De Novo Transcriptome Assembly and Population Genetic Analyses for an Endangered Chinese Endemic Acer miaotaiense (Aceraceae)


Abstract
1. Introduction
2. Materials and Methods
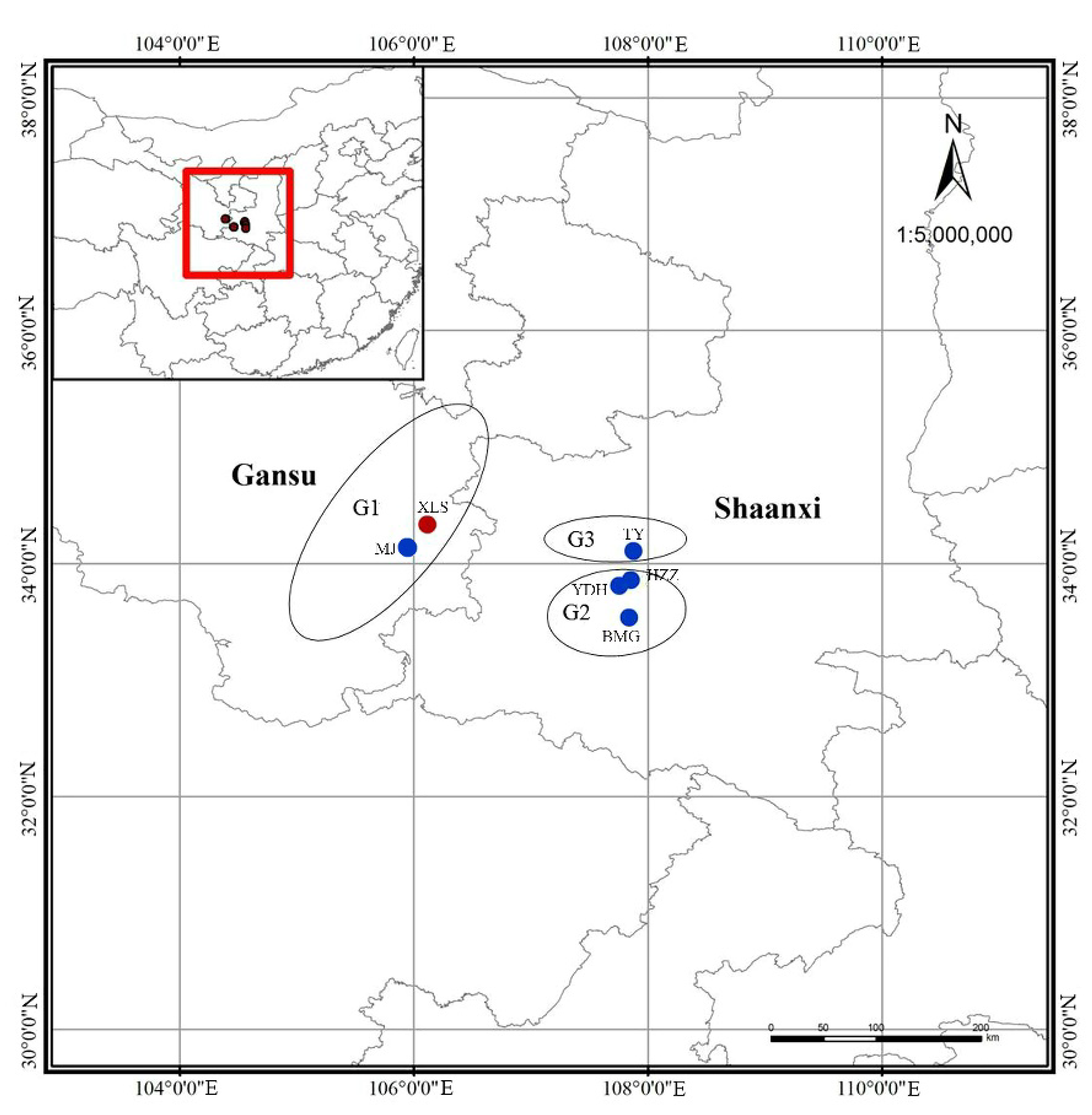
2.1. Plant Materials
2.2. RNA Extraction, Library Preparation, Sequencing and De Novo Assembly
2.3. Sequence Annotation and Classification
2.4. Simple Sequence Repeat Loci Identification and Primer Design
2.5. DNA Isolation, PCR Amplification and Simple Sequence Repeats Validation
2.6. Statistical Analyses
3. Results
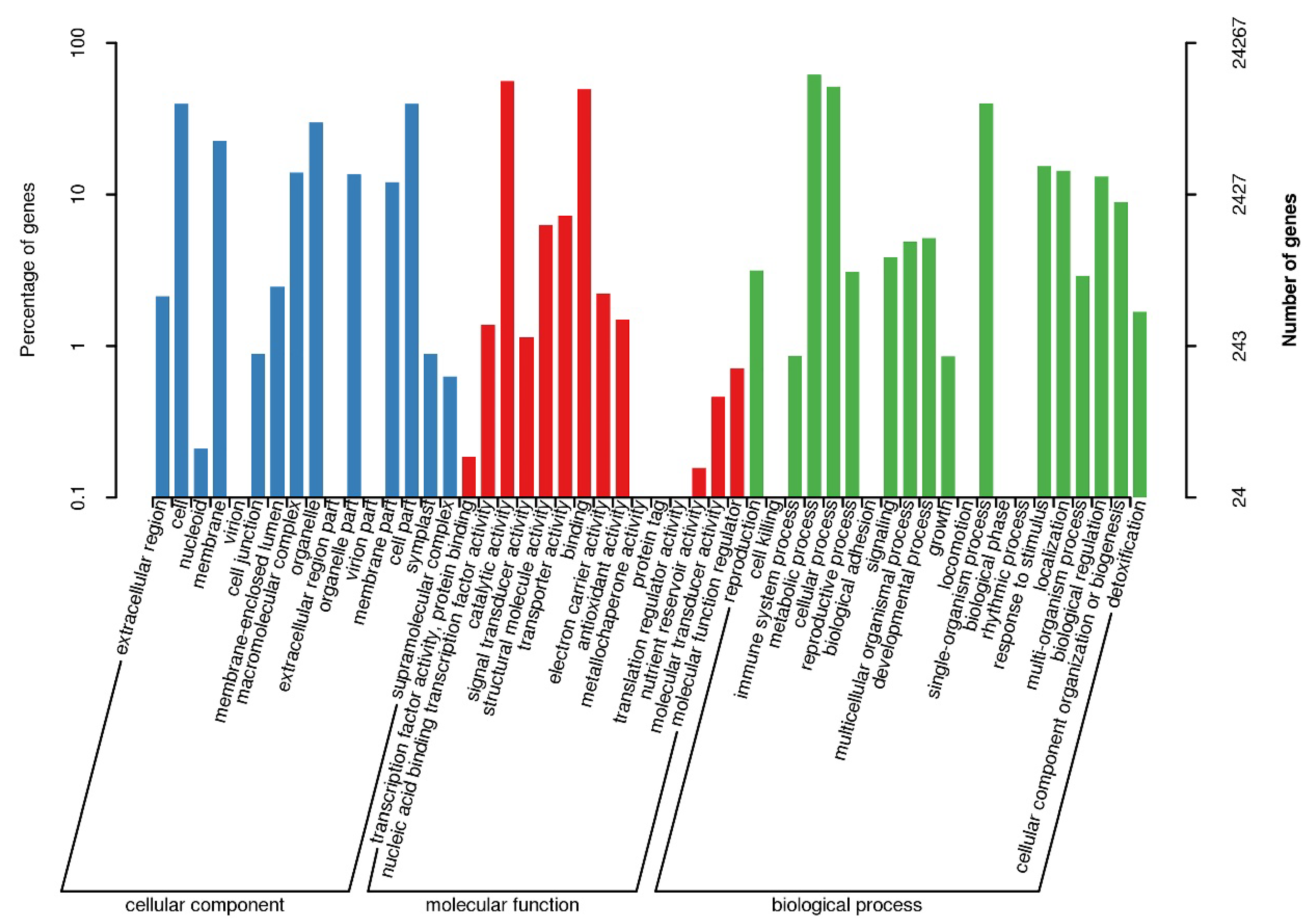
3.1. Sequencing, De Novo Assembly and Functional Annotation of Unigenes
3.2. Frequency and Distribution of Simple Sequence Repeats
3.3. Development and Polymorphism Rates of New Expressed Sequence Tag-Derived Simple Sequence Repeats Markers
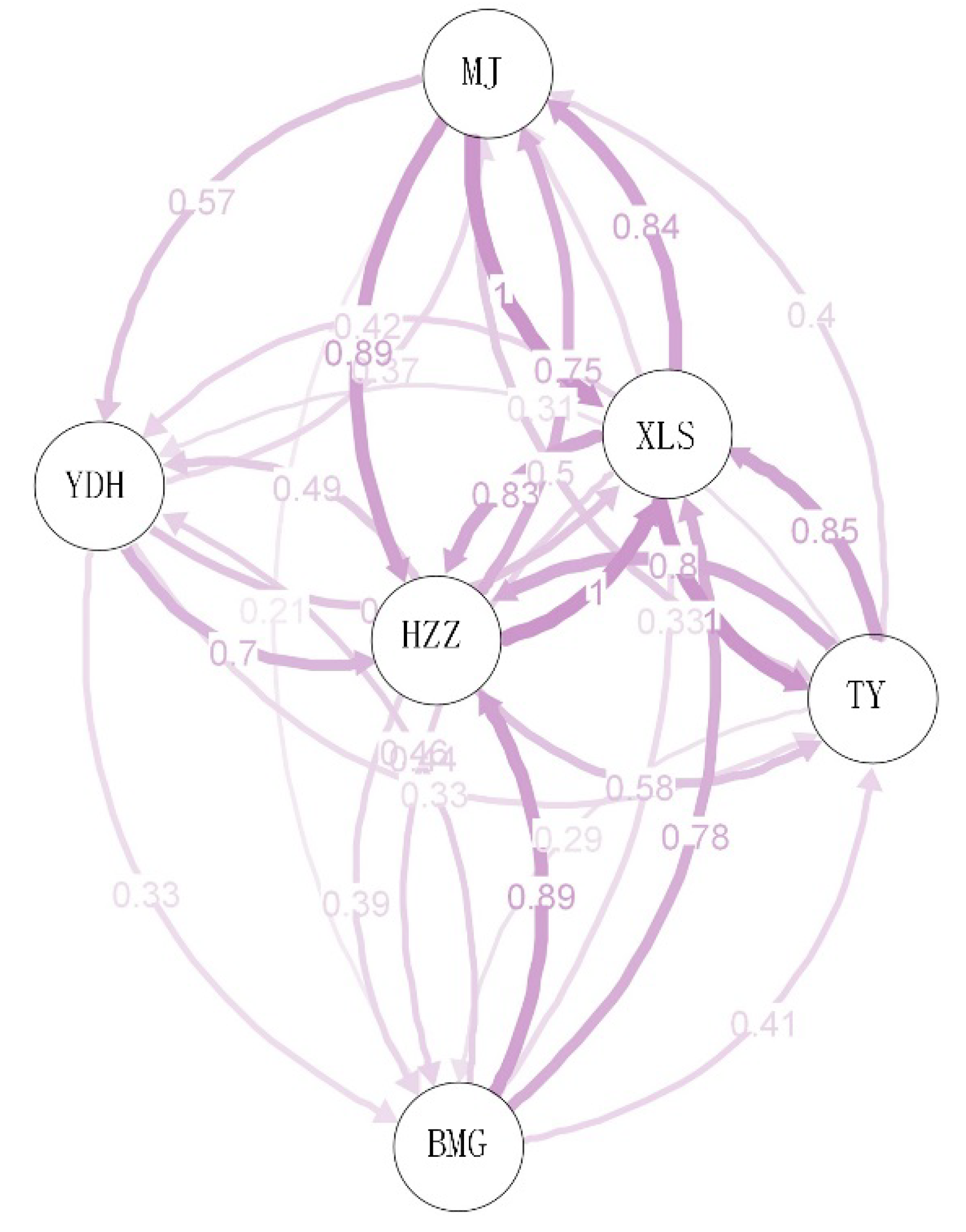
3.4. Genetic Diversity and Differentiation
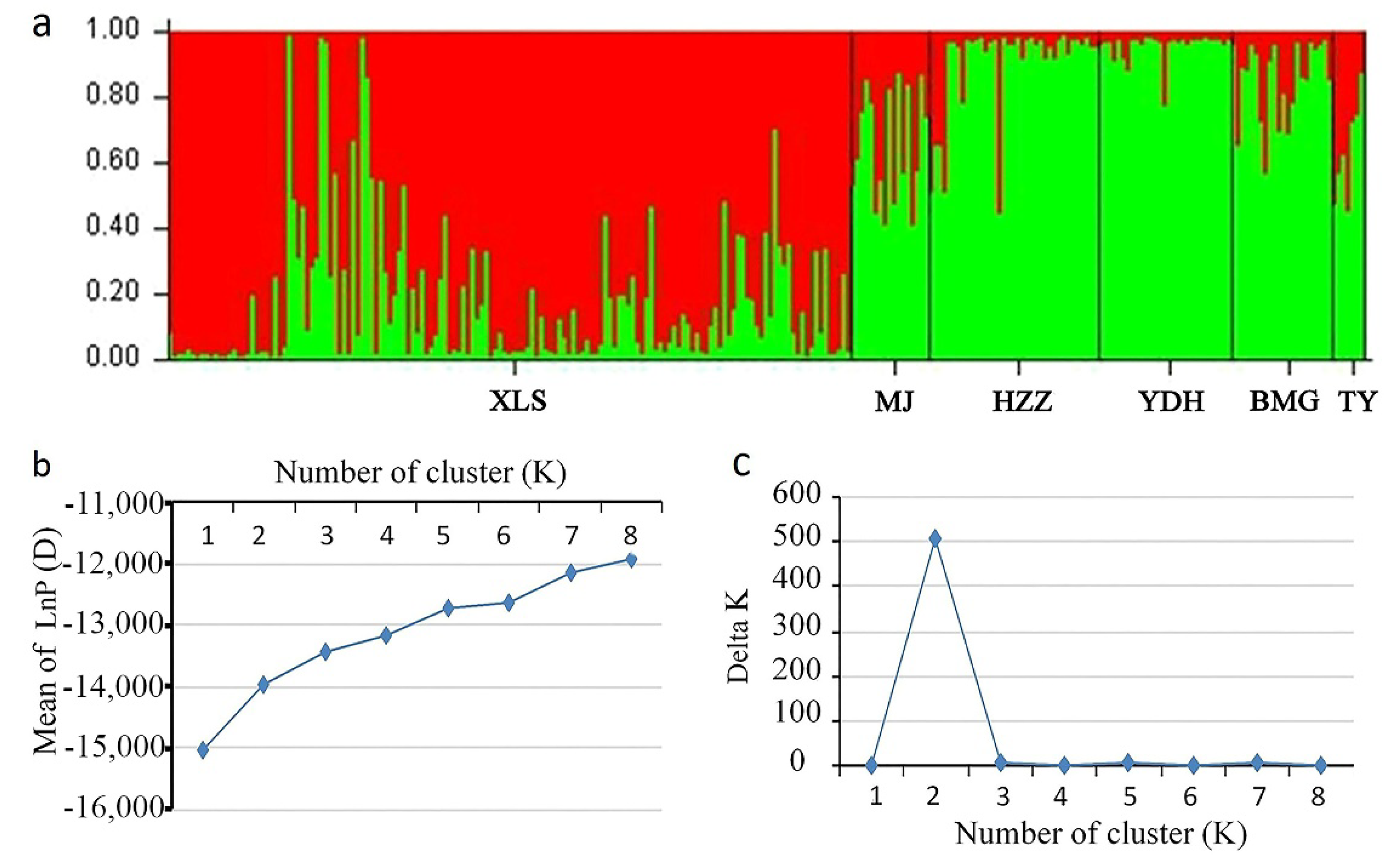
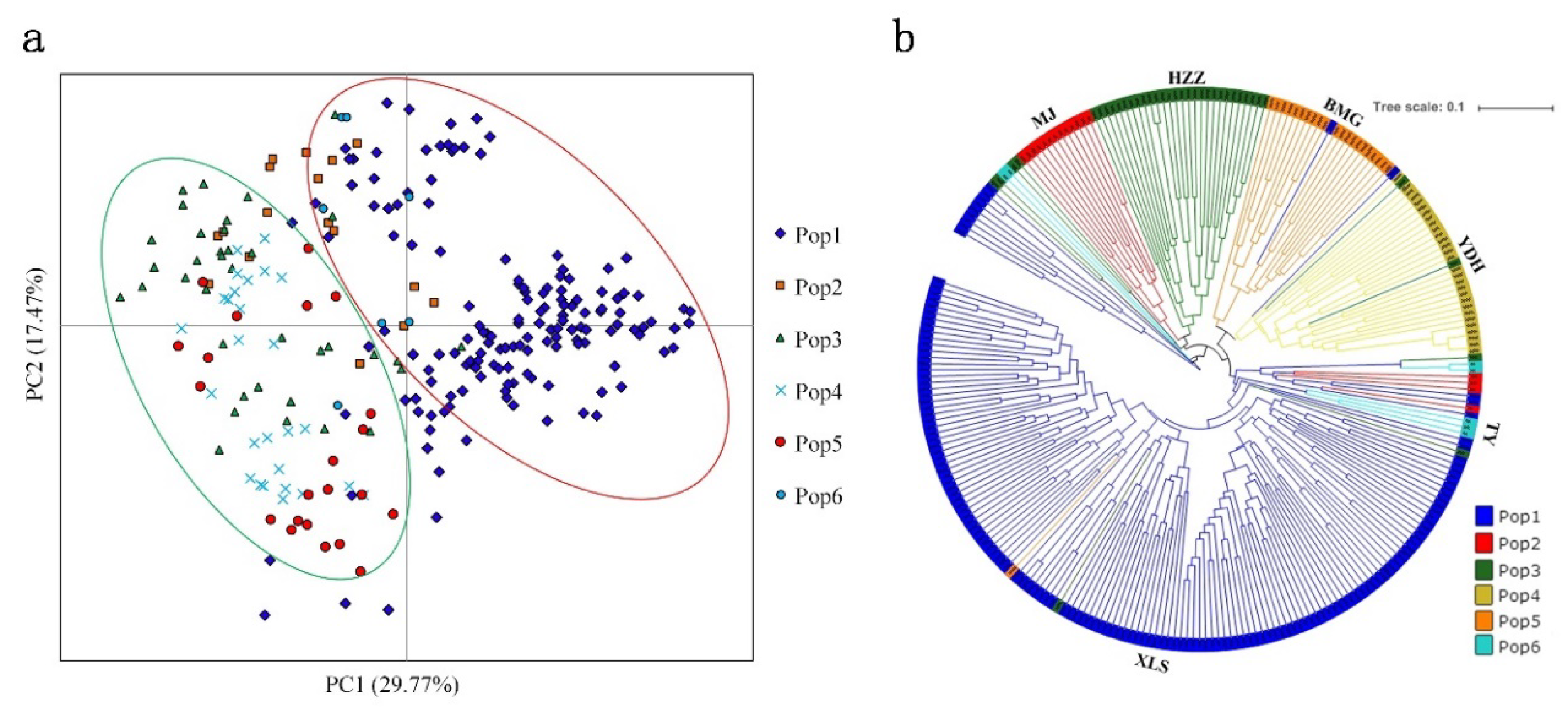
3.5. Population Structure of A. miaotaiense
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cao, X.Y.; Chen, K. Studies on the reproductive biology of a China rare plant-Acer miaotaiense1. Morphology and development of inflorescence and flower. Acta Bot. Boreali Occident. Sin. 1997, 5, 107–111. [Google Scholar]
- Zhang, Y.; Li, B.; Chen, H.; Wang, Y.C. Characterization of the complete chloroplast genome of Acer miaotaiense (Sapindales: Aceraceae), a rare and vulnerable tree species endemic to China. Conserv. Genet. Resour. 2016, 8, 1–3. [Google Scholar] [CrossRef]
- Fu, L.G. China Plant Red Data Book; Science Press: Beijing, China, 1992; pp. 107–108. [Google Scholar]
- International Union for Conservation of Nature and Natural Resources. Available online: http://dx.doi.org/10.2305/IUCN.UK.2004.RLTS.T46387A11048479.en (accessed on 12 June 2018).
- Shan, L.I.; Yan, G.Q.; Zhao, G.F. Population genetic structure and genetic diversity of Acer miaotaiense. J. Northwest Univ. 2005, 35, 71–75. [Google Scholar]
- Meng, Q.F.; Gao, H.L.; Li, H.X.; Xu, H.J. Biological and ecological characteristics of rare and endangered species of Acer miaotaiense. Henan Sci. 2016, 11, 1830–1834. [Google Scholar]
- Meng, Q.F.; Gao, H.L.; Wang, H.Y. Fruiting characteristics and sowing seedlings of rare and endangered species of Acer miaotaiense. Henan Sci. 2017, 11, 1773–1777. [Google Scholar]
- Wei, D.L. Study on sowing and seedling rising of rare species of Acer miaotaiense. J. Gansu For. Sci. Technol. 2017, 2, 37–39. [Google Scholar]
- Lu, Z.R. Rare and endangered species of Acer miaotaiense investigation and analysis in Xiaolongshan forest population distribution. Jilin Agric. 2011, 2, 165–166. [Google Scholar]
- Lei, S.G.; Ma, J.W.; Zhang, S.Z.; Hu, W.B.; Guo, X.L.; Du, Y.C.; Li, A.M. Preliminary research on community structure of Acer miaotaiense in the Xiaolongshan forest area. J. Gansu For. Sci. Technol. 2009, 34, 1–5. [Google Scholar]
- Wang, H.; Pan, G.; Ma, Q.; Zhang, J.; Pei, D. The genetic diversity and introgression of Juglans regia and Juglans sigillata in Tibet as revealed by SSR markers. Tree Genet. Genome 2015, 11, 1. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, L.; Zheng, P. Genetic diversity and evolutionary relationship analyses within and among Raphanus species using EST-SSR markers. Mol. Breed. 2015, 35, 62. [Google Scholar] [CrossRef]
- Deng, T.X.; Pang, C.Y.; Lu, X.R.; Zhu, P.; Duan, A.Q.; Tan, Z.Z.; Huang, J.; Li, H.; Chen, M.T.; Liang, X.W. De novo transcriptome assembly of the Chinese swamp buffalo by RNA sequencing and SSR marker discovery. PLoS ONE 2016, 11, e0147132. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.K.R.; Chen, H.D.; Zhou, Z.L.; Ilyas, M.K.; Wang, X.X.; Cai, X.Y.; Wang, C.Y.; Liu, F.; Wang, K.B. Genome wide SSR high density genetic map construction from an interspecific cross of Gossypium hirsutum × Gossypium tomentosum. Front. Plant Sci. 2016, 7, 436. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Dondini, L.; Franceschi, P.D.; Paris, R.; Sansavini, S.; Tartarini, S. Genetic diversity, population structure and construction of a core collection of apple cultivars from Italian germplasm. Plant Mol. Biol. Rep. 2015, 33, 458–473. [Google Scholar] [CrossRef]
- Cheng, J.W.; Zhao, Z.Z.; Bo, L.; Cheng, Q.; Wu, Z.M.; Trejosaavedra, D.L.; Luo, X.R.; Cui, J.J.; Riverabustamante, R.F.; Li, S.C. A comprehensive characterization of simple sequence repeats in pepper genomes provides valuable resources for marker development in Capsicum. Sci. Rep. 2016, 6, 18919. [Google Scholar] [CrossRef] [PubMed]
- Aboukhalid, K.; Machon, N.; Lambourdière, J.; Abdelkrim, J.; Bakha, M.; Douaik, A.; Korbecka-Glinka, G.; Gaboun, F.; Gaboun, F.; Lamiri, A. Analyses of genetic diversity and population structure of the endangered Origanum compactum from Morocco, using SSR markers: Implication for conservation. Biol. Conserv. 2017, 212, 172–182. [Google Scholar] [CrossRef]
- Yang, H.; Li, X.; Liu, D.; Chen, X.; Li, F.; Qi, X.L. Genetic diversity and population structure of the endangered medicinal plant Phellodendron amurense in China revealed by SSR markers. Biochem. Syst. Ecol. 2016, 66, 286–292. [Google Scholar] [CrossRef]
- Lopez, L.; Barreiro, R. Genetic guidelines for the conservation of the endangered polyploid Centaurea borjae (Asteraceae). J. Plant Res. 2013, 126, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Wang, Y.Y.; Xu, Y.N.; Yan, R.S.; Zhao, P.; Liu, W.Z. De novo characterization of flower bud transcriptomes and the development of EST-SSR markers for the endangered tree Tapiscia sinensis. Int. J. Mol. Sci. 2015, 16, 12855–12870. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, K.J.L.; Bryant, D.; Kalish, J.; Eng, C.; Schmidt, P.L.; Barrett, G.; Margaret, C.B. Integrating genomic data sets for knowledge discovery: An informed approach to management of captive endangered species. Int. J. Genom. 2016, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Shi, E.; Yang, Z.P.; Geng, Q.F.; Qiu, Y.X.; Wang, Z.S. Development and application of genomic resources in an endangered palaeoendemic tree, Parrotia subaequalis (Hamamelidaceae) from Eastern China. Front. Plant Sci 2018, 9, 246. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.P.; Whittaker, D.J.; Ambreth, S.; Sureshchandra, S.; Buechlein, A.; Podicheti, R.; Choi, J.H.; Lai, Z.; Mockatis, K.; Cilbourne, J.; et al. De novo transcriptome sequencing in a songbird, the dark-eyed junco (Junco hyemalis): Genomic tools for an ecological model system. BMC Genom. 2012, 13, 305. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Cao, Y.N.; Yuan, N.; Nakamura, K.; Wang, G.M.; Qiu, Y.X. Characterization of transcriptome and development of novel EST-SSR makers based on next-generation sequencing technology in Neolitsea sericea (Lauraceae) endemic to East Asian land-bridge islands. Mol. Breed. 2015, 35, 187. [Google Scholar] [CrossRef]
- Shiferaw, E.; Pè, M.E.; Porceddu, E.; Ponnaiah, M. Exploring the genetic diversity of Ethiopian grass pea (Lathyrus sativus L.) using EST-SSR markers. Mol. Breed. 2012, 30, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.Y.; Zhang, Z.X.; Wang, Z.G.; Zhang, X.P.; Wu, G.L. Transcriptome sequencing and development of EST-SSR markers in Pinus dabeshanensis, an endangered conifer endemic to China. Mol. Breed. 2015, 35, 158. [Google Scholar] [CrossRef]
- Liu, J.; Liu, H.B.; Ma, L.Y.; Wang, S.Q.; Gao, J.; Li, Y.Y.; Wu, R.L.; Pang, X.M. A Chinese jujube (Ziziphus jujuba Mill.) fruit-expressed sequence tag (EST) library: Annotation and EST-SSR characterization. Sci. Hortic. 2014, 165, 99–105. [Google Scholar] [CrossRef]
- Wu, J.; Cai, C.F.; Cheng, F.Y.; Cui, H.L.; Zhou, H. Characterisation and development of EST-SSR markers in tree peony using transcriptome sequences. Mol. Breed. 2014, 34, 1853–1866. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, J.X.; Su, L.Z.; Lv, W.; Sun, Y.H.; Li, Y. Development and evaluation of a novel set of EST-SSR markers based on transcriptome sequences of black locust (Robinia pseudoacacia L.). Genes 2017, 8, 177. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.M.; Bai, L.J.; Ke, L.X.; Xiang, X.; Zhao, J.S.; Ou, L.X. Developing a core collection of litchi (Litchi chinensis Sonn.) based on EST-SSR genotype data and agronomic traits. Sci. Hortic. 2012, 146, 29–38. [Google Scholar] [CrossRef]
- Zhou, T.; Li, Z.H.; Bai, G.Q.; Feng, L.; Chen, C.; Wei, Y.; Chang, Y.X.; Zhao, G.F. Transcriptome sequencing and development of genic SSR markers of an endangered Chinese endemic genus Dipteronia Oliver (Aceraceae). Molecules 2016, 21, 166. [Google Scholar] [CrossRef] [PubMed]
- He, Y.L.; He, Y.; Gong, L.L.; Fang, M.F.; Li, Z.H. Population genetic structure and interspecific differentiation between Acer davidii, Franchi. and A. morrisonense, Hayata (Aceraceae) based on SSR markers. Biochem. Syst. Ecol. 2017, 71, 42–49. [Google Scholar] [CrossRef]
- Ghawana, S.; Paul, A.; Kumar, H.; Kumar, A.; Singh, H.; Bhardwaj, P.K.; Rani, A.; Singh, R.S.; Raizada, J.; Singh, K. An RNA isolation system for plant tissues rich in secondary metabolites. BMC Res. Notes 2011, 4, 85. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Xian, A.; Fan, L.; Raychowdhury, R.; Zeng, Q.D. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; Garcíagómez, J.M.; Teroi, J.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Cui, B.B.; Mao, J.F.; Pang, X.M.; Liu, H.; Li, Y.Y. Novel polymorphic EST-derived microsatellite markers for the red-listed five needle pine, Pinus dabeshanensis. Conserv. Genet. Resour. 2015, 7, 191–192. [Google Scholar] [CrossRef]
- Mitchell, M.; Walther, P. GeneMarker® HID: A Reliable Software Tool for the Analyses of Forensic STR Data. J. Forensic Sci. 2011, 56, 29–35. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Nagy, S.; Poczai, P.; Cernák, I.; Gorji, A.M.; Hegedűs, G.; Taller, J. PICcalc: An online program to calculate polymorphic information content for molecular genetic studies. Biochem. Genet. 2012, 50, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [PubMed]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Nei, M.F.; Tajima, F.; Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J. Mol. Evol. 1983, 19, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analyses environment for genetic marker analyses. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. Available online: http://itol.embl.de/ (accessed on 15 July 2018). [CrossRef] [PubMed]
- Sundqvist, L.; Keenan, K.; Zackrisson, M.; Prodöhl, P.; Kleinhans, D. Directional genetic differentiation and relative migration. Ecol. Evol. 2016, 6, 3461–3475. [Google Scholar] [CrossRef] [PubMed]
- Keenan, K.; Mcginnity, P.; Cross, T.F.; Crozier, W.W.; Prodöhl, P.A. Diversity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analyses and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Masmoudi, M.B.; Chaoui, L.; Topçu, N.E.; Hammami, P.; Kara, M.H.; Aurelle, D. Contrasted levels of genetic diversity in a benthic Mediterranean octocoral: Consequences of different demographic histories? Ecol. Evol. 2016, 6, 8665–8678. [Google Scholar] [CrossRef] [PubMed]
- Senczuk, G.; Colangelo, P.; Simone, E.D.; Aloise, G.; Castiglia, R. A combination of long term fragmentation and glacial persistence drove the evolutionary history of the Italian wall lizard Podarcis siculus. BMC Evol. Biol. 2017, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Donnell, C.F.J.O.; Richter, S.; Dool, S.; Monks, J.M.; Kerth, G. Genetic diversity is maintained in the endangered New Zealand long-tailed bat (Chalinolobus tuberculatus) despite a closed social structure and regular population crashes. Conserv. Genet. 2016, 17, 91–102. [Google Scholar] [CrossRef]
- Parul, G.; Heikham, R.S.; Ravi, S.; Niharika, S.; Anita, K.; Vandna, C.; Paramvir, S.A.; Sanjay, K. De novo sequencing and characterization of Picrorhiza kurrooa transcriptome at two temperatures showed major transcriptome adjustments. BMC Genom. 2012, 13, 126. [Google Scholar]
- Tao, T.; Zhao, L.; Lv, Y.D.; Chen, J.D.; Hu, Y.; Zhang, T.Z.; Zhou, B.L. Transcriptome sequencing and differential gene expression analyses of delayed gland morphogenesis in Gossypium australe during seed germination. PLoS ONE 2013, 8, e75323. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Li, Q.; Li, S.; Tang, L.; Wen, J. De novo transcriptome sequencing of Acer palmatum and comprehensive analyses of differentially expressed genes under salt stress in two contrasting genotypes. Mol. Genet. Genom. 2016, 291, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Puniya, B.L.; Kulshreshtha, D.; Kumar, S.; Kaur, J.; Ramachandran, S.; Singh, K. De novo transcriptome sequencing reveals important molecular networks and metabolic pathways of the plant, Chlorophytum borivilianum. PLoS ONE 2013, 8, e83336. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Zhao, F.; Yan, J.Z.; Liu, C.S.; Liu, S.W.; Chen, S.Q. Transcriptome sequencing and de novo assembly of golden cuttlefish Sepia esculenta Hoyle. Int. J. Mol. Sci. 2016, 17, 1749. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wu, B.; Zhao, J.; Li, H.; Chen, W.; Zheng, Y.; Ren, X.; Chen, Y.; Zhou, X.; Lei, Y.; et al. Characterization and transferable utility of microsatellite markers in the wild and cultivated Arachis species. PLoS ONE 2016, 11, e0156633. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ma, Y.; Sun, W.; Wen, X.; Milne, R. High genetic diversity and low differentiation of Michelia coriacea (Magnoliaceae), a critically endangered endemic in Southeast Yunnan, China. Int. J. Mol. Sci. 2012, 13, 4396. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, X.; Yang, Y.; Xu, J.; Gu, J.; Fu, J.; Qian, X.; Zhang, S.; Wu, J.; Liu, K. Development and genetic mapping of microsatellite markers from whole genome shotgun sequences in Brassica oleracea. Mol. Breed. 2011, 28, 585–596. [Google Scholar] [CrossRef]
- Ipek, M.; Sahin, N.; Ipek, A.; Cansev, A.; Simon, P.W. Development and validation of new SSR markers from expressed regions in the garlic genome. Sci. Agric. 2015, 72, 41–46. [Google Scholar] [CrossRef]
- Ma, Q.Y.; Dai, X.G.; Chen, Y.N.; Zhang, D.F.; Liao, Z.Y.; Li, S.X. Characterization of microsatellites in the genome of Ziziphus jujuba. Sci. Silvae Sin. 2013, 12, 81–87. [Google Scholar]
- Zhong, R.C.; Zhou, M.L.; Zhao, C.Z.; Hou, L.; Li, C.S.; Wang, X.J.; Tang, R.H.; Xia, H. SSR marker development from peanut gynophore transcriptome sequencing. Plant Breed. 2016, 135, 111–117. [Google Scholar] [CrossRef]
- Kumpatla, S.P.; Mukhopadhyay, S. Mining and survey of simple sequence repeats in expressed sequence tags of dicotyledonous species. Genome 2005, 48, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.N.; Zhang, Y.Q.; Jin, M.D.; Li, H.K.; Song, Z.P.; Wang, Y.G.; Chen, J. Characterization and high cross-species transferability of microsatellite markers from the floral transcriptome of Aspidistra saxicola (Asparagaceae). Mol. Ecol. Resour. 2014, 14, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.Y.; Liu, G.Q.; Zong, Y.; Teng, Y.W.; Cai, D.Y. Development of genic SSR markers from transcriptome sequencing of pear buds. J. Zhejiang Univ. Sci. 2014, 15, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Huang, X.J.; Zhang, J.; Chen, Q.; Liu, Y.; Tang, H.R.; Pan, D.M.; Wang, X.R. Genetic diversity and population structure patterns in Chinese Cherry (Prunus pseudocerasus, Lindl) Landraces. Plant Mol. Biol. Rep. 2015, 34, 1–14. [Google Scholar] [CrossRef]
- Jia, H.; Yang, H.; Sun, P.; Li, J.; Zhang, J.; Guo, Y.; Han, X.; Zhang, G.; Lu, M.; Hu, J. De novo transcriptome assembly, development of EST-SSR markers and population genetic analyses for the desert biomass willow, Salix psammophila. Sci. Rep. 2016, 6, 39591. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.C.; López-Pujol, J.; Son, S.; Suh, G.U.; Yukawa, T.; Chung, M.G. Patterns of genetic diversity in rare and common orchids focusing on the Korean peninsula: Implications for conservation. Bot. Rev. 2018, 84, 1–25. [Google Scholar]
- Ferrer, M.M.; Eguiarte, L.E.; Montaña, C. Genetic structure and outcrossing rates in Flourensia cernua (Asteraceae) growing at different densities in the South-Western Chihuahuan Desert. Ann. Bot. 2004, 94, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Jin, Z.X.; Lou, W.Y.; Li, J.M. Genetic diversity of Lithocarpus harlandii populations in three forest communities with different succession stages. Chin. J. Ecol. 2008, 3, 106–111. [Google Scholar] [CrossRef]
- Hellmann, J.J.; Pineda-Krch, M. Constraints and reinforcement on adaptation under climate change: Selection of genetically correlated traits. Biol. Conserv. 2007, 140, 599–609. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Yi, T.S.; Zeng, L.Q.; Gong, X. The population genetic structure and diversification of Aristolochia delavayi (Aristolochiaceae), an endangered species of the dry hot valleys of the Jinsha River, Southwestern China. Botany 2014, 92, 579–587. [Google Scholar] [CrossRef]
- Babu, B.K.; Meena, V.; Agarwal, V.; Agrawal, P.K. Population structure and genetic diversity analyses of Indian and exotic rice (Oryza sativa L.) accessions using SSR markers. Mol. Biol. Rep. 2014, 41, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.C.; Wang, G.F.; Mao, W.H.; Hu, Q.Z.; Liu, N.; Ye, L.W.; Gong, Y.M. Genetic diversity and population structure of common bean (Phaseolus vulgaris) landraces from China revealed by a new set of EST-SSR markers. Biochem. Syst. Ecol. 2014, 57, 250–256. [Google Scholar] [CrossRef]
- Zhang, P.; Li, J.Q.; Li, X.L.; Liu, X.D.; Zhao, X.J.; Lu, Y.G. Population structure and genetic diversity in a rice core collection (Oryza sativa L.) investigated with SSR markers. PLoS ONE 2011, 6, e27565. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, S.; Shi, S.; Zhang, Z.; Liao, W.B.; Wu, W.; Zhou, R.C.; Fan, Q. High genetic diversity and weak population structure of Rhododendron jinggangshanicum, a threatened endemic species in Mount Jinggangshan of China. Biochem. Syst. Ecol. 2015, 58, 178–186. [Google Scholar] [CrossRef]
- Odat, N.; Jetschke, G.; Hellwig, F.H. Genetic diversity of Ranunculus acris L. (Ranunculaceae) populations in relation to species diversity and habitat type in grassland communities. Mol. Ecol. 2004, 13, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.P.; Cai, C.F.; Zhong, Y.; Xu, X.X.; Xian, H.L.; Cheng, F.Y.; Mao, J.F. Genetic analyses reveal independent domestication origins of the emerging oil crop Paeonia ostii, a tree peony with a long-term cultivation history. Sci. Rep. 2017, 7, 5340. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kang, M.; Liu, H.; Gao, J.; Zhang, Z.; Li, Y.; Wu, R.; Pang, X. High-level genetic diversity and complex population structure of Siberian apricot (Prunus sibirica L.) in China as revealed by nuclear SSR markers. PLoS ONE 2014, 9, e87381. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.X.; Lin, F.R.; Huang, P.; Zheng, Y.Q. Moderate genetic diversity and genetic differentiation in the relict tree Liquidambar formosana Hance revealed by genic simple sequence repeat markers. Front. Plant Sci. 2016, 7, 1411. [Google Scholar] [CrossRef] [PubMed]
- Hamrick, J.L.; Godt, M.J.W. Effects of life history traits on genetic diversity in plant species. Philos. Trans. Biol. Sci. 1996, 351, 1291–1298. [Google Scholar] [CrossRef]
- Wright, S. Evolution and the Genetics of Population, Variability within and among Natural Populations; The University of Chicago Press: Chicago, IL, USA, 1978. [Google Scholar]
- Schaal, B.A.; Hayworth, D.A.; Olsen, K.M.; Rauscher, J.T.; Smith, W.A. Phylogeographic studies in plants: Problems and prospects. Mol. Ecol. 1998, 7, 465–474. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regions | Sampling Locality | Sample Size | Population Codes | Latitude (N)/Longitude (E) | Altitude (m) |
|---|---|---|---|---|---|
| G1 | Tianshui, Gansu | 149 | XLS | N: 34°18.405′/E: 106°08.323′ | 1504 |
| Maiji, Gansu | 17 | MJ | N: 34°07.052′/E: 105°54.214′ | 1520 | |
| G2 | Zhouzhi, Shaanxi | 37 | HZZ | N: 33°50.680′/E: 107°48.068′ | 1502 |
| Zhouzhi, Shaanxi | 29 | YDH | N: 34°50.057′/E: 107°47.395′ | 1500 | |
| Foping, Shaanxi | 22 | BMG | N: 33°33.382′/E: 107°48.699′ | 1260 | |
| G3 | Lantian, Shaanxi | 7 | TY | N: 34°02.577′/E: 107°53.075′ | 1293 |
| Primer Sequences (5′ds-3′) | Repeat Motif | Ta (°C) | Expected Size (bp) | Na | Ho | He | PIC | HWE | NRA | |
|---|---|---|---|---|---|---|---|---|---|---|
| BFAM-2 | F:AGGAGATGGAAAGCAGGGAT R:GGGACTTTCGGACAAACAAA | (CT)17 | 60 | 224 | 10 | 0.449 | 0.777 | 0.752 | *** | 1 |
| BFAM-74 | F:CTAAGACACCGTGCAAGCAA R:ACGCATGATAGGGCTCGTAA | (AT)8 | 60 | 196 | 9 | 0.591 | 0.779 | 0.747 | *** | 3 |
| BFAM-50 | F:TCTCCATCTCCCCTTGGTAA R:GGAGGTTTCATGGACACGAT | (TA)10 | 60 | 269 | 8 | 0.397 | 0.800 | 0.772 | *** | 2 |
| BFAM-88 | F:GAACTCCTTGTCGGATTCCA R:ATTTCCACGGACCGTACTCA | (TCT)8 | 60 | 243 | 5 | 0.289 | 0.35 | 0.329 | *** | 0 |
| BFAM-34 | F:GCGGTGATGAACTGATGATG R:CACCCAAATCACCTTCGTCT | (GA)7 | 60 | 140 | 6 | 0.506 | 0.566 | 0.517 | * | 3 |
| BFAM-109 | F:GTCATCACATGCCTTTCCCT R:GCAACTCGGCTAAAGATTGC | (GAT)8 | 60 | 244 | 4 | 0.238 | 0.238 | 0.223 | *** | 1 |
| BFAM-91 | F:TGACTTGTCCCTCAAATCCA R:GAAAATGGGGTGTCTCCAAA | (AT)13 | 60 | 206 | 10 | 0.753 | 0.82 | 0.796 | *** | 2 |
| BFAM-219 | F:AGAGTGCAAAGCAGCAAATG R:CGTTGGGACTCATGTCAATG | (AT)12 | 60 | 235 | 8 | 0.813 | 0.841 | 0.820 | *** | 1 |
| BFAM-107 | F:CAAACAGAGTCGGCTGTGAA R:TTTTGTCGGCCAGTTAAACC | (GTT)7 | 60 | 203 | 3 | 0.240 | 0.247 | 0.229 | ns | 0 |
| BFAM-241 | F:ATCAGAGAGGCGGACTTGAA R:TCATTTGCCCTCCTAATTGC | (CAA)8 | 60 | 153 | 8 | 0.708 | 0.735 | 0.699 | ** | 2 |
| BFAM-254 | F:GGACGAATGGAGATTGGAGA R:AAAGAGCTCCAAGAGGGTGA | (AAG)7 | 59 | 244 | 3 | 0.076 | 0.153 | 0.147 | *** | 0 |
| BFAM-255 | F:CGCACCTTTCAGTAATGCCT R:AGAACATGCCCACAACCTGT | (AT)10 | 60 | 264 | 13 | 0.882 | 0.822 | 0.801 | *** | 2 |
| BFAM-136 | F:GTGGGGAAATGAAGGGAAAT R:GCATCTGTCCCGAATCAAAT | (TTC)7 | 60 | 273 | 4 | 0.455 | 0.448 | 0.372 | *** | 0 |
| BFAM-123 | F:AGGCAGGTGTCAGTGTTTCA R:GCCAGGTGGGTTCTACAAAA | (GAA)10 | 60 | 208 | 8 | 0.564 | 0.675 | 0.621 | *** | 3 |
| BFAM-262 | F:TGTTGTGTTCGTTCCATCGT R:CCTTTGGTTCCCTGACCTTT | (AT)7 | 60 | 243 | 9 | 0.571 | 0.777 | 0.745 | *** | 3 |
| BFAM-166 | F:CAAATCCCGACAATCTCTCC R:TGGAGGAAGCAGTCAAGGTT | (CT)10 | 60 | 225 | 19 | 0.698 | 0.812 | 0.791 | *** | 6 |
| BFAM-263 | F:TATGGGCAGTCTTGGGTTTC R:CCAGGAACAAGCATGGATTT | (TG)10 | 60 | 223 | 13 | 0.523 | 0.774 | 0.743 | *** | 4 |
| BFAM-178 | F:ACCAAACCAGAGATCCAACG R:AATCTCTCACGCCCCTTTCT | (GA)11 | 60 | 176 | 5 | 0.667 | 0.675 | 0.631 | ** | 1 |
| BFAM-220 | F:GCTCAACCATCCAACGATTT R:TCCAGTGGCATCAGATTGAA | (TA)13 | 60 | 273 | 7 | 0.620 | 0.775 | 0.740 | *** | 1 |
| Total | 152 | 10.04 | 12.06 | 11.47 | 35 | |||||
| Mean | 8 | 0.528 | 0.635 | 0.604 | 1.842 | |||||
| Sample Size | Na | Ne | Ho | He | F | Private Alleles | |
|---|---|---|---|---|---|---|---|
| XLS | 149 | 6.053 | 2.965 | 0.512 | 0.573 | 0.088 | 13 |
| MJ | 17 | 4.263 | 2.783 | 0.586 | 0.561 | −0.046 | 2 |
| HZZ | 37 | 5.737 | 3.249 | 0.55 | 0.602 | 0.106 | 9 |
| YDH | 29 | 4.211 | 2.755 | 0.584 | 0.557 | −0.046 | 3 |
| BMG | 22 | 4.053 | 2.561 | 0.464 | 0.504 | 0.056 | 6 |
| TY | 7 | 4.105 | 3.173 | 0.496 | 0.585 | 0.147 | 2 |
| Mean | 44 | 4.737 | 2.914 | 0.532 | 0.564 | 0.051 | 5.833 |
| XLS | MJ | HZZ | YDH | BMG | TY | |
|---|---|---|---|---|---|---|
| XLS | **** | 0.065 | 0.074 | 0.089 | 0.078 | 0.065 |
| MJ | 0.181 | **** | 0.059 | 0.095 | 0.116 | 0.093 |
| HZZ | 0.251 | 0.218 | **** | 0.082 | 0.083 | 0.077 |
| YDH | 0.293 | 0.325 | 0.291 | **** | 0.103 | 0.091 |
| BMG | 0.227 | 0.401 | 0.267 | 0.313 | **** | 0.096 |
| TY | 0.173 | 0.334 | 0.286 | 0.304 | 0.327 | **** |
| Degrees of Freedom | Sum of Squares | Variance Components | Percentage of Variation (%) | Genetic Differentiation Index | |
|---|---|---|---|---|---|
| Variance partition a | |||||
| Among populations | 5 | 633.767 | 3.414 | 18.99 | Fst = 0.190 |
| Within populations | 255 | 3712.486 | 14.559 | 81.01 | |
| Total | 260 | 4346.253 | 17.973 | ||
| Variance partition b | |||||
| Among groups | 2 | 307.470 | 0.260 | 1.44 | Fst = 0.014 |
| Among populations within groups | 3 | 326.298 | 3.215 | 17.83 | |
| within populations | 255 | 3712.486 | 14.559 | 80.73 | |
| Total | 260 | 4346.253 | 18.034 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Li, M.; Hou, L.; Zhang, Z.; Pang, X.; Li, Y. De Novo Transcriptome Assembly and Population Genetic Analyses for an Endangered Chinese Endemic Acer miaotaiense (Aceraceae). Genes 2018, 9, 378. https://doi.org/10.3390/genes9080378
Li X, Li M, Hou L, Zhang Z, Pang X, Li Y. De Novo Transcriptome Assembly and Population Genetic Analyses for an Endangered Chinese Endemic Acer miaotaiense (Aceraceae). Genes. 2018; 9(8):378. https://doi.org/10.3390/genes9080378
Chicago/Turabian StyleLi, Xiang, Meng Li, Lu Hou, Zhiyong Zhang, Xiaoming Pang, and Yingyue Li. 2018. "De Novo Transcriptome Assembly and Population Genetic Analyses for an Endangered Chinese Endemic Acer miaotaiense (Aceraceae)" Genes 9, no. 8: 378. https://doi.org/10.3390/genes9080378
APA StyleLi, X., Li, M., Hou, L., Zhang, Z., Pang, X., & Li, Y. (2018). De Novo Transcriptome Assembly and Population Genetic Analyses for an Endangered Chinese Endemic Acer miaotaiense (Aceraceae). Genes, 9(8), 378. https://doi.org/10.3390/genes9080378