Specific LTR-Retrotransposons Show Copy Number Variations between Wild and Cultivated Sunflowers

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Genotypes and Illumina Sequences Used in the Analyses
2.2. Long Terminal Repeats-Retrotransposon Redundancy Estimation
2.3. Retrotransposon Distribution along the Sunflower (HanXRQInbred Line) Genome
2.4. Analysis of Proximity of Long Terminal Repeats-Retrotransposons to Genes
3. Results
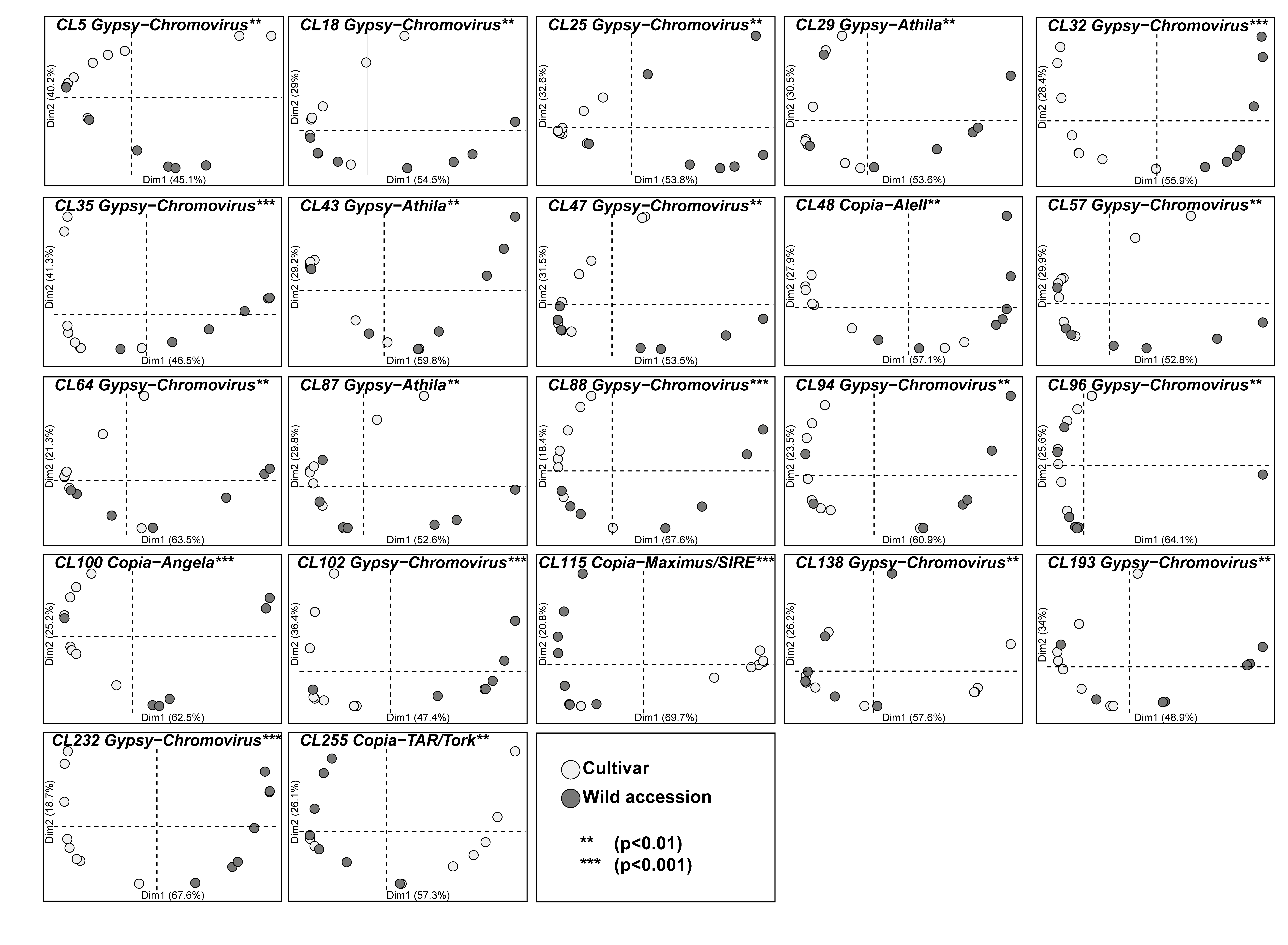
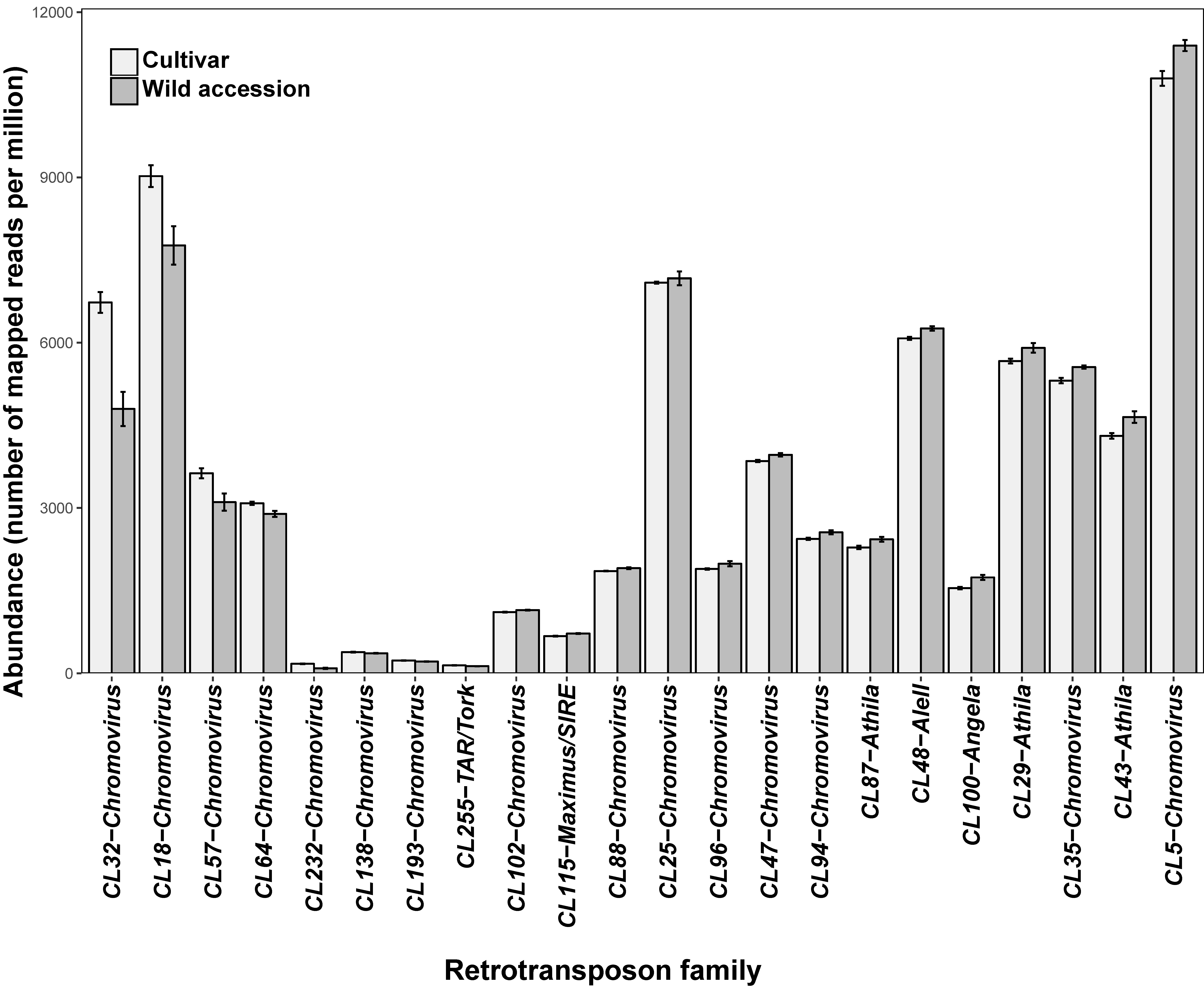
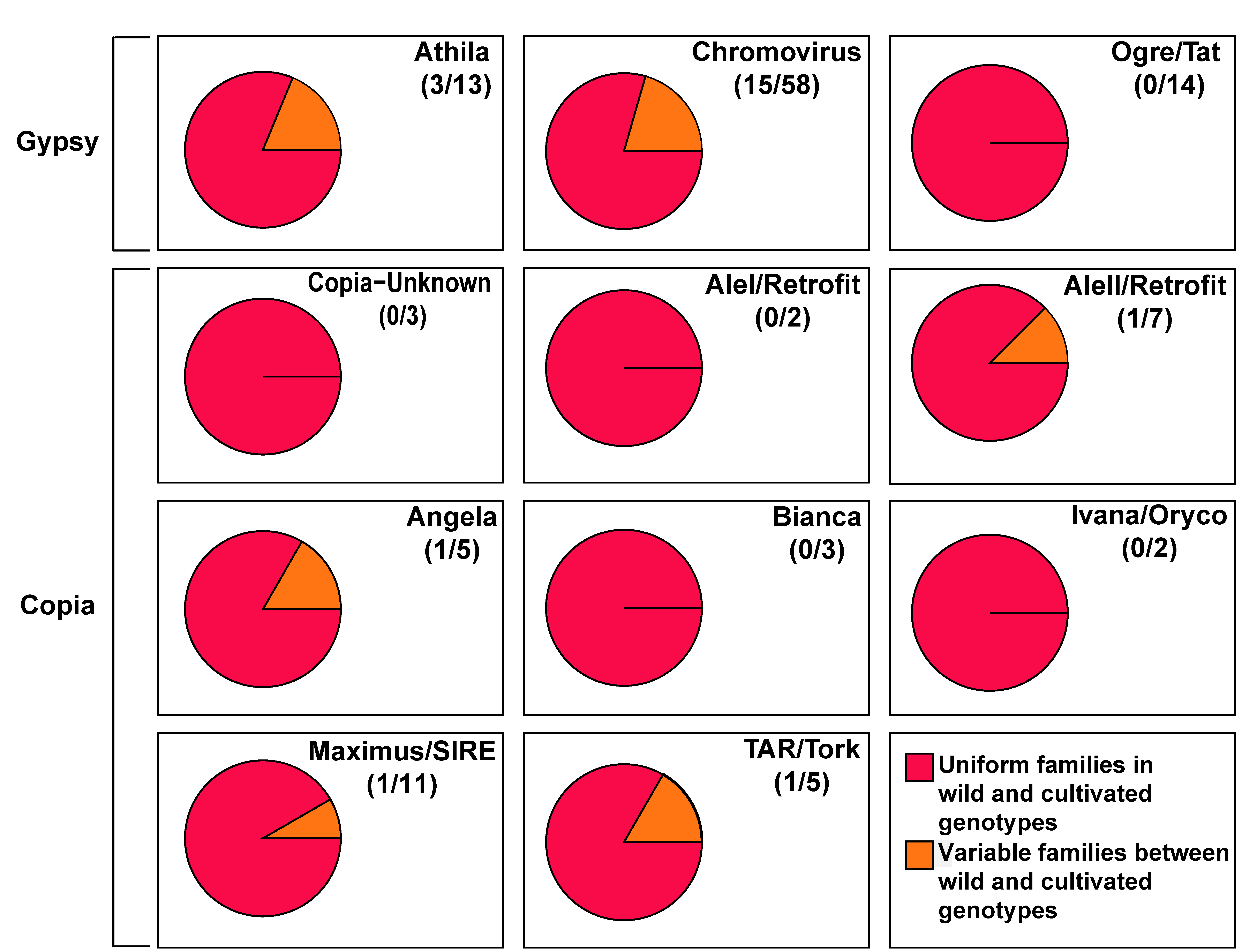
3.1. Some Long Terminal Repeats-Retrotransposon Families Show Significant Differences in Abundance between Wild and Cultivated Genotypes
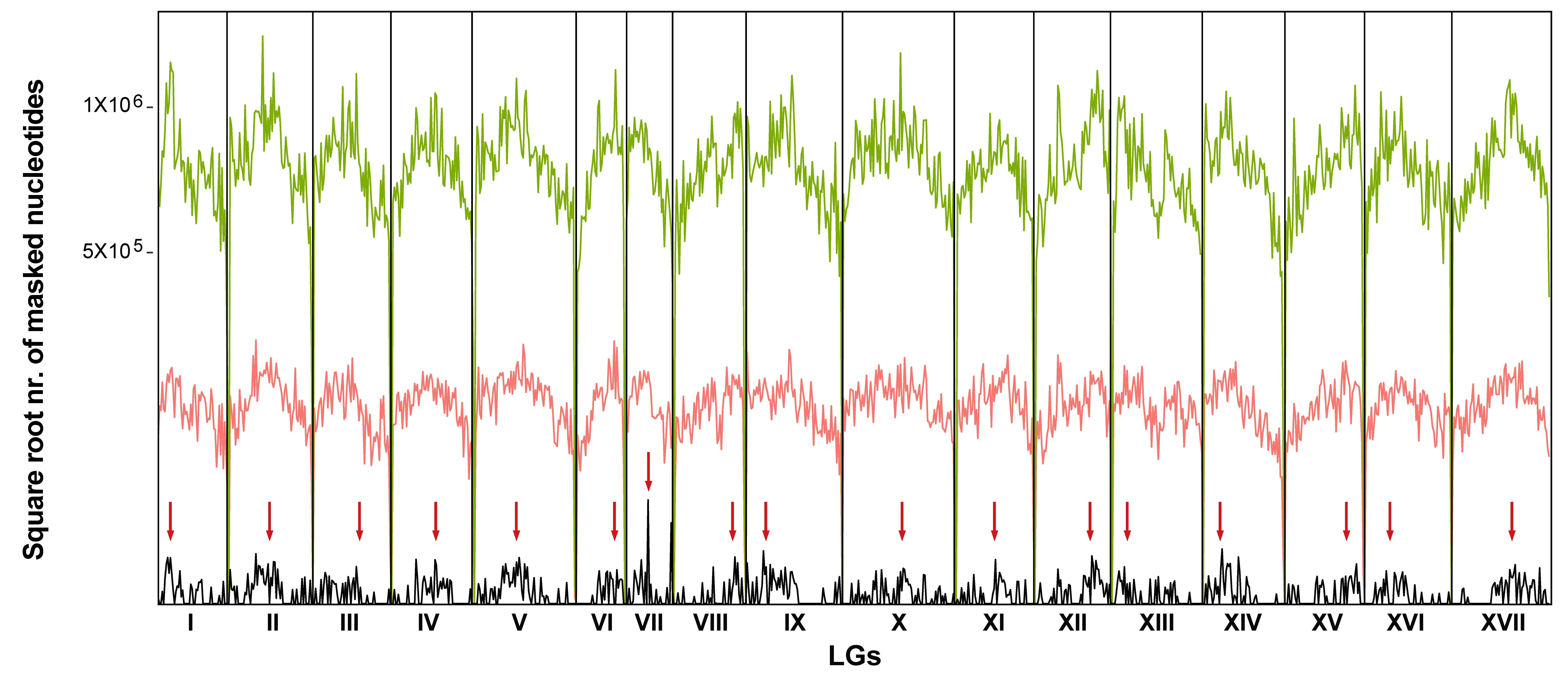
3.2. Chromosomal Localization of Long Terminal Repeats-Retrotransposons Families
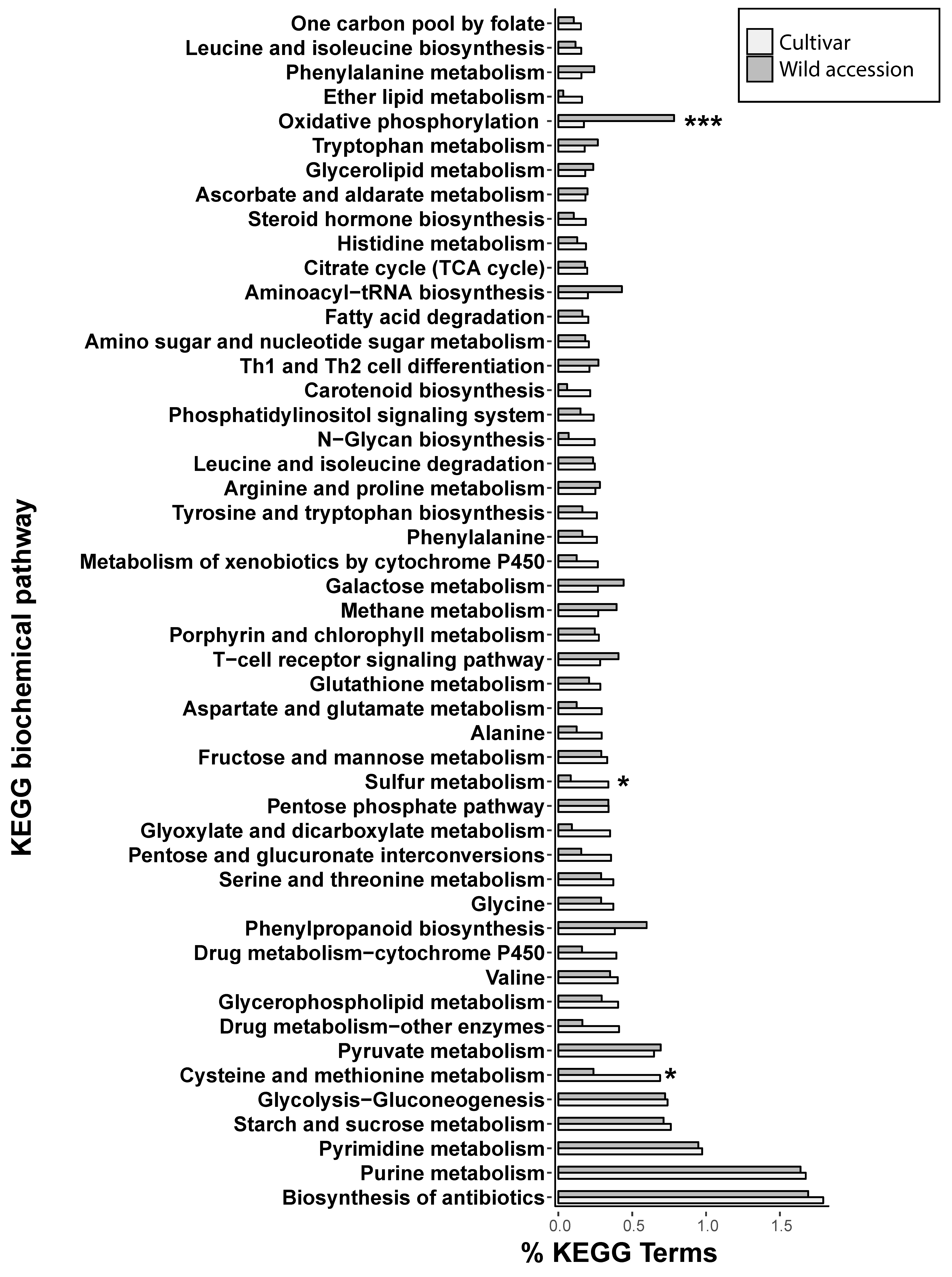
3.3. Proximity of Retrotransposons to Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Keller, B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007, 17, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Llorens, C.; Futami, R.; Covelli, L.; Domínguez-Escribá, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodríguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The Gypsy Database (GyDB) of mobile genetic elements: Release 2.0. Nucl. Acids Res. 2011, 39, D70–D74. [Google Scholar] [CrossRef] [PubMed]
- Natali, L.; Cossu, R.M.; Mascagni, F.; Giordani, T.; Cavallini, A. A survey of Gypsy and Copia LTR-retrotransposon superfamilies and lineages and their distinct dynamics in the Populustrichocarpa (L.) genome. Tree Genet. Genomes 2015, 11, 107. [Google Scholar] [CrossRef]
- Vitte, C.; Bennetzen, J.L. Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc. Natl. Acad. Sci. USA 2006, 103, 17638–17643. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Bennetzen, J.L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 12404–12410. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Dooner, H.K. Remarkable variation in maize genome structure inferred from haplotype diversity at the bz locus. Proc. Natl. Acad. Sci. USA 2006, 103, 17644–17649. [Google Scholar] [CrossRef] [PubMed]
- Devos, K.M.; Brown, J.K.; Bennetzen, J.L. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 2002, 12, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Vitte, C.; Panaud, O. Formation of solo-LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice (Oryza sativa L.). Mol. Biol. Evol. 2003, 20, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Brunner, S.; Fengler, K.; Morgante, M.; Tingey, S.; Rafalski, A. Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell 2005, 17, 343–360. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Luo, X.; Tian, F.; Li, K.; Zhu, Z.; Su, W.; Qian, X.; Fu, Y.; Wang, X.; Sun, C.; et al. Haplotype variation in structure and expression of a gene cluster associated with a quantitative trait locus for improved yield in rice. Genome Res. 2006, 16, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Dubin, M.J.; Mittelsten Scheid, O.; Becker, C. Transposons: A blessing curse. Curr. Opin. Plant Biol. 2018, 42, 23–29. [Google Scholar] [CrossRef] [PubMed]
- VanDriel, R.; Fransz, P.F.; Verschure, P.J. The eukaryotic genome: A system regulated at different hierarchical levels. J. Cell Sci. 2003, 116, 4067–4075. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Sui, A.; Garen, A. Binding of mouse VL30 retrotransposon RNA to PSF protein induces genes repressed by PSF: Effects on steroidogenesis and oncogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Hollister, J.D.; Smith, L.M.; Guo, Y.L.; Ott, F.; Weigel, D.; Gaut, B.S. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2011, 108, 2322–2327. [Google Scholar] [CrossRef] [PubMed]
- Vitte, C.; Fustier, M.A.; Alix, K.; Tenaillon, M.I. The bright side of transposons in crop evolution. Brief. Funct. Genom. 2014, 13, 276–295. [Google Scholar] [CrossRef] [PubMed]
- Springer, N.M.; Ying, K.; Fu, Y.; Ji, T.; Yeh, C.T.; Jia, Y.; Wu, W.; Richmond, T.; Kitzman, J.; Rosenbaum, H.; et al. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet. 2009, 5, e1000734. [Google Scholar] [CrossRef] [PubMed]
- Albert, P.S.; Gao, Z.; Danilova, T.V.; Birchler, J.A. Diversity of chromosomal karyotypes in maize and its relatives. Cytogenet. Genome Res. 2010, 129, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Naito, K.; Cho, E.; Yang, G.; Campbell, M.A.; Yano, K.; Okumoto, Y.; Tanisaka, T.; Wessler, S.R. Dramatic amplification of a rice transposable element during recent domestication. Proc. Natl. Acad. Sci. USA 2006, 103, 17620–17625. [Google Scholar] [CrossRef] [PubMed]
- Mascagni, F.; Barghini, E.; Giordani, T.; Rieseberg, L.H.; Cavallini, A.; Natali, L. Repetitive DNA and plant domestication: Variation in copy number and proximity to genes of LTR-retrotransposons among wild and cultivated sunflower (Helianthus annuus) genotypes. Genome Biol. Evol. 2015, 7, 3368–3382. [Google Scholar] [CrossRef] [PubMed]
- Schilling, E.E. Phylogenetic analysis of Helianthus (Asteraceae) based on chloroplast DNA restriction site data. Theor. Appl. Genet. 1997, 94, 925–933. [Google Scholar] [CrossRef]
- Schilling, E.E.; Linder, C.R.; Noyes, R.D.; Rieseberg, L.H. Phylogenetic relationships in Helianthus (Asteraceae) based on nuclear ribosomal DNA internal transcribed spacer region sequence data. Syst Bot. 1998, 23, 177–187. [Google Scholar] [CrossRef]
- Lentz, D.L.; Pohl, M.D.; Alvarado, J.L.; Tarighat, S.; Bye, R. Sunflower (Helianthus annuus L.) as a pre-Columbian domesticate in Mexico. Proc. Natl. Acad. Sci. USA 2008, 105, 6232–6237. [Google Scholar] [CrossRef] [PubMed]
- Harter, A.V.; Gardner, K.A.; Falush, D.; Lentz, D.L.; Bye, R.A.; Rieseberg, L.H. Origin of extant domesticated sunflowers in eastern North America. Nature 2004, 430, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Blackman, B.K.; Scascitelli, M.; Kane, N.C.; Luton, H.H.; Rasmussen, D.A.; Bye, R.A.; Lentz, D.L.; Rieseberg, L.H. Sunflower domestication alleles support single domestication center in eastern North America. Proc. Natl. Acad. Sci. USA 2011, 108, 14360–14365. [Google Scholar] [CrossRef] [PubMed]
- Zukovsky, P.M. Cultivated Plants and Their Wild Relatives; Commonwealth Agriculture Bureau: Farnham Royal, UK, 1950. [Google Scholar]
- Meyer, R.S.; Purugganan, M.D. Evolution of crop species: Genetics of domestication and diversification. Nat. Rev. Genet. 2013, 14, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Olsen, K.M.; Wendel, J.F. A bountiful harvest: Genomic insights into crop domestication phenotypes. Ann. Rev. Plant Biol. 2013, 64, 47–70. [Google Scholar] [CrossRef] [PubMed]
- Semelczi-Kovacs, A. Acclimatization and dissemination of the sunflower in Europe. Acta Ethnogr. Acad. Sci. Hung. 1975, 24, 47–88. [Google Scholar]
- Korell, M.; Mosges, G.; Friedt, W. Construction of a sunflower pedigree map. Helia 1992, 15, 7–16. [Google Scholar]
- Burke, J.M.; Tang, S.; Knapp, S.J.; Rieseberg, L.H. Genetic analysis of sunflower domestication. Genetics 2002, 161, 1257–1267. [Google Scholar] [PubMed]
- Rogers, C.; Thompson, T.; Seiler, G.J. Sunflower Species of the United States; National Sunflower Association: Bismarck, ND, USA, 1982. [Google Scholar]
- Leclercq, P. Une stérilité male cytoplasmique chez le tournesol. Annales de l’Amelioration des Plantes 1969, 19, 99–106. (In French) [Google Scholar]
- Blackman, B.K.; Rasmussen, D.A.; Strasburg, J.L.; Raduski, A.R.; Burke, J.M.; Knapp, S.J.; Michaels, S.D.; Rieseberg, L.H. Contributions of flowering time genes to sunflower domestication and improvement. Genetics 2011, 187, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Burke, J.M. Evidence of selection on fatty acid biosynthetic genes during the evolution of cultivated sunflower. Theor. Appl. Genet. 2012, 125, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Mandel, J.R.; Nambeesan, S.; Bowers, J.E.; Marek, L.F.; Ebert, D.; Rieseberg, L.H.; Knapp, S.J.; Burke, J.M. Association mapping and the genomic consequences of selection in sunflower. PLoS Genet. 2013, 9, e1003378. [Google Scholar] [CrossRef] [PubMed]
- Mandel, J.R.; McAssey, E.V.; Nambeesan, S.; Garcia-Navarro, E.; Burke, J.M. Molecular evolution of candidate genes for crop-related traits in sunflower (Helianthus annuus L.). PLoS ONE 2014, 9, e99620. [Google Scholar] [CrossRef] [PubMed]
- Baute, G.J.; Kane, N.C.; Grassa, C.; Lai, Z.; Rieseberg, L.H. Genome scans reveal candidate domestication and improvement genes in cultivated sunflower, as well as post-domestication introgression with wild relatives. New Phytol. 2015, 206, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.R.; Tittes, S.; Mendieta, J.P.; Collier-zans, E.; Rowe, H.C.; Rieseberg, L.H.; Kane, N.C. Genetics of alternative splicing evolution during sunflower domestication. Proc. Natl. Acad. Sci. USA 2018, 115, 6768–6773. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Cavallini, A.; Natali, L.; Minelli, S.; Maggini, F.; Cionini, P.G. Ty1/copia-and Ty3/gypsy-like DNA sequences in Helianthus species. Chromosoma 2002, 111, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Natali, L.; Santini, S.; Giordani, T.; Minelli, S.; Maestrini, P.; Cionini, P.G.; Cavallini, A. Distribution of Ty3-gypsy- and Ty1-copia-like DNA sequences in the genus Helianthus and other Asteraceae. Genome 2006, 49, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Natali, L.; Cossu, R.M.; Barghini, E.; Giordani, T.; Buti, M.; Mascagni, F.; Morgante, M.; Gill, N.; Kane, N.C.; Rieseberg, L.H.; et al. The repetitive component of the sunflower genome as shown by different procedures for assembling next generation sequencing reads. BMC Genom. 2013, 14, 686. [Google Scholar] [CrossRef] [PubMed]
- Staton, S.E.; Bakken, B.H.; Blackman, B.K.; Chapman, M.A.; Kane, N.C.; Tang, S.; Ungerer, M.C.; Knapp, S.J.; Rieseberg, L.H.; Burke, J.M. The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 2012, 72, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Giordani, T.; Cavallini, A.; Natali, L. The repetitive component of the sunflower genome. Curr. Plant Biol. 2014, 1, 45–54. [Google Scholar] [CrossRef]
- Vukich, M.; Schulman, A.H.; Giordani, T.; Natali, L.; Kalendar, R.; Cavallini, A. Genetic variability in sunflower (Helianthus annuus L.) and in the Helianthus genus as assessed by retrotransposon-based molecular markers. Theor. Appl. Genet. 2009, 119, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.C.; Strakosh, S.C.; Stimpson, K.M. Proliferation of Ty3/Gypsy-like retrotransposons in hybrid sunflower taxa inferred from phylogenetic data. BMC Biol. 2009, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Buti, M.; Giordani, T.; Cattonaro, F.; Cossu, R.M.; Pistelli, L.; Vukich, M.; Morgante, M.; Cavallini, A.; Natali, L. Temporal dynamics in the evolution of the sunflower genome as revealed by sequencing and annotation of three large genomic regions. Theor. Appl. Genet. 2011, 123, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Vukich, M.; Giordani, T.; Natali, L.; Cavallini, A. Copia and Gypsy retrotransposons activity in sunflower (Helianthus annuus L.). BMC Plant Biol. 2009, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Neumann, P.; Macas, J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinform. 2010, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Mascagni, F.; Giordani, T.; Ceccarelli, M.; Cavallini, A.; Natali, L. Genome-wide analysis of LTR-retrotransposon diversity and its impact on the evolution of the genus Helianthus (L.). BMC Genom. 2017, 18, 634. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Package ‘Vegan’, Community Ecology Package. R Package, version 2.0-10. 2013.
- Cavallini, A.; Natali, L.; Zuccolo, A.; Giordani, T.; Jurman, I.; Ferrillo, V.; Vitacolonna, N.; Sarri, V.; Cattonaro, F.; Ceccarelli, M.; et al. Analysis of transposons and repeat composition of the sunflower (Helianthus annuus L.) genome. Theor. Appl. Genet. 2010, 120, 491–508. [Google Scholar] [CrossRef] [PubMed]
- Rowe, H.C.; Rieseberg, L.H. Genome-scale transcriptional analyses of first-generation interspecific sunflower hybrids reveals broad regulatory compatibility. BMC Genom. 2013, 14, 342. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, K.; Varala, K.; Hudson, M.E. Global repeat discovery and estimation of genomic copy number in a large, complex genome using a high-throughput 454 sequence survey. BMC Genom. 2007, 8, 132. [Google Scholar] [CrossRef] [PubMed]
- Tenaillon, M.I.; Hufford, M.B.; Gaut, B.S.; Ross-Ibarra, J. Genome size and transposable element content as determined by high-throughput sequencing in maize and Zealuxurians. Genome Biol. Evol. 2011, 3, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Barghini, E.; Natali, L.; Cossu, R.M.; Giordani, T.; Pindo, M.; Cattonaro, F.; Scalabrin, S.; Velasco, R.; Morgante, M.; Cavallini, A. The peculiar landscape of repetitive sequences in the olive (Olea europaea L.) genome. Genome Biol. Evol. 2014, 6, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Barghini, E.; Natali, L.; Giordani, T.; Cossu, R.M.; Scalabrin, S.; Cattonaro, F.; Šimková, H.; Vrána, J.; Doležel, J.; Morgante, M.; et al. LTR retrotransposon dynamics in the evolution of the olive (Olea europaea) genome. DNA Res. 2015, 22, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Mascagni, F.; Cavallini, A.; Giordani, T.; Natali, L. Different histories of two highly variable LTR retrotransposons in sunflower species. Gene 2017, 634, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Navrátilová, A.; Koblížková, A.; Kejnovský, E.; Hřibová, E.; Hobza, R.; Widmer, A.; Doležel, J.; Macas, J. Plant centromeric retrotransposons: A structural and cytogenetic perspective. Mob. DNA 2011, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.L.; Stec, A.; Hey, J.; Lukens, L.; Doebley, J. The limits of selection during maize domestication. Nature 1999, 398, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Gepts, P.; Papa, R. Evolution during domestication. In Encyclopedia of Life Sciences; Nature Publishing Group: London, UK, 2002. [Google Scholar]
- Olsen, K.M.; Purugganan, M.D. Molecular evidence on the origin and evolution of glutinous rice. Genetics 2002, 162, 941–950. [Google Scholar] [PubMed]
- Doebley, J. The genetics of maize evolution. Annu. Rev. Genet. 2004, 38, 37–59. [Google Scholar] [CrossRef] [PubMed]
- Innan, H.; Kim, Y. Pattern of polymorphism after strong artificial selection in a domestication event. Proc. Natl. Acad. Sci. USA 2004, 101, 10667–10672. [Google Scholar] [CrossRef] [PubMed]
- Eyre-Walker, A.; Gaut, R.L.; Hilton, H.; Feldman, D.L.; Gaut, B.S. Investigation of the bottleneck leading to the domestication of maize. Proc. Natl. Acad. Sci. USA 1998, 95, 4441–4446. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, E.; Bitocchi, E.; Ferrarini, A.; Benazzo, A.; Biagetti, E.; Klie, S.; Minio, A.; Rau, D.; Rodriguez, M.; Panziera, A.; et al. Decreased nucleotide and expression diversity and modified coexpression patterns characterize domestication in the common bean. Plant Cell 2014, 26, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 2012, 24, 1242–1255. [Google Scholar] [CrossRef] [PubMed]
- Falchi, R.; Vendramin, E.; Zanon, L.; Scalabrin, S.; Cipriani, G.; Verde, I.; Vizzotto, G.; Morgante, M. Three distinct mutational mechanisms acting on a single gene underpin the origin of yellow flesh in peach. Plant J. 2013, 76, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Millar, A.H.; Eubel, H.; Jansch, L.; Kruft, V.; Heazlewood, J.L.; Braun, H.P. Mitochondrial cytochrome c oxidase and succinate dehydrogenase complexes contain plant specific subunits. Plant Mol. Biol. 2004, 56, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L. Biochemistry of sulfur-containing amino acids. Ann. Rev. Biochem. 1983, 52, 187–222. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Wachter, A. Sulfur metabolism: A versatile platform for launching defence operations. Trends Plant Sci. 2005, 10, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.X.; Wirtz, M.; Phua, S.Y.; Estavillo, G.M.; Pogson, B.J. Balancing metabolites in drought: The sulfur assimilation conundrum. Trends Plant Sci. 2013, 18, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Tanksley, S.D.; McCouch, S.R. Seed banks and molecular maps: Unlocking genetic potential from the wild. Science 1997, 277, 1063–1066. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Name | Id Code | Area of Cultivation | Raw Reads | Trimmed Reads (as Single Ends, 90 nt) | Trimmed Reads (as Paired Ends) |
|---|---|---|---|---|---|---|
| Domesticated | Hata | Ames 22503 | Argentina | 32,100,390 | 31,085,284 | 31,624,960 |
| Dussol | Ames 22499 | France | 25,678,406 | 24,988,640 | 25,375,912 | |
| Argentario | Ames 1842 | Italy | 10,759,866 | 10,134,402 | 10,566,652 | |
| Karlik | Ames 3454 | Spain | 23,499,752 | 22,938,364 | 23,087,458 | |
| Zelenka | Ames 22530 | Russia | 9,048,276 | 8,824,270 | 8,858,154 | |
| Roman “A” | PI531386 | Romania | 19,408,888 | 18,621,244 | 19,095,974 | |
| HOPI | PI369359 | USA | 15,768,198 | 15,254,502 | 15,437,790 | |
| Seneca | PI369360 | USA | 13,911,506 | 13,334,732 | 13,667,436 | |
| Wild | Arizona (AZ) | Ames14400 | - | 14,641,510 | 14,013,588 | 14,357,666 |
| Colorado (CO) | PI586840 | - | 23,335,576 | 21,965,694 | 22,916,284 | |
| Illinois (IL) | PI 435540 | - | 18,577,580 | 17,366,768 | 18,145,470 | |
| Kentucky (KY) | PI 435613 | - | 14,853,802 | 13,845,748 | 14,580,828 | |
| Mississippi (MS) | PI 435608 | - | 22,921,544 | 21,376,594 | 22,226,864 | |
| North Dakota (ND) | PI586811 | - | 51,681,332 | 47,906,352 | 49,574,892 | |
| Washington (WA) | PI 531018 | - | 6,996,658 | 6,479,624 | 6,724,410 |
| Superfamily | Lineage | Family | Mean nr. of Gene-RE Mapping Paired Reads per Million Reads | ||
|---|---|---|---|---|---|
| Cultivars | Wild accessions | PERMANOVA | |||
| Gypsy | Chromovirus | CL5 | 2.95 | 4.05 | |
| Gypsy | Chromovirus | CL18 | 1.61 | 1.64 | |
| Gypsy | Chromovirus | CL25 | 4.37 | 6.53 | * |
| Gypsy | Chromovirus | CL32 | 1.07 | 0.80 | |
| Gypsy | Chromovirus | CL35 | 0.80 | 0.90 | |
| Gypsy | Chromovirus | CL47 | 0.84 | 1.57 | *** |
| Gypsy | Chromovirus | CL57 | 0.73 | 1.04 | * |
| Gypsy | Chromovirus | CL64 | 0.47 | 0.65 | |
| Gypsy | Chromovirus | CL88 | 0.47 | 0.81 | * |
| Gypsy | Chromovirus | CL94 | 0.25 | 0.32 | |
| Gypsy | Chromovirus | CL96 | 0.76 | 0.82 | |
| Gypsy | Chromovirus | CL102 | 0.14 | 0.18 | |
| Gypsy | Chromovirus | CL138 | 0.17 | 0.14 | |
| Gypsy | Chromovirus | CL193 | 0.03 | 0.05 | |
| Gypsy | Chromovirus | CL232 | 0.03 | 0.01 | |
| Gypsy | Athila | CL29 | 1.16 | 1.34 | |
| Gypsy | Athila | CL43 | 1.31 | 1.90 | * |
| Gypsy | Athila | CL87 | 0.42 | 0.64 | * |
| Mean Gypsy | 0.98 | 1.38 | |||
| Copia | AleII | CL48 | 1.08 | 1.08 | |
| Copia | Maximus/SIRE | CL115 | 0.12 | 0.21 | |
| Copia | Angela | CL100 | 0.27 | 0.29 | |
| Copia | TAR/Tork | CL255 | 0.18 | 0.17 | |
| Mean Copia | 0.41 | 0.44 | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mascagni, F.; Vangelisti, A.; Giordani, T.; Cavallini, A.; Natali, L. Specific LTR-Retrotransposons Show Copy Number Variations between Wild and Cultivated Sunflowers. Genes 2018, 9, 433. https://doi.org/10.3390/genes9090433
Mascagni F, Vangelisti A, Giordani T, Cavallini A, Natali L. Specific LTR-Retrotransposons Show Copy Number Variations between Wild and Cultivated Sunflowers. Genes. 2018; 9(9):433. https://doi.org/10.3390/genes9090433
Chicago/Turabian StyleMascagni, Flavia, Alberto Vangelisti, Tommaso Giordani, Andrea Cavallini, and Lucia Natali. 2018. "Specific LTR-Retrotransposons Show Copy Number Variations between Wild and Cultivated Sunflowers" Genes 9, no. 9: 433. https://doi.org/10.3390/genes9090433
APA StyleMascagni, F., Vangelisti, A., Giordani, T., Cavallini, A., & Natali, L. (2018). Specific LTR-Retrotransposons Show Copy Number Variations between Wild and Cultivated Sunflowers. Genes, 9(9), 433. https://doi.org/10.3390/genes9090433