, metal ion and natural organic matter on identification and determination of reduced glutathione by cathodic stripping voltammetry. Int. J. Environ. Anal. Chem. 2017, 97, 330–334. [Google Scholar] [CrossRef]
- Krznarić, D.; Ciglenečki, I.; Ćosović, B. Voltammetric investigations of 2-dimethylarsinyl-ethanol sulphide in NaCl and seawater. Anal. Chim. Acta 2001, 431, 269–278. [Google Scholar] [CrossRef]
- Milanović, I.; Krznarić, D.; Bura-Nakić, E.; Ciglenečki, I. Deposition and dissolution of metal sulfide layers at a Hg electrode surface in seawater electrolyte conditions. Environ. Chem. 2013, 11, 167–172. [Google Scholar] [CrossRef]
- Florence, T.M. Cathodic stripping voltammetry: Part I. Determination of organic sulfur compounds, flavins and porphyrins at the sub-micromolar level. J. Electroanal. Chem. 1979, 97, 219–236. [Google Scholar] [CrossRef]
- Orlović-Leko, P.; Omanović, D.; Ciglenečki, I.; Vidović, K.; Brenko, T. Application of electrochemical methods in the physico-chemical characterization of atmospheric precipitation. Bulg. Chem. Commun. 2017, 49, 211–217. [Google Scholar]
- Williams, K.D.; Jones, A.; Roberts, D.L.; Senior, C.A.; Woodage, M.J. The response of the climate system to the indirect effects of anthropogenic sulfate aerosol. Clim. Dyn. 2001, 17, 845–856. [Google Scholar] [CrossRef]
- Burnett, R.T.; Dales, R.; Krewski, D.; Vincent, R.; Dann, T.; Brook, J.R. Associations between ambient particulate sulfate and admissions to Ontario hospitals for cardiac and respiratory diseases. Am. J. Epidemiol. 1995, 142, 15–22. [Google Scholar] [CrossRef]
- Pardo, M.; Porat, Z.; Rudich, A.; Schauer, J.J.; Rudich, Y. Repeated exposures to roadside particulate matter extracts suppresses pulmonary defense mechanisms, resulting in lipid and protein oxidative damage. Environ. Pollut. 2016, 210, 227–237. [Google Scholar] [CrossRef]
- Galloway, J.N. Acid deposition: Perspectives in time and space. Water Air Soil Pollut. 1995, 85, 15–24. [Google Scholar] [CrossRef]
- Charlson, R.J.; Lovelock, J.E.; Andreae, M.O.; Warren, S.G. Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate. Nature 1987, 326, 655–661. [Google Scholar] [CrossRef]
- Quinn, P.K.; Bates, T.S. The case against climate regulation via oceanic phytoplankton sulphur emissions. Nature 2011, 480, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Cullis, C.F.; Hirschler, M.M. Atmospheric sulfur: Natural and man-made sources. Atmos. Environ. 1980, 14, 1263–1278. [Google Scholar] [CrossRef]
- Chin, M.; Jacob, D.J.; Gardner, G.M.; Foreman-Fowler, M.S.; Spiro, P.A.; Savoie, D.L. A global three-dimensional model of tropospheric sulfate. J. Geophys. Res. 1996, 101, 18667–18690. [Google Scholar] [CrossRef]
- Smith, S.J.; Pitcher, H.; Wigley, T.M.L. Global and Regional Anthropogenic Sulfur Dioxide Emissions. Glob. Planet. Change 2001, 29, 99–119. [Google Scholar] [CrossRef]
- Graf, H.F.; Langmann, B.; Feichter, J. The contribution of Earth degassing to the atmospheric sulfur budget. Chem. Geol. 1998, 147, 131–145. [Google Scholar] [CrossRef]
- Bhugwant, C.; Siéja, B.; Bessafi, M.; Staudacher, T.; Ecormier, J. Atmospheric sulfur dioxide measurements during the 2005 and 2007 eruptions of the Piton de La Fournaise volcano: Implications for human health and environmental changes. J. Volcanol. Geotherm. Res. 2009, 184, 208–224. [Google Scholar] [CrossRef]
- Kristmannsdottir, H.; Sigurgeirsson, M.; Armannsson, H.; Hjartarson, H.; Olafsson, M. Sulfur gas emissions from geothermal power plants in Iceland. Geothermics 2000, 29, 525–538. [Google Scholar] [CrossRef]
- Andreae, M.O. Ocean-atmosphere Interactions in the global biogeochemical sulfur cycle. Mar. Chem. 1990, 30, 1–29. [Google Scholar] [CrossRef]
- Malin, G.; Kirst, G.O. Algal production of dimethyl sulfide and its atmospheric role. J. Phycol. 1997, 33, 889–896. [Google Scholar] [CrossRef]
- Stefels, J.; Steinke, M.; Turner, S.; Malin, G.; Belviso, S. Environmental constraints on the production and removal of the climatically active gas dimethylsulphide (DMS) and implications for ecosystem modeling. Biogeochemistry 2007, 83, 245–275. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics from Air Pollution to Climate Change, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2006. [Google Scholar]
- Neubauer, K.R.; Sum, S.T.; Johnston, M.V.; Wexler, A.S. Sulfur speciation in individual aerosol particles. J. Geophys. Res. 1996, 101, 18701–18707. [Google Scholar] [CrossRef]
- Graham, B.; Guyon, P.; Maenhaut, W.; Taylor, P.E.; Ebert, M.; Matthias-Maser, S.; Mayol-Bracero, O.L.; Godoi, R.H.M.; Artaxo, P.; Meixner, F.X.; et al. Composition and diurnal variability of the natural Amazonian aerosol. J. Geophys. Res. 2003, 108, 4765–4782. [Google Scholar] [CrossRef]
- Cozzi, F.; Pellergrini, I.; Adami, G.; Reisenhofer, E.; Bovenzi, M.; Barbieri, P. Sulphur speciation of PM10 samples by XANES spectroscopy. Cent. Eur. J. Chem. 2009, 7, 395–401. [Google Scholar] [CrossRef]
- Frka, S.; Dautović, J.; Kozarac, Z.; Ćosović, B.; Hoffer, A.; Kiss, G. Surface-active substances in atmospheric aerosol: An electrochemical. Tellus B Chem. Phys. Meteorol. 2012, 64, 18490–18503. [Google Scholar] [CrossRef][Green Version]
- Luther, G.W.; Church, T.M.; Giblin, A.E.; Howarth, R.W. Speciation of Dissolved Sulfur in Salt Marshes by Polarographic Methods. In Organic Marine Geochemistry; Symposium Series; Mary, L.J., Ed.; Marine Chemistry in the Coastal Environment, American Chemical Society: Washington, DC, USA; Volume 305, pp. 340–355, Chapter 20.
- Renard, J.J.; Kubes, G.; Bolker, H.I. Polarographic determination of sulfur compounds in pulping liquors. Anal. Chem. 1975, 47, 1347–1352. [Google Scholar] [CrossRef]
- Wang, F.; Tessier, A. Voltammetric determination of elemental sulfur in pore waters. Limnol. Oceanogr. 1998, 43, 1353–1361. [Google Scholar] [CrossRef]
- Laglera, L.M.; Tovar-Sanchez, A. Direct recognition and quantification by voltammetry of thiol/ thioamide mixes in seawater. Talanta 2012, 89, 496–504. [Google Scholar] [CrossRef]
- Pernet-Coudrier, B.; Waeles, M.; Filella, M.; Quentel, F.; Riso, R.D. Simple and simultaneous determination of glutathione, thioacetamide and refractory organic matter in natural waters by DP-CSV. Sci. Total Environ. 2013, 463, 997–1005. [Google Scholar] [CrossRef]
- Banica, F.-G.; Galik, M.; Švancar, I.; Vytras, K. Electrochemical Investigation of Metal Sulfides at Mercury Electrodes Using Thiourea as a Source of Sulfide Ion. Electroanalysis 2009, 21, 332–341. [Google Scholar] [CrossRef]
- Omanović, D.; Branica, M. Automation of voltammetric measurements by polarographic analyser PAR 384B. Croat. Chem. Acta 1998, 71, 421–433. [Google Scholar]
- Ciglenečki, I.; Ljubešić, Z.; Janeković, I.; Batistić, M. Rogoznica Lake, A Euxinic Marine Lake on the ADRIATIC Coast (Croatia) that Fluctuates Between Anoxic Holomictic and Meromictic Conditions; Ramesh, D.G., Zadereev, E.S., Degermendzhi, A.G., Eds.; Springer: Wageningen, The Netherlands, 2017; pp. 125–154. [Google Scholar]
- Ciglenečki, I.; Kodba, Z.; Ćosović, B. Sulfur Species in Rogoznica Lake. Mar. Chem. 1996, 53, 101–110. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Ceburnis, D.; Cavalli, F.; Jourdan, O.; Putaud, J.P.; Facchini, M.C.; Decesari, S.; Fuzzi, S.; Sellegri, K.; Jennings, S.G.; et al. Seasonal characteristics of the physicochemical properties of North Atlantic marine atmospheric aerosols. J. Geophys. Res. Atoms. Geophys. Union 2007, 112, D04206. [Google Scholar] [CrossRef]
- Shakya, K.M.; Peltier, R.E. Non-sulfate sulfur in fine aerosols across the United States: Insight for organosulfate prevalence. Atmos. Environ. 2015, 100, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Casotto, R.; Cvitešić Kušan, A.; Bhattu, D.; Ciglenečki, I.; Frka, S.; Kroflič, A.; Grgić, I.; Baltensperger, U.; Slowik, J.; Prévôt, A.S.H. Combined analysis using AMS and EESI measures for organic aerosol source apportionment of the Adriatic coast, poster presentation (P1-054). In Proceedings of the European Aerosol Conference, Gothenburg, Sweden, 25–30 August 2019. [Google Scholar]
- Kiss, G.; Tombacz, E.; Hansson, H.C. Surface tension effects of humic-like substances in the aqueous extract of tropospheric fine aerosol. J. Atmos. Chem. 2005, 50, 279–294. [Google Scholar] [CrossRef]
- Facchini, M.C.; Decesari, S.; Mircea, M.; Fuzzi, S.; Loglio, G. Surface tension of atmospheric wet aerosol and cloud/fog droplets in relation to their organic carbon content and chemical composition. Atmos. Environ. 2000, 34, 4853–4857. [Google Scholar] [CrossRef]
- Kiss, G.; Varga, B.; Galambos, I.; Ganszky, I. Characterization of water-soluble organic matter isolated from atmospheric fine aerosol. J. Geophys. Res. Atmos. 2002, 107, D21. [Google Scholar] [CrossRef]
- Gašparović, B.; Ćosović, B. Electrochemical estimation of the dominant type of surface active substances in seawater samples using o-nitrophenol as a probe. Mar. Chem. 1994, 46, 179–188. [Google Scholar] [CrossRef]
- Kroflič, A.; Frka, S.; Simmel, M.; Wex, H.; Grgić, I. Size-resolved surface active substances of atmospheric aerosol: Reconsideration of 3 the impact on sloud droplet formation. Environ. Sci. Technol. 2018, 52, 9179–9187. [Google Scholar] [CrossRef]
- Jang, K.-S.; Choi, A.Y.; Choi, M.; Kang, H.; Kim, T.-W.K.; Park, K.T. Size-Segregated chemical compositions of HULISs in ambient aerosols collected during the winter season in Songdo, South Korea. Atmosphere 2019, 10, 226–240. [Google Scholar] [CrossRef]
- Jung, J.; Hong, S.-B.; Chen, M.; Hur, J.; Jiao, L.; Lee, Y.; Park, K.; Hahm, D.; Choi, Y.-O.; Yang, E.J.; et al. Characteristics of biogenically-derived aerosols over the Amundsen Sea, Antarctica. Atmos. Chem. Phys. Discuss. 2019, 3. [Google Scholar] [CrossRef]
- Ciglenečki, I.; Plavšić, M.; Vojvodić, V.; Ćosović, B.; Pepi, M.; Baldi, F. Mucopolysaccharide transformation by sulfide in diatom culture and natural mucilage. Mar. Ecol. Prog. Ser. 2003, 263, 17–27. [Google Scholar] [CrossRef]
- Strmečki, S.; Plavšić, M.; Steigenberger, S.; Passow, U. Characterization of phytoplankton exudates and carbohydrates in relation to their complexation of copper, cadmium and iron. Mar. Ecol. Prog. Ser. 2010, 408, 33–46. [Google Scholar] [CrossRef]
- Strmečki, S.; Paleček, E. Adsorption/desorption of biomacromolecules involved in catalytic hydrogen evolution. Bioelectrochemistry 2018, 120, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Cvitešić Kušan, A.; Kroflič, A.; Grgić, I.; Ciglenečki, I.; Frka, S. Chemical characterization of fine aerosols in respect to water-soluble ions at the eastern Middle Adriatic coast. Environ. Sci. Pollut. Res. 2019. Submitted to. [Google Scholar]
- Shea, D.; MaCcre, W.A. Determination of hydrophilic thiols in sediment porewater using ion-pair liquid chromatography coupled with electrochemical detection. Anal. Chem. 1988, 60, 1449–1454. [Google Scholar] [CrossRef]
- Saltzman, E.S.; Cooper, W.J. Biogenic Sulfur in the Environment; American Chemical Society: Washington, DC, USA, 1989; Volume 39. [Google Scholar]
- Eitel, E.M.; Taillefert, M. Mechanistic investigation of Fe(III) oxide reduction by low molecular weight organic sulfur species. Geochim. Cosmochim. Acta 2017, 215, 173–188. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aerosol Sample | Sampling Period | WSOC (µg m−3) | SAS eq. T-X-100 (µg m−3) | RSS (ng m−3) Mercapto Type | RSS (ng m−3) Inorganic Type | SO42− (µg m−3) |
|---|---|---|---|---|---|---|
| S1 | 29.03–31.03.2016. | 2.36 | 0.18 | 2.62 | - | 3.00 |
| S2 | 31.03–02.04.2016. | 2.22 | 0.30 | 9.42 | - | 2.33 |
| S3 | 02.04–04.04.2016. | 2.20 | 0.23 | 3.68 | - | 4.22 |
| A1 | 08.10–10.08.2016. | 1.84 | 0.23 | 2.23 | 0.26 | 2.03 |
| A2 | 10.10–12.08.2016. | 1.18 | 0.21 | 1.46 | 0.05 | 1.58 |
| A3 | 12.10–14.10.2016. | 0.97 | 0.21 | 1.00 | 0.04 | 1.77 |
| A6 | 18.10–20.10.2016. | 1.34 | 0.23 | 1.02 | 0.03 | 1.50 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cvitešić Kušan, A.; Frka, S.; Ciglenečki, I. Electrochemical Evidence of non-Volatile Reduced Sulfur Species in Water-Soluble Fraction of Fine Marine Aerosols. Atmosphere 2019, 10, 674. https://doi.org/10.3390/atmos10110674
Cvitešić Kušan A, Frka S, Ciglenečki I. Electrochemical Evidence of non-Volatile Reduced Sulfur Species in Water-Soluble Fraction of Fine Marine Aerosols. Atmosphere. 2019; 10(11):674. https://doi.org/10.3390/atmos10110674
Chicago/Turabian StyleCvitešić Kušan, Ana, Sanja Frka, and Irena Ciglenečki. 2019. "Electrochemical Evidence of non-Volatile Reduced Sulfur Species in Water-Soluble Fraction of Fine Marine Aerosols" Atmosphere 10, no. 11: 674. https://doi.org/10.3390/atmos10110674
APA StyleCvitešić Kušan, A., Frka, S., & Ciglenečki, I. (2019). Electrochemical Evidence of non-Volatile Reduced Sulfur Species in Water-Soluble Fraction of Fine Marine Aerosols. Atmosphere, 10(11), 674. https://doi.org/10.3390/atmos10110674