Deciphering the Adaptation of Corynebacterium glutamicum in Transition from Aerobiosis via Microaerobiosis to Anaerobiosis

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strain and Media
2.2. Cultivation Conditions
2.3. Analytical Methods
2.3.1. Optical Density
2.3.2. Cell Dry Weight
2.3.3. Enzyme Assays
2.3.4. Protein Quantification
2.3.5. High-Performance Liquid Chromatography
2.4. RNA-Sequencing
2.4.1. Sample Harvest and RNA Isolation
2.4.2. Complementary DNA Library Preparation and Sequencing
2.4.3. Data Analysis, Read Mapping, Data Visualization and Analysis of Differential Gene Expression
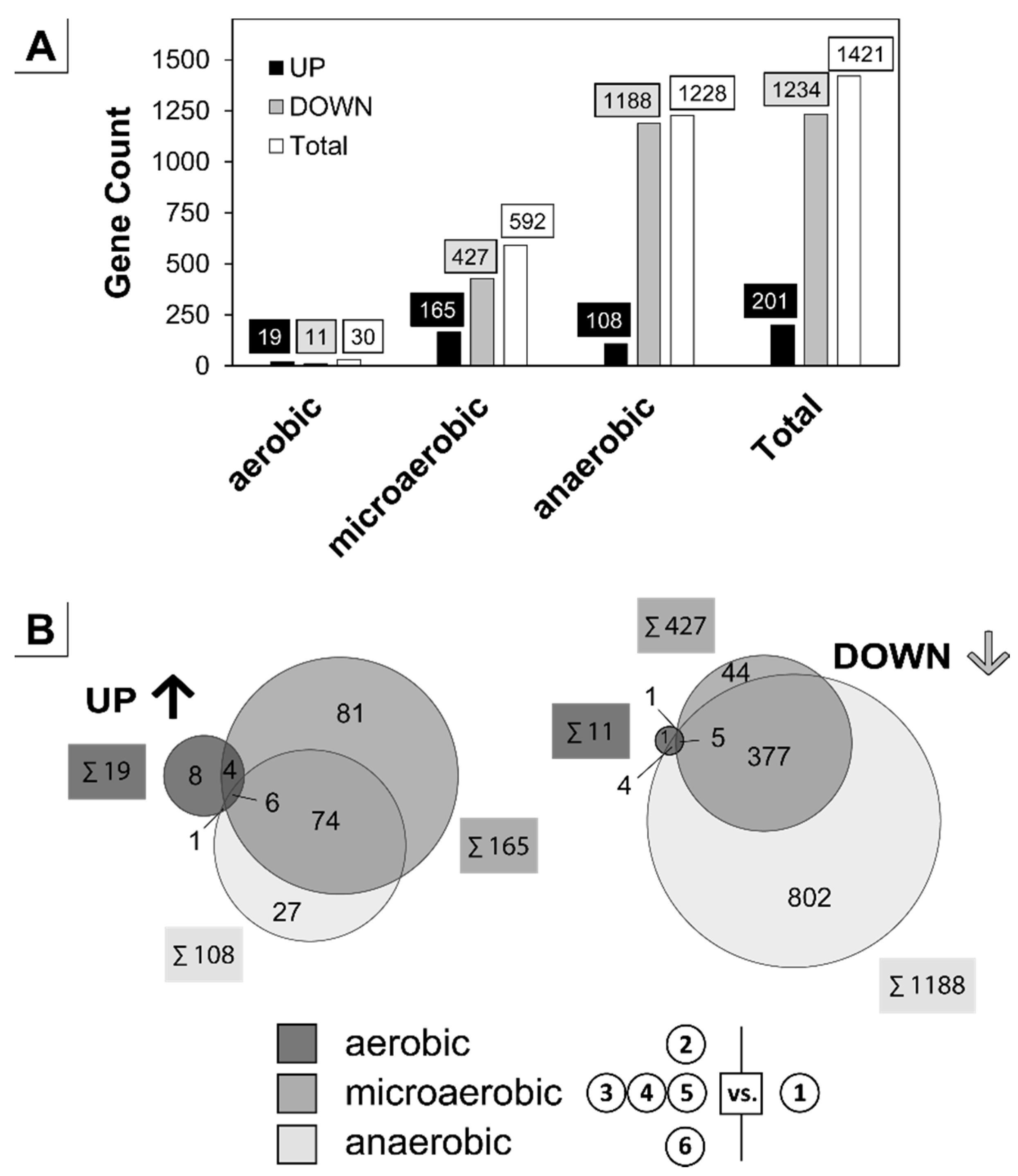
2.4.4. Differential Gene Expression Cut-Off Definition
2.5. Calculations
3. Results
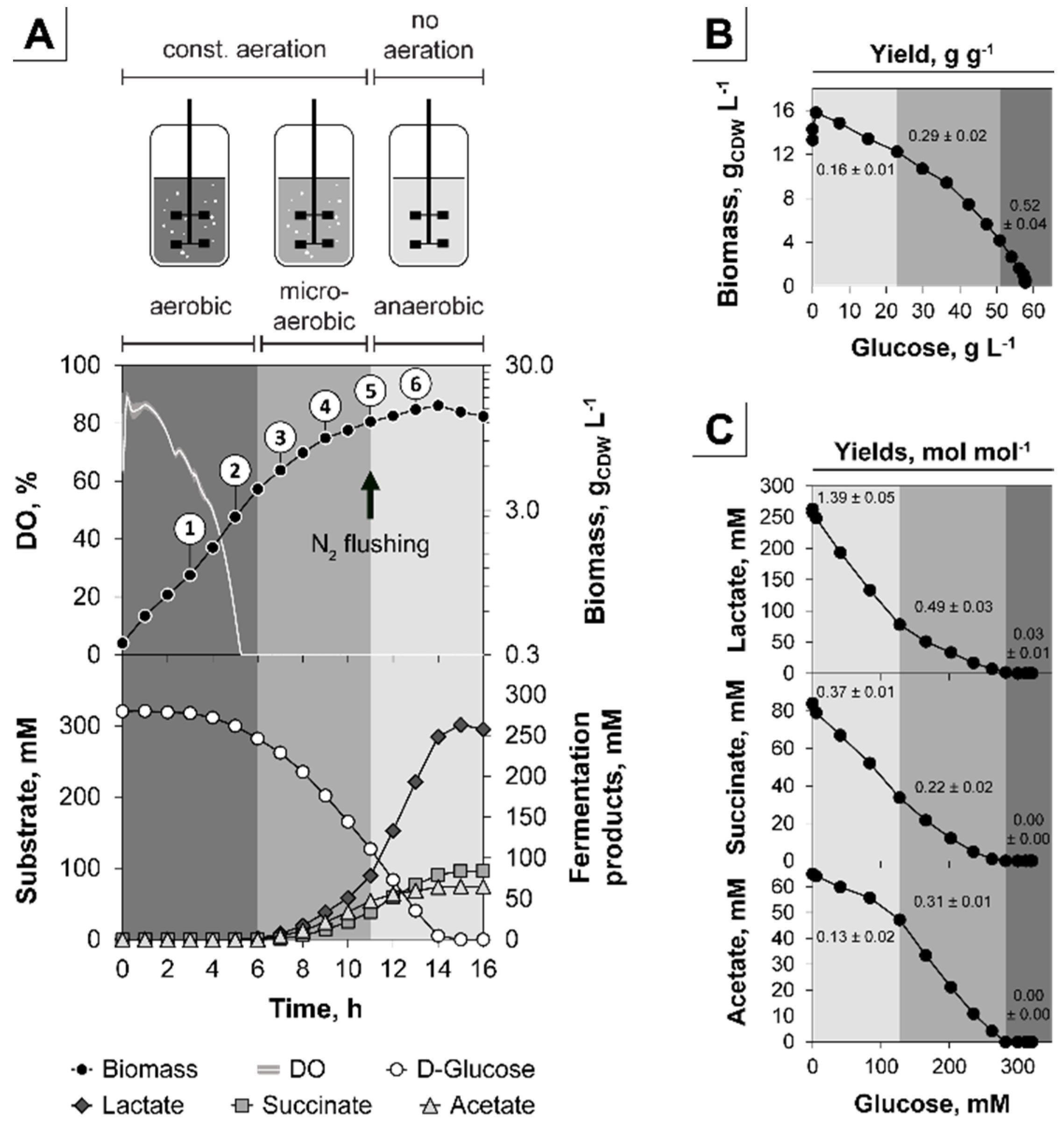
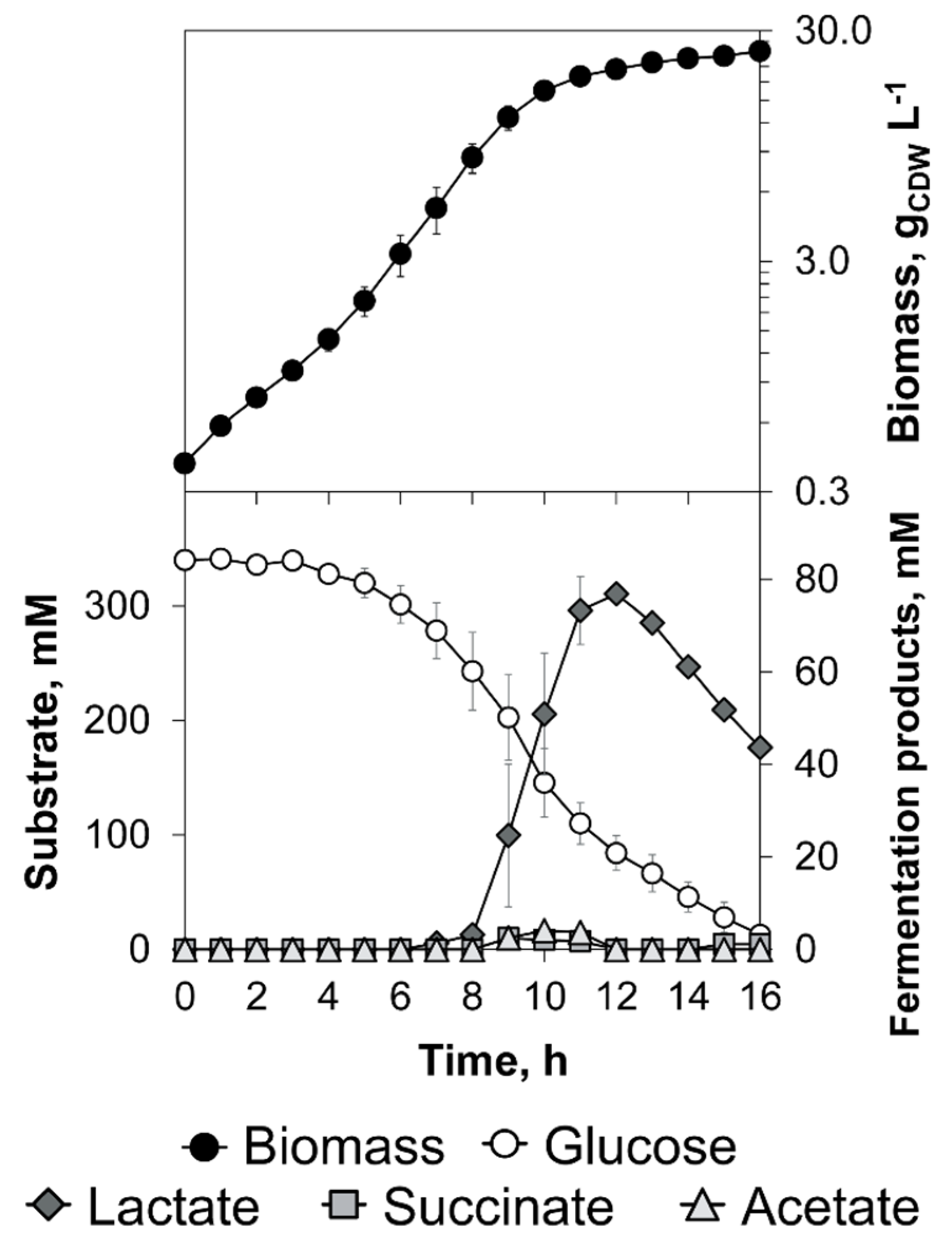
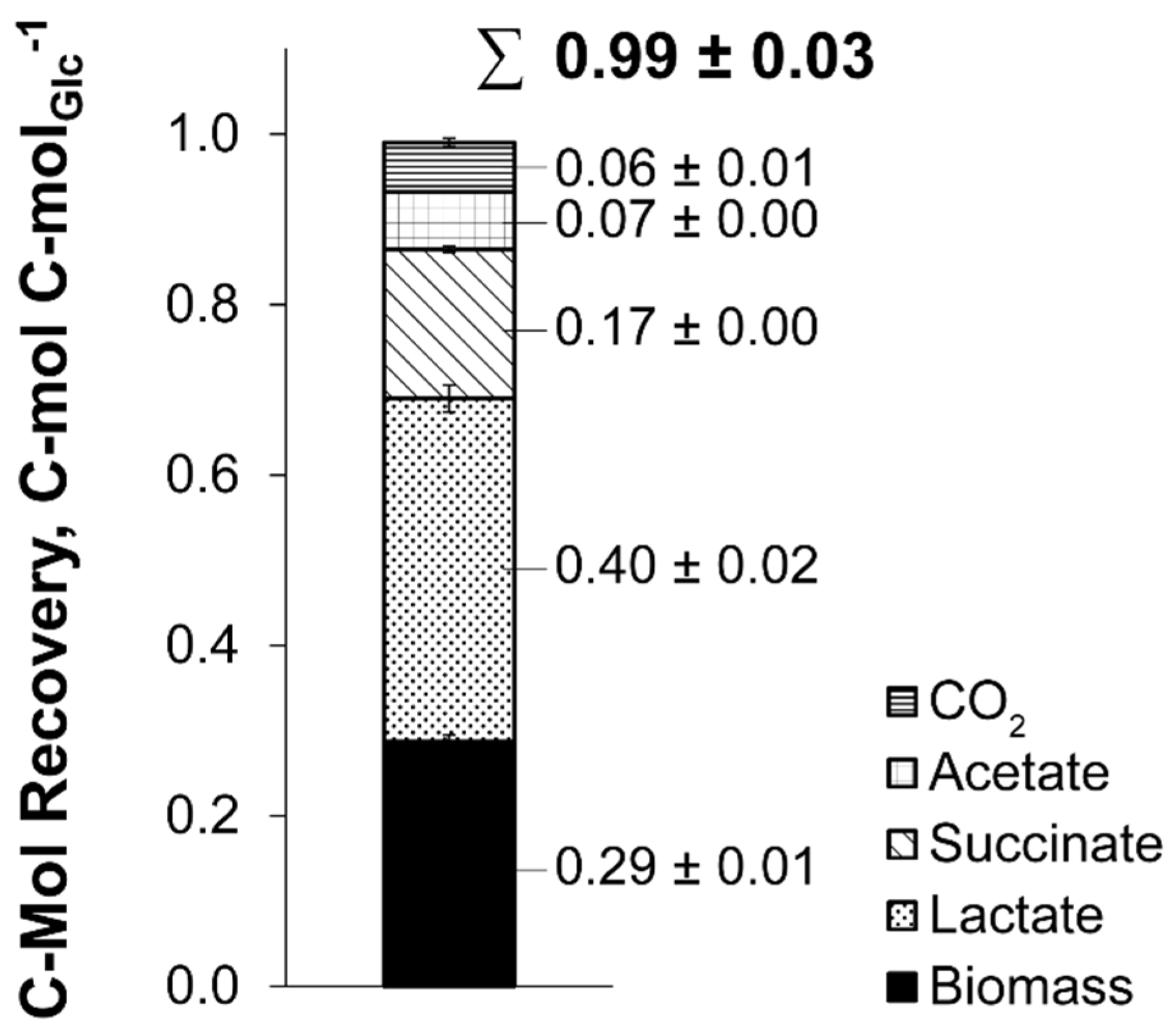
3.1. The Triple-Phase Batch Fermentation
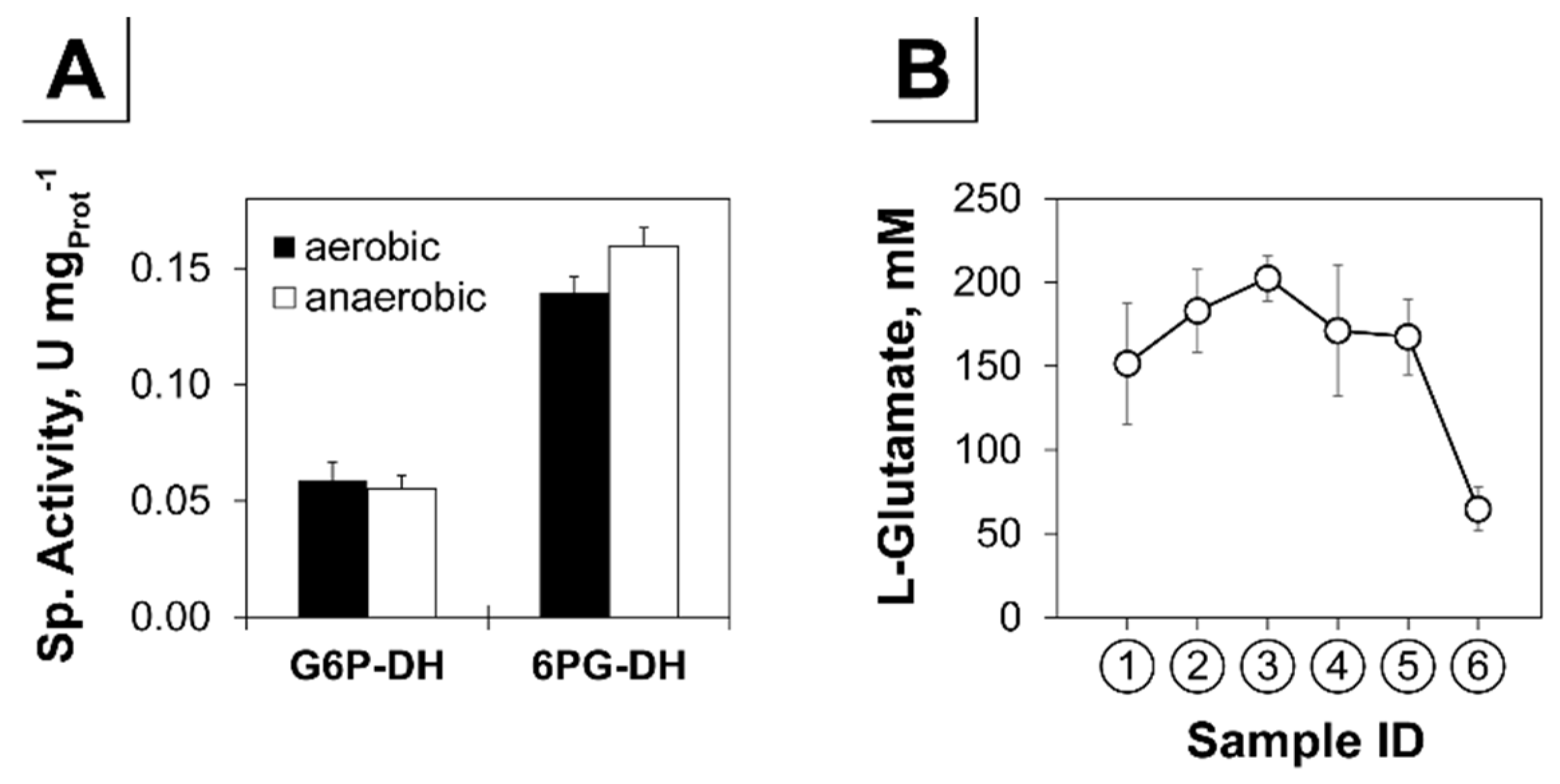
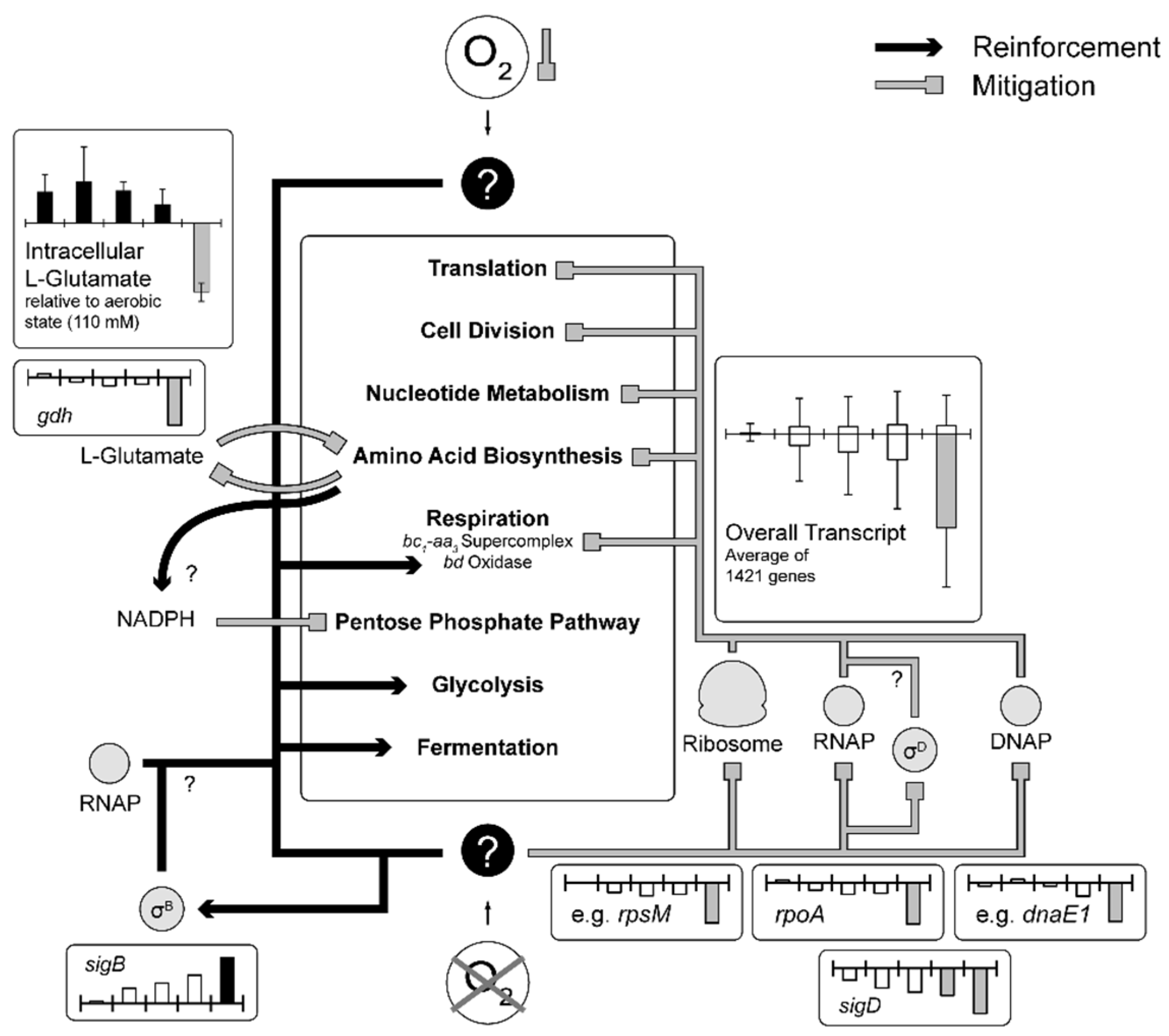
3.2. Analysis of the Transient Transcriptional Adaptation in Response to Decreasing Oxygen Availability
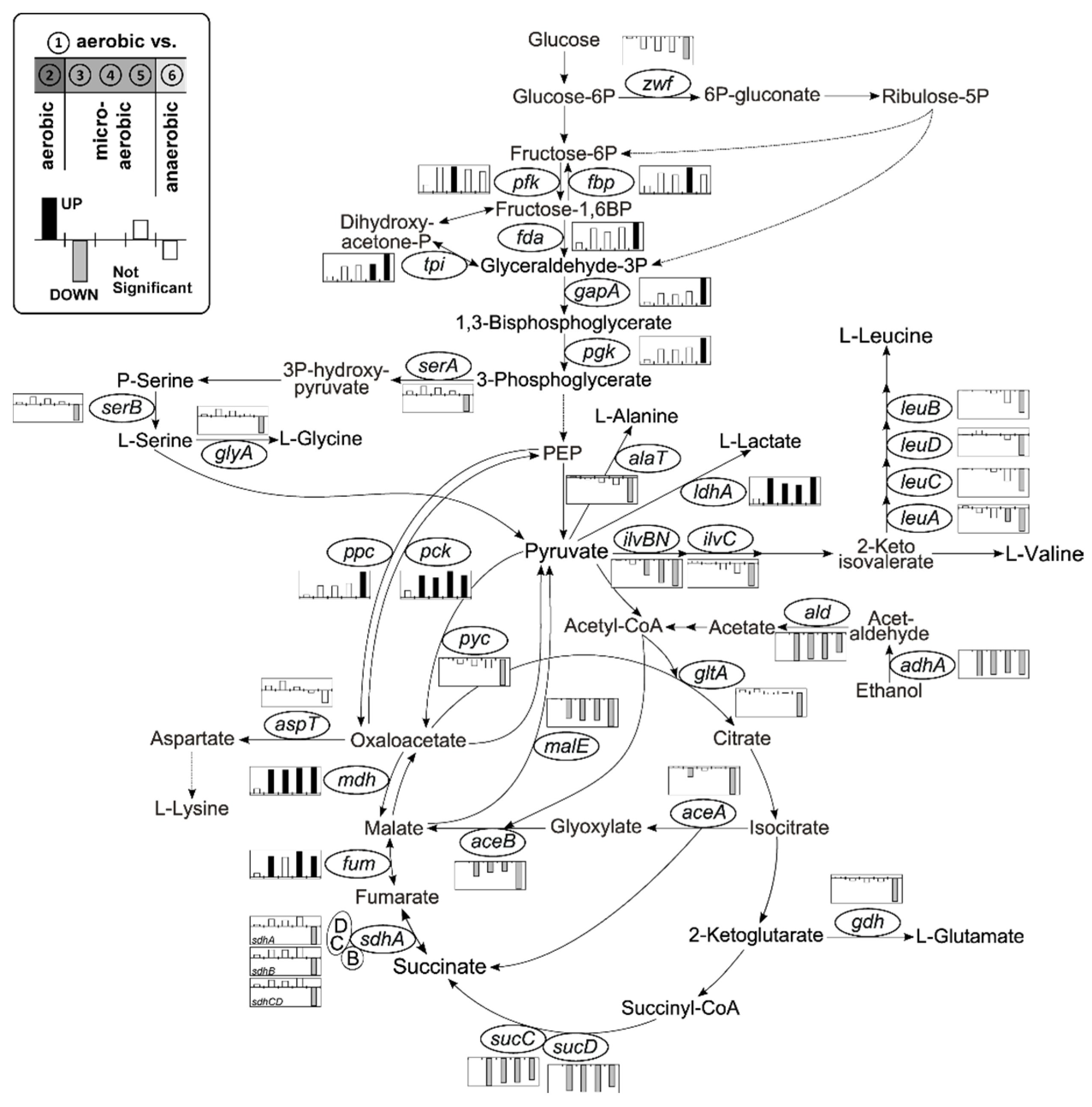
3.2.1. Central Metabolism and Amino Acid Biosynthesis
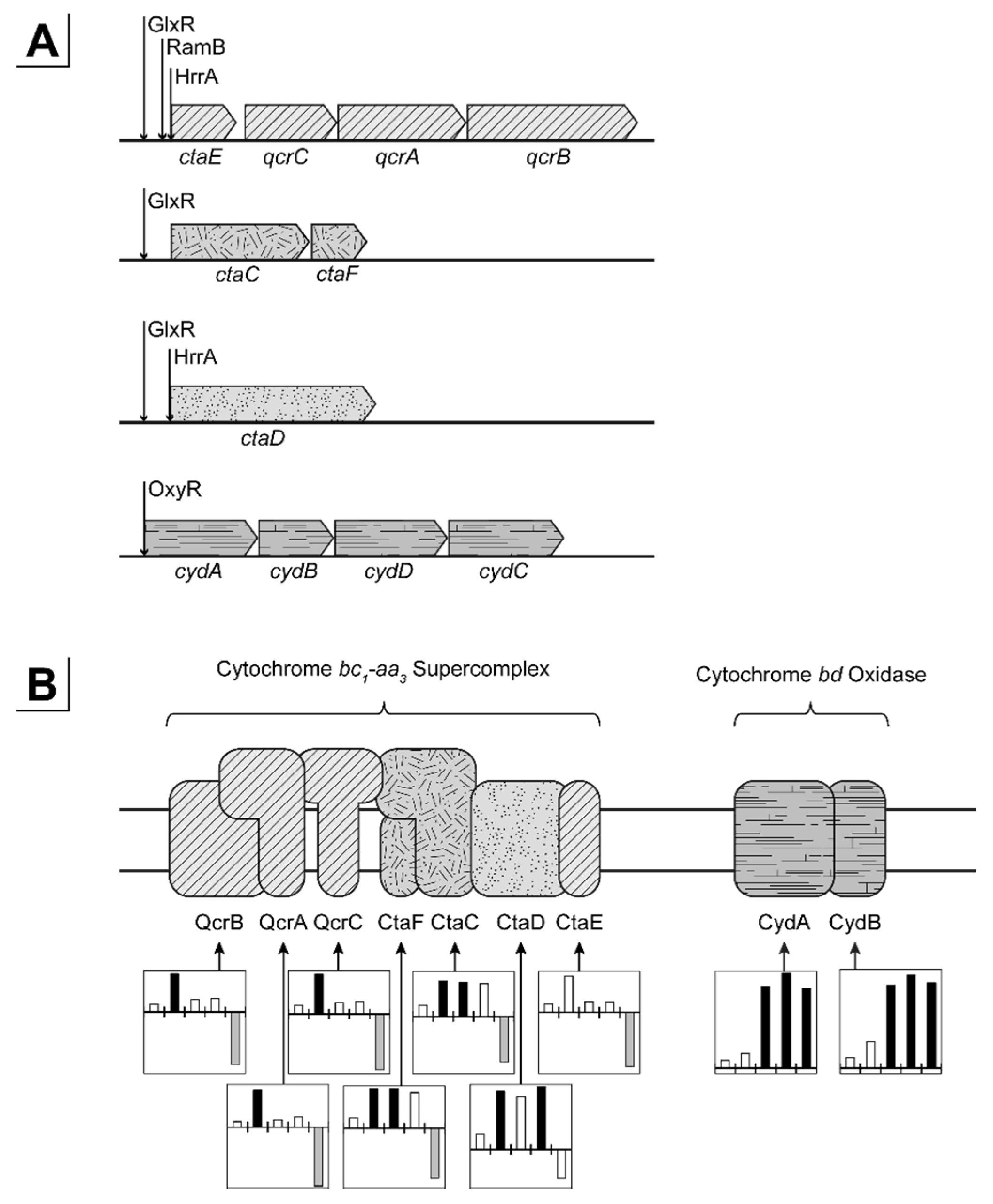
3.2.2. Respiratory Chain and Energy Metabolism
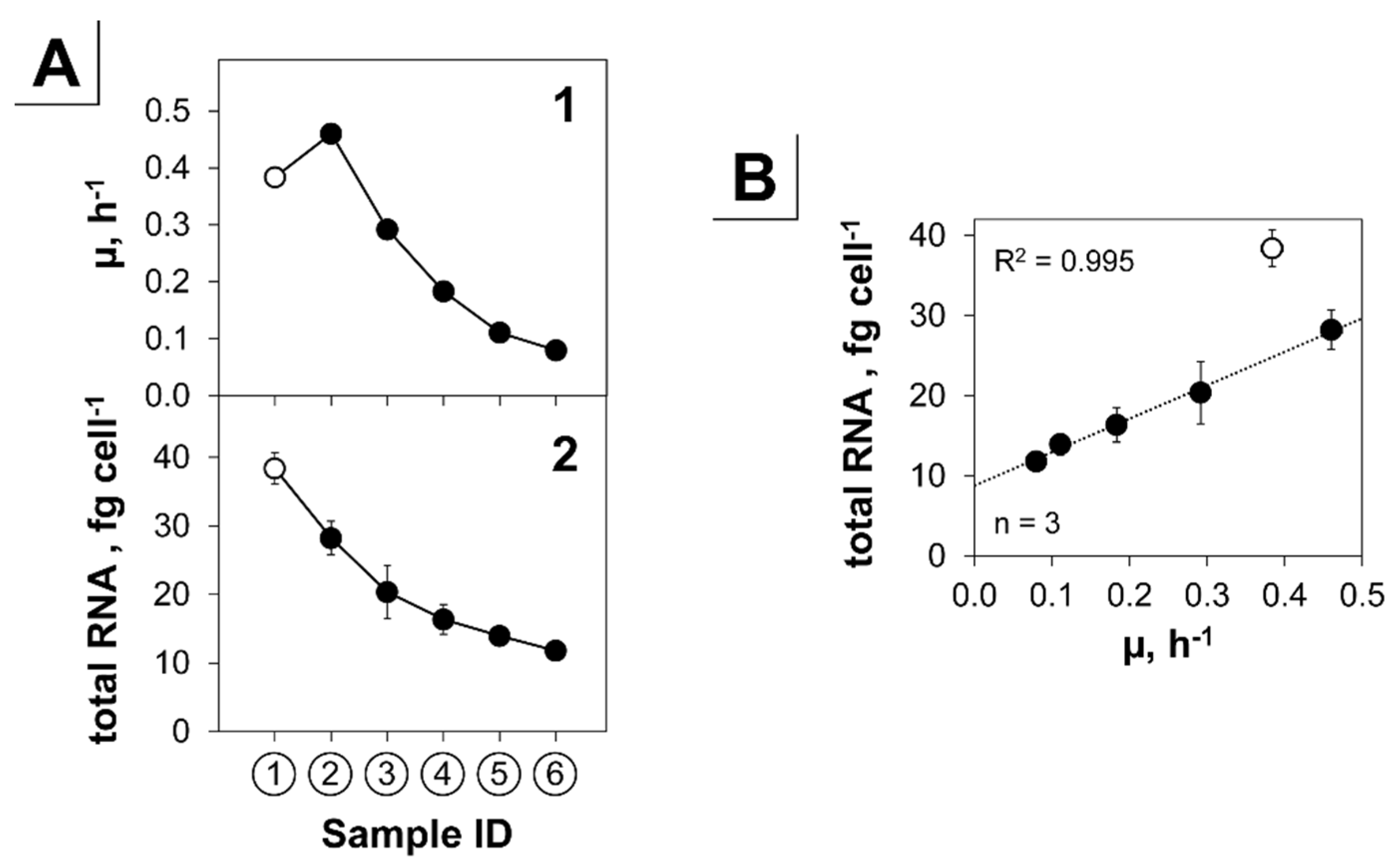
3.2.3. Translation, Transcription and Replication
3.2.4. Sigma Factors and Transcriptional Regulators
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



Deletion of Transcriptional Regulators
Standard Molecular Biology Methods
Cloning of Plasmids and Deletion of Putatively Oxygen Responsive Regulators
- Plasmid assembly. The plasmid pK19mobsacB was linearized by restriction with HindIII/NheI, PstI/NheI, BamHI/NheI or BamHI/EcoRI, as given in Table A1, following general protocols of Thermo Fisher Scientific Inc. (Waltham, MA, USA). Cloning was performed based on the isothermal assembly principle [107]. The experimental procedure was rooted on published recommendations [108]. DNA fragments were amplified with designed primers via polymerase chain reaction (PCR) [109,110] in a Biometra TAdvanced thermocycler (Biometra GmbH, Göttingen, Germany) and applying Phusion Hot Start II HF DNA Polymerase (Thermo Fisher Scientific Inc., Waltham, MA, USA). Oligonucleotides were manufactured by the biomers.net GmbH (Ulm, Germany). Adjacent fragments granted ≥15 bps homologous overlaps by specifically designed oligonucleotides (Table A1). Where fragment size varied dramatically, overlap extension PCR [111] was used to approximate fragment size and to lower total fragment number prior to assembly. Deletion plasmids pJULΔcg3303, pJULΔcg2320, pJULΔcg2965, pJULΔcg2746, pJULΔsutR, pJULΔcg1327, pJULΔznr, pJULΔzur, pJULΔfarR, pJULΔripA, pJULΔcg2648, pJULΔiclR, pJULΔcspA, pJULΔrbsR, pJULΔgenR, pJULΔcg0150 and pJULΔmmpLR harbor a Flank1 and a Flank2 of >500 bp homology to up- or downstream regions of the targeted regulator. Flank1 and Flank2 were amplified from the C. glutamicum chromosome with respective primer pairs Δ*-1/Δ*-2 and Δ*-3/Δ*-4 (asterisk stands for targeted gene; Table A1) and assembled with linearized pK19mobsacB.
- Transformation of E. coli. Electrocompetent E. coli DH5α were manufactured and transformation with the above described isothermal assembly batches or pK19ΔramB was achieved according to literature [112].
- Sequencing. Sequencing of plasmids with the primers pK19seqfw and pK19seqrv was conducted by the GATC Biotech AG (Konstanz, Germany).
- Transformation of C. glutamicum. Electrocompetent C. glutamicum were produced as described in literature [113]. For transformation, the plasmids were isolated from E. coli and transferred to C. glutamicum pursuing the protocols available in literature [112,114]. An additional heat shock was implemented after electroporation as recommended previously [115].
- Deletion selection and verification. Selection of rare double-crossover events for gene deletion was conducted as described elsewhere [116]. Markerless deletion of the regulators via pJULΔ* and pK19ΔramB were verified through colony PCR (Taq DNA Polymerase S, Genaxxon BioScience GmbH, Ulm, Germany) using the outside primer pair Δ*-1/Δ*-4 or ΔramB1/ΔramB2. Bacterial strains, cloned plasmids and applied oligonucleotides are given in Table A1.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain, Plasmid, or Oligo-Nucleotide | Relevant Characteristics or Sequence | Source, Reference or Purpose |
|---|---|---|
| Strains | ||
| Escherichia coli DH5α | F- Φ80lacZΔM15 Δ(lacZYA-argF) U169 endA1 recA1 hsdR17 (rk−, mk+) supE44 thi-1 gyrA96 relA1 phoA | [117] |
| C. glutamicum ΔoxyR | Markerless deletion of OxyR (cg2109) | [100] |
| C. glutamicum Δcg3303 | Markerless deletion of cg3303 by homologous recombination with pJULΔcg3303 | This study |
| C. glutamicum Δcg2320 | Markerless deletion of cg2320 by homologous recombination with pJULΔcg2320 | This study |
| C. glutamicum Δcg2965 | Markerless deletion of g2965 by homologous recombination with pJULΔcg2965 | This study |
| C. glutamicum Δcg2746 | Markerless deletion of cg2746 by homologous recombination with pJULΔcg2746 | This study |
| C. glutamicum ΔsutR | Markerless deletion SutR (cg0993) by homologous recombination with pJULΔsutR | This study |
| C. glutamicum Δcg1327 | Markerless deletion of cg1327 by homologous recombination with pJULΔcg1327 | This study |
| C. glutamicum Δznr | Markerless deletion of Znr (cg2500) by homologous recombination with pJULΔznr | This study |
| C. glutamicum Δzur | Markerless deletion of Zur (cg2502) by homologous recombination with pJULΔzur | This study |
| C. glutamicum ΔfarR | Markerless deletion of FarR (cg3202) by homologous recombination with pJULΔfarR | This study |
| C. glutamicum ΔripA | Markerless deletion RipA (cg1120) by homologous recombination with pJULΔripA | This study |
| C. glutamicum Δcg2648 | Markerless deletion of cg2648 by homologous recombination with pJULΔcg2648 | This study |
| C. glutamicum ΔiclR | Markerless deletion of IclR (cg3388) by homologous recombination with pJULΔiclR | This study |
| C. glutamicum ΔcspA | Markerless deletion of CspA (cg0215) by homologous recombination with pJULΔcspA | This study |
| C. glutamicum ΔrbsR | Markerless deletion of RbsR (cg1410) by homologous recombination with pJULΔrbsR | This study |
| C. glutamicum ΔgenR | Markerless deletion of GenR (cg3352) by homologous recombination with pJULΔgenR | This study |
| C. glutamicum Δcg0150 | Markerless deletion of cg0150 by homologous recombination with pJULΔcg0150 | This study |
| C. glutamicum ΔmmpLR | Markerless deletion of MmpLR (cg1053) by homologous recombination with pJULΔmmpLR | This study |
| C. glutamicum ΔramB | Markerless deletion of RamB (cg0444) by homologous recombination with pK19ΔramB | This study |
| Plasmids | ||
| pK19mobsacB | For chromosomal integration and deletion of genetic information (lacZα, RP4 mob, oriVE. coli, sacBB. subtilis, KanR) | [106] |
| pJULΔcg3303 | For deletion of cg3303, pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔcg2320 | For deletion of cg2320, pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔcg2965 | For deletion of cg2965, pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔcg2746 | For deletion of cg2746, pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔsutR | For deletion of sutR (cg0993), pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔcg1327 | For deletion of cg1327, pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔznr | For deletion of znr (cg2500), pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔzur | For deletion of zur (cg2502), pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔfarR | For deletion of farR (cg3202), pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔripA | For deletion of ripA (cg1120), pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔcg2648 | For deletion of cg2648, pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔiclR | For deletion of iclR (cg3388), pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔcspA | For deletion of cspA (cg0215), pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔrbsR | For deletion of rbsR (cg1410), pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔgenR | For deletion of genR (cg3352), pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔcg0150 | For deletion of cg0150, pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pJULΔmmpLR | For deletion of mmpLR (cg1053), pK19mobsacB::(Flank1-Flank2), KanR | This study |
| pK19ΔramB | For deletion of ramB (cg0444), based on pK19mobsacB, KanR | [102] |
| Oligonucleotides | 5′ → 3′ | |
| pK19seqfw | TAATGCAGCTGGCACGAC | Fw Sequencing primer pK19mobsaB derivatives [118] |
| pK19seqrv | TAATGGTAGCTGACATTCATCCG | Rv Sequencing primer pK19mobsaB derivatives |
| Δcg3303-1 | GAAACAGCTATGACCATGATTACGCCAAGCTTGCCGTCGCAGCACATTGG | Fw primer Flank1 in pJULΔcg3303 (pK19mobsacB, HindIII) |
| Δcg3303-2 | CCCCAGTACCATGCAGCTG | Rv primer Flank1 in pJULΔcg3303 (Flank2) |
| Δcg3303-3 | CTTCGCGCAGCTGCATGGTACTGGGGATCATTATCTCCTGTTCTTGAACTGAAG | Fw primer Flank2 in pJULΔcg3303 (Flank1) |
| Δcg3303-4 | GTGCTTGCGGCAGCGTGAAGCTAGCCCGAGTTCTCCCGTCAGC | Rv primer Flank2 in pJULΔcg3303 (pK19mobsacB, NheI) |
| Δcg2320-1 | AACAGCTATGACCATGATTACGCCAAGCTTGCTGCGGGGATCACTAAA | Fw primer Flank1 in pJULΔcg2320 (pK19mobsacB, HindIII) |
| Δcg2320-2 | AAAGAGCTTTTCAGAACACTTGGCAAACCTCAC | Rv primer Flank1 in pJULΔcg2320 (Flank2) |
| Δcg2320-3 | TTGCCAAGTGTTCTGAAAAGCTCTTTCATCC | Fw primer Flank2 in pJULΔcg2320 (Flank1) |
| Δcg2320-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCGCAACAGTAGATGGAGCTG | Rv primer Flank2 in pJULΔcg2320 (pK19mobsacB, NheI) |
| Δcg2965-1 | ATTACGCCAAGCTTGCATGCCTGCAGGGCGCACACGTATGGGCAGA | Fw primer Flank1 in pJULΔcg2965 (pK19mobsacB, PstI) |
| Δcg2965-2 | GAGAACAAAAACCGGTGCGTACCACAATAGAGTCTTAG | Rv primer Flank1 in pJULΔcg2965 (Flank2) |
| Δcg2965-3 | TGTGGTACGCACCGGTTTTTGTTCTCAGGCGGA | Fw primer Flank2 in pJULΔcg2965 (Flank1) |
| Δcg2965-4 | GAGTGCTTGCGGCAGCGTGAAGCTAGCCCATCGGAAATTCACTGATGTGC | Rv primer Flank2 in pJULΔcg2965 (pK19mobsacB, NheI) |
| Δcg2746-1 | AACAGCTATGACCATGATTACGCCAAGCTTGCGTGGATCCTGACCTGAAG | Fw primer Flank1 in pJULΔcg2746 (pK19mobsacB, HindIII) |
| Δcg2746-2 | AACCTGGGATTCCAAAATTGCACCTATATATATGGTGCAAAAC | Rv primer Flank1 in pJULΔcg2746 (Flank2) |
| Δcg2746-3 | TAGGTGCAATTTTGGAATCCCAGGTTAGCGGGG | Fw primer Flank2 in pJULΔcg2746 (Flank1) |
| Δcg2746-4 | GTGCTTGCGGCAGCGTGAAGCTAGCCGTCGTCGTGCTGGATGC | Rv primer Flank2 in pJULΔcg2746 (pK19mobsacB, NheI) |
| ΔsutR-1 | AACAGCTATGACCATGATTACGCCAAGCTTCACAATCATGATCGCAGCGG | Fw primer Flank1 in pJULΔsutR (pK19mobsacB, HindIII) |
| ΔsutR-2 | TCACGGAGGAATACCTTTTACCCTCTAGAGACGACTATCAG | Rv primer Flank1 in pJULΔsutR (Flank2) |
| ΔsutR-3 | AGAGGGTAAAAGGTATTCCTCCGTGACTAGGCTAGATGACGGATCC | Fw primer Flank2 in pJULΔsutR (Flank1) |
| ΔsutR-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCGCATGCGGGTGTTTGCGCGG | Rv primer Flank2 in pJULΔsutR (pK19mobsacB, NheI) |
| Δcg1327-1 | GCATGCCTGCAGGTCGACTCTAGAGGATCCGCTCGGCAACTGAGGTGCCC | Fw primer Flank1 in pJULΔcg1327 (pK19mobsacB, BamHI) |
| Δcg1327-2 | CAAGCGGAAAGTAAAGCCCATGCTACCCAGGATATTTTC | Rv primer Flank1 in pJULΔcg1327 (Flank2) |
| Δcg1327-3 | GTAGCATGGGCTTTACTTTCCGCTTGTTTGATCTAG | Fw primer Flank2 in pJULΔcg1327 (Flank1) |
| Δcg1327-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCCAGGTCTTCCACGTTTTCATG | Rv primer Flank2 in pJULΔcg1327 (pK19mobsacB, NheI) |
| Δznr-1 | AACAGCTATGACCATGATTACGCCAAGCTTGTCCGCACGGTAACGCTTGTG | Fw primer Flank1 in pJULΔznr (pK19mobsacB, HindIII) |
| Δznr-2 | GTACTTCGATAGTGGGGAAGTCCTTCCGTCCTTAG | Rv primer Flank1 in pJULΔznr (Flank2) |
| Δznr-3 | GAAGGACTTCCCCACTATCGAAGTACATTTTGTGTC | Fw primer Flank2 in pJULΔznr (Flank1) |
| Δznr-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCCAAGGCTATTTTTCGAAATAG | Rv primer Flank2 in pJULΔznr (pK19mobsacB, NheI) |
| Δzur-1 | AACAGCTATGACCATGATTACGCCAAGCTTGTCATTTTGCGGTCTTCGCG | Fw primer Flank1 in pJULΔzur (pK19mobsacB, HindIII) |
| Δzur-2 | ATATGTCCTTGAACGTTGATCCTCCTCAATGACAC | Rv primer Flank1 in pJULΔzur (Flank2) |
| Δzur-3 | AGGAGGATCAACGTTCAAGGACATATGAAGCTGTCGAAC | Fw primer Flank2 in pJULΔzur (Flank1) |
| Δzur-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCCATCGCCGGAGTCGTCATCA | Rv primer Flank2 in pJULΔzur (pK19mobsacB, NheI) |
| ΔfarR-1 | AACAGCTATGACCATGATTACGCCAAGCTTGATCCTTTGGCTCGAAATCAAAAG | Fw primer Flank1 in pJULΔfarR (pK19mobsacB, HindIII) |
| ΔfarR-2 | AAATGGGTTCACGGGTGTTCATTTTAGCCGATCTG | Rv primer Flank1 in pJULΔfarR (Flank2) |
| ΔfarR-3 | TAAAATGAACACCCGTGAACCCATTTTGGTGGC | Fw primer Flank2 in pJULΔfarR (Flank1) |
| ΔfarR-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCCTTCCGCAGGTGGCAGGATC | Rv primer Flank2 in pJULΔfarR (pK19mobsacB, NheI) |
| ΔripA-1 | AACAGCTATGACCATGATTACGCCAAGCTTGACCCCTATTTTCCAGGGATC | Fw primer Flank1 in pJULΔripR (pK19mobsacB, HindIII) |
| ΔripA-2 | ACCTTTACTACCTATCTCATCCTCACTACAAGCAAATTT | Rv primer Flank1 in pJULΔripR (Flank2) |
| ΔripA-3 | GTGAGGATGAGATAGGTAGTAAAGGTGTGAAAATAGTTCCTCACG | Fw primer Flank2 in pJULΔripR (Flank1) |
| ΔripA-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCGTGCCAAGGACTGCCTGGCC | Rv primer Flank2 in pJULΔripR (pK19mobsacB, NheI) |
| Δcg2648-1 | AACAGCTATGACCATGATTACGCCAAGCTTGTGATCTTTGAACGGGTGTC | Fw primer Flank1 in pJULΔcg2648 (pK19mobsacB, HindIII) |
| Δcg2648-2 | TTCTTTAATCTCAAAATTTAAAATTCCATAAATTTAGACAATC | Rv primer Flank1 in pJULΔcg2648 (Flank2) |
| Δcg2648-3 | GAATTTTAAATTTTGAGATTAAAGAAGCAGCTTCTTG | Fw primer Flank2 in pJULΔcg2648 (Flank1) |
| Δcg2648-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCGTAGTGAAATTCTCCGCGCG | Rv primer Flank2 in pJULΔcg2648 (pK19mobsacB, NheI) |
| ΔiclR-1 | GCATGCCTGCAGGTCGACTCTAGAGGATCCGTGTCATAGCCGAAGAGAAG | Fw primer Flank1 in pJULΔiclR (pK19mobsacB, BamHI) |
| ΔiclR-2 | GTCAATGAATTGCATTTGATCCGTTTTTCTAAAG | Rv primer Flank1 in pJULΔiclR (Flank2) |
| ΔiclR-3 | AAACGGATCAAATGCAATTCATTGACGTACAAAGTGATG | Fw primer Flank2 in pJULΔiclR (Flank1) |
| ΔiclR-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCCGATTCAGACAGGCGGACGT | Rv primer Flank2 in pJULΔiclR (pK19mobsacB, NheI) |
| ΔcspA-1 | GCATGCCTGCAGGTCGACTCTAGAGGATCCGGTTACTTTTTCGGGGCCTTTTG | Fw primer Flank1 in pJULΔcspA (pK19mobsacB, BamHI) |
| ΔcspA-2 | TAGCAGTTAGAGCATTTGTACCTTTTCCTAATCAGGTGATG | Rv primer Flank1 in pJULΔcspA (Flank2) |
| ΔcspA-3 | AAAAGGTACAAATGCTCTAACTGCTAGCTAAAAATTCCGC | Fw primer Flank2 in pJULΔcspA (Flank1) |
| ΔcspA-4 | CGACGTTGTAAAACGACGGCCAGTGAATTCGGAAGGCTTGCTCCCACTGC | Rv primer Flank2 in pJULΔcspA (pK19mobsacB, EcoRI) |
| ΔrbsR-1 | GCATGCCTGCAGGTCGACTCTAGAGGATCCGACCTTCACGGGAATTGGAC | Fw primer Flank1 in pJULΔrbsR (pK19mobsacB, BamHI) |
| ΔrbsR-2 | ATGAAGCGCTTGTCTCCTCACCAACTTTCTGGAAG | Rv primer Flank1 in pJULΔrbsR (Flank2) |
| ΔrbsR-3 | AGTTGGTGAGGAGACAAGCGCTTCATCAGCATG | Fw primer Flank2 in pJULΔrbsR (Flank1) |
| ΔrbsR-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCCAATTTCACGACCAGTCAACG | Rv primer Flank2 in pJULΔrbsR (pK19mobsacB, NheI) |
| ΔgenR-1 | AACAGCTATGACCATGATTACGCCAAGCTTCCACAGGGTAGGGGAGATG | Fw primer Flank1 in pJULΔgenR (pK19mobsacB, HindIII) |
| ΔgenR-2 | GGAAAGAGTGATTATGGGGGGAATTTTCAGAGC | Rv primer Flank1 in pJULΔgenR (Flank2) |
| ΔgenR-3 | AAATTCCCCCCATAATCACTCTTTCCAGATAGCG | Fw primer Flank2 in pJULΔgenR (Flank1) |
| ΔgenR-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCGGTCTTACGTGGAACCAAATC | Rv primer Flank2 in pJULΔgenR (pK19mobsacB, NheI) |
| Δcg0150-1 | GCATGCCTGCAGGTCGACTCTAGAGGATCCCTCGGACTGCGGGGTGTAC | Fw primer Flank1 in pJULΔcg0150 (pK19mobsacB, BamHI) |
| Δcg0150-2 | CACAATCGATGAACTCCATAACGAGAACTTAATCGAGCAAC | Rv primer Flank1 in pJULΔcg0150 (Flank2) |
| Δcg0150-3 | TCTCGTTATGGAGTTCATCGATTGTGAGTGAGCGGTAATAATG | Fw primer Flank2 in pJULΔcg0150 (Flank1) |
| Δcg0150-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCGAAATTGTGCGAGGCCCCCG | Rv primer Flank2 in pJULΔcg0150 (pK19mobsacB, NheI) |
| ΔmmpLR-1 | GCATGCCTGCAGGTCGACTCTAGAGGATCCGAACAAGACAACCTCTACATCTTCG | Fw primer Flank1 in pJULΔmmpLR (pK19mobsacB, BamHI) |
| ΔmmpLR-2 | AAGGAAAATGTAGAAATTGTGGCGTGTGAACCTC | Rv primer Flank1 in pJULΔmmpLR (Flank2) |
| ΔmmpLR-3 | CACGCCACAATTTCTACATTTTCCTTCAGTTCCTCGGTGC | Fw primer Flank2 in pJULΔmmpLR (Flank1) |
| ΔmmpLR-4 | CCTGAGTGCTTGCGGCAGCGTGAAGCTAGCCATTGATCGCGGCTCTGGGC | Rv primer Flank2 in pJULΔmmpLR (pK19mobsacB, NheI) |
| ΔramB1 | CCACGCCGGGCACCTG | Fw primer ΔramB verification |
| ΔramB2 | GGCGCGATAGTGGATTCGTG | Rv primer ΔramB verification |
| Gene ID | Name | Description | Rel. Diff. Expression |
|---|---|---|---|
| cg2092 | sigA | Primary (housekeeping) sigma factor |  |
| cg2102 | sigB | Nonessential primary-like sigma factor involved in gene expression during the transition phase, under oxygen deprivation and during environmental stress responses |  |
| cg0309 | sigC | Regulates expression of a branched quinol oxidation pathway |  |
| cg0696 | sigD | ECF sigma factor probably involved in the adaptation to micro-aerobic environments |  |
| cg1271 | sigE | ECF sigma factor involved in responses to cells surface stresses |  |
| cg0876 | sigH | ECF sigma factor controlling the heat and oxidative stress response |  |
| cg3420 | sigM | ECF sigma factor controlling the expression of disulfide stress-related genes |  |
| No. | Gene ID | Name | Description | Rel. Diff. Expression |
|---|---|---|---|---|
| 1 | cg0993 | sutR | Bacterial regulatory protein |  |
| 2 | cg3303 | - | Putative transcriptional regulator, PadR-family |  |
| 3 | cg1327 | - | Putative transcriptional regulator, Crp-family |  |
| 4 | cg2500 | znr | Putative transcriptional regulator, ArsR-family |  |
| 5 | cg2502 | zur | Putative transcriptional regulator, Fur-family |  |
| 6 | cg3202 | farR | Transcriptional regulator, GntR-family |  |
| 7 | cg1120 | ripA | Repressor of iron protein genes |  |
| 8 | cg2965 | - | Putative transcriptional regulator, AraC-family |  |
| 9 | cg0444 | ramB | Master regulator of carbon metabolism |  |
| 10 | cg2320 | - | Putative transcriptional regulator, ArsR-family |  |
| 11 | cg2746 | - | Putative sugar diacid utilization regulator |  |
| 12 | cg2648 | - | Putative transcriptional regulator, ArsR-family |  |
| 13 | cg3388 | iclR | Activator of putative hydroxyquinol pathway genes |  |
| 14 | cg0215 | cspA | Cold-shock protein A |  |
| 15 | cg1410 | rbsR | Repressor of ribose uptake and uridine utilization genes |  |
| 16 | cg3352 | genR | Transcriptional activator of gentisate catabolism |  |
| 17 | cg0150 | - | Putative transcriptional regulatory protein, Fic/Doc family |  |
| 18 | cg1053 | mmpLR | Putative transcriptional regulator, TetR-family |  |
| 19 | cg2109 | oxyR | Hydrogen peroxide sensing regulator |  |
References
- Ajinomoto Co., Inc. FY2015 Market and Other Information. Available online: https://www.ajinomoto.com/en/ir/event/presentation/main/09/teaserItems1/00/linkList/02/link/FY15_Data_E.pdf (accessed on 7 September 2017).
- Becker, J.; Wittmann, C. Advanced biotechnology: Metabolically engineered cells for the bio-based production of chemicals and fuels, materials, and health-care products. Angew. Chem. Int. Ed. 2015, 54, 3328–3350. [Google Scholar] [CrossRef] [PubMed]
- Liebl, W. Corynebacterium taxonomy. In Handbook of Corynebacterium Glutamicum; Eggeling, L., Bott, M., Eds.; CRC Press: Boca Raton, FL, USA, 2005; pp. 9–34. [Google Scholar]
- Nishimura, T.; Vertès, A.A.; Shinoda, Y.; Inui, M.; Yukawa, H. Anaerobic growth of Corynebacterium glutamicum using nitrate as a terminal electron acceptor. Appl. Microbiol. Biotechnol. 2007, 75, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Wieschalka, S.; Blombach, B.; Eikmanns, B.J. Engineering Corynebacterium glutamicum for the production of pyruvate. Appl. Microbiol. Biotechnol. 2012, 94, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Jojima, T.; Inui, M.; Yukawa, H. Biotechnological application of Corynebacterium glutamicum under oxygen deprivation. In Corynebacterium Glutamicum: From Systems Biology to Biotechnological Applications; Burkovski, A., Ed.; Caister Academic Press: Norfolk, UK, 2015; pp. 151–160. [Google Scholar]
- Blombach, B.; Riester, T.; Wieschalka, S.; Ziert, C.; Youn, J.-W.; Wendisch, V.F.; Eikmanns, B.J. Corynebacterium glutamicum tailored for efficient isobutanol production. Appl. Environ. Microbiol. 2011, 77, 3300–3310. [Google Scholar] [CrossRef] [PubMed]
- Blombach, B.; Eikmanns, B.J. Current knowledge on isobutanol production with Escherichia coli, Bacillus subtilis and Corynebacterium glutamicum. Bioeng. Bugs 2011, 2, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Suda, M.; Niimi, S.; Inui, M.; Yukawa, H. Strain optimization for efficient isobutanol production using Corynebacterium glutamicum under oxygen deprivation. Biotechnol. Bioeng. 2013, 110, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.; Takors, R.; Blombach, B. Zero-growth bioprocesses—A challenge for microbial production strains and bioprocess engineering. Eng. Life Sci. 2017, 17, 27–35. [Google Scholar] [CrossRef]
- Zhu, J.; Thakker, C.; San, K.-Y.; Bennett, G. Effect of culture operating conditions on succinate production in a multiphase fed-batch bioreactor using an engineered Escherichia coli strain. Appl. Microbiol. Biotechnol. 2011, 92, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, M.; Zhang, X.; Yang, P.; Liang, Q.; Qi, Q. A novel whole-phase succinate fermentation strategy with high volumetric productivity in engineered Escherichia coli. Bioresour. Technol. 2013, 149, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Martínez, I.; Bennett, G.N.; San, K.-Y. Metabolic impact of the level of aeration during cell growth on anaerobic succinate production by an engineered Escherichia coli strain. Metab. Eng. 2010, 12, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Wieschalka, S.; Blombach, B.; Bott, M.; Eikmanns, B.J. Bio-based production of organic acids with Corynebacterium glutamicum. Microb. Biotechnol. 2013, 6, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Kaboré, A.-K.; Olmos, E.; Fick, M.; Blanchard, F.; Guedon, E.; Delaunay, S. Aerobiosis–anaerobiosis transition has a significant impact on organic acid production by Corynebacterium glutamicum. Process Biochem. 2016, 52, 10–21. [Google Scholar] [CrossRef]
- Lara, A.R.; Leal, L.; Flores, N.; Gosset, G.; Bolívar, F.; Ramírez, O.T. Transcriptional and metabolic response of recombinant Escherichia coli to spatial dissolved oxygen tension gradients simulated in a scale—Down system. Biotechnol. Bioeng. 2006, 93, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, J.; Graf, M.; Freund, A.; Busche, T.; Kalinowski, J.; Blombach, B.; Takors, R. CO2/HCO3- perturbations of simulated large scale gradients in a scale-down device cause fast transcriptional responses in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2014, 98, 8563–8572. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, A.; Maya Martίnez-Iturralde, N.; Spann, R.; Neubauer, P.; Junne, S. Response of Corynebacterium glutamicum exposed to oscillating cultivation conditions in a two- and a novel three-compartment scale-down bioreactor. Biotechnol. Bioeng. 2015, 112, 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Limberg, M.H.; Schulte, J.; Aryani, T.; Mahr, R.; Baumgart, M.; Bott, M.; Wiechert, W.; Oldiges, M. Metabolic profile of 1,5-diaminopentane producing Corynebacterium glutamicum under scale-down conditions: Blueprint for robustness to bioreactor inhomogeneities. Biotechnol. Bioeng. 2017, 114, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Bylund, F.; Collet, E.; Enfors, S.-O.; Larsson, G. Substrate gradient formation in the large-scale bioreactor lowers cell yield and increases by-product formation. Bioprocess Eng. 1998, 18, 171. [Google Scholar] [CrossRef]
- Sandoval-Basurto, E.A.; Gosset, G.; Bolívar, F.; Ramírez, O.T. Culture of Escherichia coli under dissolved oxygen gradients simulated in a two-compartment scale-down system: Metabolic response and production of recombinant protein. Biotechnol. Bioeng. 2005, 89, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xiu, Z.; Wang, J.; Zhang, D.; Xu, P. Stoichiometric analysis and experimental investigation of glycerol bioconversion to 1,3-propanediol by Klebsiella pneumoniae under microaerobic conditions. Enzyme Microb. Technol. 2003, 33, 386–394. [Google Scholar] [CrossRef]
- Alfenore, S.; Cameleyre, X.; Benbadis, L.; Bideaux, C.; Uribelarrea, J.-L.; Goma, G.; Molina-Jouve, C.; Guillouet, S.E. Aeration strategy: A need for very high ethanol performance in Saccharomyces cerevisiae fed-batch process. Appl. Microbiol. Biotechnol. 2004, 63, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Biswas, R.; Yamaoka, M.; Nakayama, H.; Kondo, T.; Yoshida, K.; Bisaria, V.S.; Kondo, A. Enhanced production of 2,3-butanediol by engineered Bacillus subtilis. Appl. Microbiol. Biotechnol. 2012, 94, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Huang, D.; Li, Y.; Wen, J.; Jia, X. Rational improvement of the engineered isobutanol-producing Bacillus subtilis by elementary mode analysis. Microb. Cell. Fact. 2012, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Hirasawa, T.; Nishii, M.; Furusawa, C.; Shimizu, H. Enhanced acetic acid and succinic acid production under microaerobic conditions by Corynebacterium glutamicum harboring Escherichia coli transhydrogenase gene pntAB. J. Gen. Appl. Microbiol. 2014, 60, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.-K.; Chen, F.; Xia, Q. Monitoring microaerobic denitrification of Pseudomonas aeruginosa by online NAD(P)H fluorescence. J. Ind. Microbiol. Biotechnol. 2005, 32, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, L.F. Oxidation-Reduction Potentials in Bacteriology and Biochemistry, 6th ed.; Williams and Wilkins: Baltimore, MD, USA; Edinburgh, UK, 1950. [Google Scholar]
- Goncharuk, V.V.; Bagrii, V.A.; Mel’nik, L.A.; Chebotareva, R.D.; Bashtan, S.Y. The use of redox potential in water treatment processes. J. Water Chem. Technol. 2010, 32, 1–9. [Google Scholar] [CrossRef]
- Inui, M.; Suda, M.; Okino, S.; Nonaka, H.; Puskas, L.G.; Vertes, A.A.; Yukawa, H. Transcriptional profiling of Corynebacterium glutamicum metabolism during organic acid production under oxygen deprivation conditions. Microbiology 2007, 153, 2491–2504. [Google Scholar] [CrossRef] [PubMed]
- Wimpenny, J.W.; Firth, A. Levels of nicotinamide adenine dinucleotide and reduced nicotinamide adenine dinucleotide in facultative bacteria and the effect of oxygen. J. Bacteriol. 1972, 111, 24–32. [Google Scholar] [PubMed]
- Stolper, D.A.; Revsbech, N.P.; Canfield, D.E. Aerobic growth at nanomolar oxygen concentrations. Proc. Natl. Acad. Sci. USA 2010, 107, 18755–18760. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, O.; Tauch, A. Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J. Biotechnol. 2003, 104, 287–299. [Google Scholar] [CrossRef]
- Wendisch, V.F.; Bott, M.; Kalinowski, J.; Oldiges, M.; Wiechert, W. Emerging Corynebacterium glutamicum systems biology. J. Biotechnol. 2006, 124, 74–92. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Inui, M. Genome engineering of Corynebacterium glutamicum. In Corynebacterium Glutamicum: Biology and Biotechnology; Yukawa, H., Inui, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 89–105. ISBN 978-3-642-29857-8. [Google Scholar]
- Burkovski, A. Corynebacterium Glutamicum: From Systems Biology to Biotechnological Applications, 1st ed.; Caister Academic Press: Norfolk, UK, 2015; ISBN 9781910190050. [Google Scholar]
- Wendisch, V.F.; Bott, M.; Eikmanns, B.J. Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for biotechnological production of organic acids and amino acids. Curr. Opin. Microbiol. 2006, 9, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.S.; Choi, K.R.; Prabowo, C.P.S.; Shin, J.H.; Yang, D.; Jang, J.; Lee, S.Y. CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab. Eng. 2017, 42, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Wendisch, V.F. Production of amino acids—Genetic and metabolic engineering approaches. Bioresour. Technol. 2017, 245, 1575–1587. [Google Scholar] [CrossRef] [PubMed]
- Khoroshilova, N.; Popescu, C.; Münck, E.; Beinert, H.; Kiley, P.J. Iron-sulfur cluster disassembly in the FNR protein of Escherichia coli by O2: [4Fe-4S] to [2Fe-2S] conversion with loss of biological activity. Proc. Natl. Acad. Sci. USA 1997, 94, 6087–6092. [Google Scholar] [CrossRef] [PubMed]
- Jordan, P.A.; Thomson, A.J.; Ralph, E.T.; Guest, J.R.; Green, J. FNR is a direct oxygen sensor having a biphasic response curve. FEBS Lett. 1997, 416, 349–352. [Google Scholar] [CrossRef]
- Bekker, M.; Alexeeva, S.; Laan, W.; Sawers, G.; Teixeira de Mattos, J.; Hellingwerf, K. The ArcBA two-component system of Escherichia coli is regulated by the redox state of both the ubiquinone and the menaquinone pool. J. Bacteriol. 2010, 192, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Sawers, G. The aerobic/anaerobic interface. Curr. Opin. Microbiol. 1999, 2, 181–187. [Google Scholar] [CrossRef]
- Bibikov, S.I.; Biran, R.; Rudd, K.E.; Parkinson, J.S. A signal transducer for aerotaxis in Escherichia coli. J. Bacteriol. 1997, 179, 4075–4079. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Weber, K.D.; Qiu, Y.; Kiley, P.J.; Blattner, F.R. Genome-wide expression analysis indicates that FNR of Escherichia coli K-12 regulates a large number of genes of unknown function. J. Bacteriol. 2005, 187, 1135–1160. [Google Scholar] [CrossRef] [PubMed]
- Georgellis, D.; Kwon, O.; Lin, E.C. Quinones as the redox signal for the arc two-component system of bacteria. Science 2001, 292, 2314–2316. [Google Scholar] [CrossRef] [PubMed]
- Patschkowski, T.; Bates, D.M.; Kiley, P.J. Mechanisms for sensing and responding to oxygen deprivation. In Bacterial Stress Responses; Storz, G., Hengge-Aronis, R., Eds.; ASM Press: Washington, DC, USA, 2000; pp. 61–78. ISBN 978-1-555-81621-6. [Google Scholar]
- Partridge, J.D.; Sanguinetti, G.; Dibden, D.P.; Roberts, R.E.; Poole, R.K.; Green, J. Transition of Escherichia coli from aerobic to micro-aerobic conditions involves fast and slow reacting regulatory components. J. Biol. Chem. 2007, 282, 11230–11237. [Google Scholar] [CrossRef] [PubMed]
- Berríos-Rivera, S.J.; Bennett, G.N.; San, K.-Y. The effect of increasing NADH availability on the redistribution of metabolic fluxes in Escherichia coli chemostat cultures. Metab. Eng. 2002, 4, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Tolla, D.A.; Savageau, M.A. Regulation of aerobic-to-anaerobic transitions by the FNR cycle in Escherichia coli. J. Mol. Biol. 2010, 397, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Ederer, M.; Steinsiek, S.; Stagge, S.; Rolfe, M.D.; Ter Beek, A.; Knies, D.; Teixeira de Mattos, M.J.; Sauter, T.; Green, J.; Poole, R.K.; et al. A mathematical model of metabolism and regulation provides a systems-level view of how Escherichia coli responds to oxygen. Front. Microbiol. 2014, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Sakai, M.; Inui, M.; Yukawa, H. Diversity of metabolic shift in response to oxygen deprivation in Corynebacterium glutamicum and its close relatives. Appl. Microbiol. Biotechnol. 2011, 90, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Shinfuku, Y.; Sorpitiporn, N.; Sono, M.; Furusawa, C.; Hirasawa, T.; Shimizu, H. Development and experimental verification of a genome-scale metabolic model for Corynebacterium glutamicum. Microb. Cell Fact. 2009, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Limberg, M.H.; Joachim, M.; Klein, B.; Wiechert, W.; Oldiges, M. pH fluctuations imperil the robustness of C. glutamicum to short term oxygen limitation. J. Biotechnol. 2017, 259, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Käß, F.; Junne, S.; Neubauer, P.; Wiechert, W.; Oldiges, M. Process inhomogeneity leads to rapid side product turnover in cultivation of Corynebacterium glutamicum. Microb. Cell Fact. 2014, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Käß, F.; Hariskos, I.; Michel, A.; Brandt, H.-J.; Spann, R.; Junne, S.; Wiechert, W.; Neubauer, P.; Oldiges, M. Assessment of robustness against dissolved oxygen/substrate oscillations for C. glutamicum DM1933 in two-compartment bioreactor. Bioprocess Biosyst. Eng. 2014, 37, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Junker, B.H. Scale-up methodologies for Escherichia coli and yeast fermentation processes. J. Biosci. Bioeng. 2004, 97, 347–364. [Google Scholar] [CrossRef]
- Lara, A.R.; Galindo, E.; Ramírez, O.T.; Palomares, L.A. Living with heterogeneities in bioreactors: Understanding the effects of environmental gradients on cells. Mol. Biotechnol. 2006, 34, 355–382. [Google Scholar] [CrossRef]
- Takors, R. Scale-up of microbial processes: Impacts, tools and open questions. J. Biotechnol. 2012, 160, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Volume 3, ISBN 0-87969-577-3. [Google Scholar]
- Eikmanns, B.J.; Metzger, M.; Reinscheid, D.; Kircher, M.; Sahm, H. Amplification of three threonine biosynthesis genes in Corynebacterium glutamicum and its influence on carbon flux in different strains. Appl. Microbiol. Biotechnol. 1991, 34, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Keilhauer, C.; Eggeling, L.; Sahm, H. Isoleucine synthesis in Corynebacterium glutamicum: Molecular analysis of the ilvB-ilvN-ilvC operon. J. Bacteriol. 1993, 175, 5595–5603. [Google Scholar] [CrossRef] [PubMed]
- Follmann, M.; Ochrombel, I.; Krämer, R.; Trötschel, C.; Poetsch, A.; Rückert, C.; Hüser, A.; Persicke, M.; Seiferling, D.; Kalinowski, J.; et al. Functional genomics of pH homeostasis in Corynebacterium glutamicum revealed novel links between pH response, oxidative stress, iron homeostasis and methionine synthesis. BMC Genom. 2009, 10, 621. [Google Scholar] [CrossRef] [PubMed]
- Moritz, B.; Striegel, K.; De Graaf, A.A.; Sahm, H. Kinetic properties of the glucose-6-phosphate and 6-phosphogluconate dehydrogenases from Corynebacterium glutamicum and their application for predicting pentose phosphate pathway flux in vivo. Eur. J. Biochem. 2000, 267, 3442–3452. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, J.; Schwentner, A.; Brunnenkan, B.; Gabris, C.; Grimm, S.; Gerstmeir, R.; Takors, R.; Eikmanns, B.J.; Blombach, B. Platform engineering of Corynebacterium glutamicum with reduced pyruvate dehydrogenase complex activity for improved production of L-lysine, l-valine, and 2-ketoisovalerate. Appl. Environ. Microbiol. 2013, 79, 5566–5575. [Google Scholar] [CrossRef] [PubMed]
- Cserjan-Puschmann, M.; Kramer, W.; Duerrschmid, E.; Striedner, G.; Bayer, K. Metabolic approaches for the optimisation of recombinant fermentation processes. Appl. Microbiol. Biotechnol. 1999, 53, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Löffler, M.; Simen, J.D.; Jäger, G.; Schäferhoff, K.; Freund, A.; Takors, R. Engineering E. coli for large-scale production—Strategies considering ATP expenses and transcriptional responses. Metab. Eng. 2016, 38, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Krömer, J.O.; Sorgenfrei, O.; Klopprogge, K.; Heinzle, E.; Wittmann, C. In-depth profiling of lysine-producing Corynebacterium glutamicum by combined analysis of the transcriptome, metabolome, and fluxome. J. Bacteriol. 2004, 186, 1769–1784. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Hilker, R.; Stadermann, K.B.; Schwengers, O.; Anisiforov, E.; Jaenicke, S.; Weisshaar, B.; Zimmermann, T.; Goesmann, A. ReadXplorer 2—Detailed read mapping analysis and visualization from one single source. Bioinformatics 2016, 32, 3702–3708. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Gerosa, L.; Kochanowski, K.; Heinemann, M.; Sauer, U. Dissecting specific and global transcriptional regulation of bacterial gene expression. Mol. Syst. Biol. 2013, 9, 658. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, J.; Graf, M.; Blombach, B.; Takors, R. Improving the carbon balance of fermentations by total carbon analyses. Biochem. Eng. J. 2014, 90, 162–169. [Google Scholar] [CrossRef]
- Venn, J. On the diagrammatic and mechanical representation of propositions and reasonings. Lond. Edinb. Dublin Philos. Mag. J. Sci. Ser. 5 1880, 10, 1–18. [Google Scholar] [CrossRef]
- Kalinowski, J.; Bathe, B.; Bartels, D.; Bischoff, N.; Bott, M.; Burkovski, A.; Dusch, N.; Eggeling, L.; Eikmanns, B.J.; Gaigalat, L.; et al. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of l-aspartate-derived amino acids and vitamins. J. Biotechnol. 2003, 104, 5–25. [Google Scholar] [CrossRef]
- Bartek, T.; Blombach, B.; Lang, S.; Eikmanns, B.J.; Wiechert, W.; Oldiges, M.; Nöh, K.; Noack, S. Comparative 13C metabolic flux analysis of pyruvate dehydrogenase complex-deficient, l-valine-producing Corynebacterium glutamicum. Appl. Environ. Microbiol. 2011, 77, 6644–6652. [Google Scholar] [CrossRef] [PubMed]
- Radoš, D.; Turner, D.L.; Fonseca, L.L.; Carvalho, A.L.; Blombach, B.; Eikmanns, B.J.; Neves, A.R.; Santos, H. Carbon flux analysis by 13C nuclear magnetic resonance to determine the effect of CO2 on anaerobic succinate production by Corynebacterium glutamicum. Appl. Environ. Microbiol. 2014, 80, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Marienhagen, J.; Kennerknecht, N.; Sahm, H.; Eggeling, L. Functional analysis of all aminotransferase proteins inferred from the genome sequence of Corynebacterium glutamicum. J. Bacteriol. 2005, 187, 7639–7646. [Google Scholar] [CrossRef] [PubMed]
- Eikmanns, B.J.; Blombach, B. The pyruvate dehydrogenase complex of Corynebacterium glutamicum: An attractive target for metabolic engineering. J. Biotechnol. 2014, 192 Pt B, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Kusumoto, K.; Sakiyama, M.; Sakamoto, J.; Noguchi, S.; Sone, N. Menaquinol oxidase activity and primary structure of cytochrome bd from the amino-acid fermenting bacterium Corynebacterium glutamicum. Arch. Microbiol. 2000, 173, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Niebisch, A. The respiratory chain of Corynebacterium glutamicum. J. Biotechnol. 2003, 104, 129–153. [Google Scholar] [CrossRef]
- Niebisch, A. Purification of a cytochrome bc1-aa3 supercomplex with quinol oxidase activity from Corynebacterium glutamicum. J. Biol. Chem. 2002, 278, 4339–4346. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, D.; van der Rest, M.E.; Drysch, A.; Yücel, R. Functions of the membrane-associated and cytoplasmic malate dehydrogenases in the citric acid cycle of Corynebacterium glutamicum. J. Bacteriol. 2000, 182, 6884–6891. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Niebisch, A. Respiratory energy metabolism. In Handbook of Corynebacterium Glutamicum; Eggeling, L., Bott, M., Eds.; CRC Press: Boca Raton, FL, USA, 2005; pp. 305–332. ISBN 0849318211. [Google Scholar]
- Pauling, J.; Röttger, R.; Tauch, A.; Azevedo, V.; Baumbach, J. CoryneRegNet 6.0—Updated database content, new analysis methods and novel features focusing on community demands. Nucl. Acids Res. 2012, 40, D610–D614. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucl. Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Goesmann, A.; McHardy, A.C.; Bartels, D.; Bekel, T.; Clausen, J.; Kalinowski, J.; Linke, B.; Rupp, O.; Giegerich, R.; et al. GenDB—An open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003, 31, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Martín, J.F.; Barreiro, C.; González-Lavado, E.; Barriuso, M. Ribosomal RNA and ribosomal proteins in corynebacteria. J. Biotechnol. 2003, 104, 41–53. [Google Scholar] [CrossRef]
- Bremer, H.; Dennis, P.P. Modulation of chemical composition and other parameters of the cell at different exponential growth rates. EcoSal Plus 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer-Sancar, K.; Mentz, A.; Rückert, C.; Kalinowski, J. Comprehensive analysis of the Corynebacterium glutamicum transcriptome using an improved RNAseq technique. BMC Genom. 2013, 14, 888. [Google Scholar] [CrossRef] [PubMed]
- Gourse, R.L.; Ross, W.; Rutherford, S.T. General pathway for turning on promoters transcribed by RNA polymerases containing alternative σ factors. J. Bacteriol. 2006, 188, 4589–4591. [Google Scholar] [CrossRef] [PubMed]
- Landini, P.; Egli, T.; Wolf, J.; Lacour, S. sigmaS, a major player in the response to environmental stresses in Escherichia coli: Role, regulation and mechanisms of promoter recognition. Environ. Microbiol. Rep. 2014, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ehira, S.; Shirai, T.; Teramoto, H.; Inui, M.; Yukawa, H. Group 2 sigma factor SigB of Corynebacterium glutamicum positively regulates glucose metabolism under conditions of oxygen deprivation. Appl. Environ. Microbiol. 2008, 74, 5146–5152. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Baba, M.; Tsukamoto, N.; Komatsu, T.; Mitsuhashi, S.; Takeno, S. Elucidation of genes relevant to the microaerobic growth of Corynebacterium glutamicum. Biosci. Biotechnol. Biochem. 2009, 73, 2806–2808. [Google Scholar] [CrossRef] [PubMed]
- Compan, I.; Touati, D. Anaerobic activation of arcA transcription in Escherichia coli: Roles of Fnr and ArcA. Mol. Microbiol. 1994, 11, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Schröder, J.; Tauch, A. Transcriptional regulation of gene expression in Corynebacterium glutamicum: The role of global, master and local regulators in the modular and hierarchical gene regulatory network. FEMS Microbiol. Rev. 2010, 34, 685–737. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, H.; Inui, M.; Yukawa, H. OxyR acts as a transcriptional repressor of hydrogen peroxide-inducible antioxidant genes in Corynebacterium glutamicum R. FEBS J. 2013, 280, 3298–3312. [Google Scholar] [CrossRef] [PubMed]
- Milse, J.; Petri, K.; Rückert, C.; Kalinowski, J. Transcriptional response of Corynebacterium glutamicum ATCC 13032 to hydrogen peroxide stress and characterization of the OxyR regulon. J. Biotechnol. 2014, 190, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Kabus, A.; Niebisch, A.; Bott, M. Role of cytochrome bd oxidase from Corynebacterium glutamicum in growth and lysine production. Appl. Environ. Microbiol. 2007, 73, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Gerstmeir, R.; Cramer, A.; Dangel, P.; Schaffer, S.; Eikmanns, B.J. RamB, a novel transcriptional regulator of genes involved in acetate metabolism of Corynebacterium glutamicum. J. Bacteriol. 2004, 186, 2798–2809. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, K.; Inui, M. The extracytoplasmic function σ factor σC regulates expression of a branched quinol oxidation pathway in Corynebacterium glutamicum. Mol. Microbiol. 2016, 100, 486–509. [Google Scholar] [CrossRef] [PubMed]
- Green, J.; Paget, M.S. Bacterial redox sensors. Nat. Rev. Microbiol. 2004, 2, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Busche, T.; Patschkowski, T.; Niehaus, K.; Pátek, M.; Kalinowski, J.; Wendisch, V.F. Physiological roles of sigma factor SigD in Corynebacterium glutamicum. BMC Microbiol. 2017, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Tauch, A.; Jäger, W.; Kalinowski, J.; Thierbach, G.; Pühler, A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: Selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 1994, 145, 69–73. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol. 2011, 498, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Mullis, K.B.; Faloona, F.A. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 1987, 155, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Horton, R.M.; Cai, Z.L.; Ho, S.N.; Pease, L.R. Gene splicing by overlap extension: Tailor-made genes using the polymerase chain reaction. Biotechniques 1990, 8, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Dower, W.J.; Miller, J.F.; Ragsdale, C.W. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 1988, 16, 6127–6145. [Google Scholar] [CrossRef] [PubMed]
- Tauch, A.; Kirchner, O.; Löffler, B.; Götker, S.; Pühler, A.; Kalinowski, J. Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr. Microbiol. 2002, 45, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Liebl, W.; Bayerl, A.; Schein, B.; Stillner, U.; Schleifer, K.H. High efficiency electroporation of intact Corynebacterium glutamicum cells. FEMS Microbiol. Lett. 1989, 53, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Van der Rest, M.E.; Lange, C.; Molenaar, D. A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA. Appl. Microbiol. Biotechnol. 1999, 52, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Eggeling, L.; Reyes, O. Experiments. In Handbook of Corynebacterium Glutamicum; Eggeling, L., Bott, M., Eds.; CRC Press: Boca Raton, FL, USA, 2005; Volume 535–566, pp. 421–422. ISBN 0849318211. [Google Scholar]
- Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983, 166, 557–580. [Google Scholar] [CrossRef]
- Shah, A.; Eikmanns, B.J.; Yukawa, H.; Marin, K.; Wendisch, V.; Eikmanns, B.; Prieto, M. Transcriptional regulation of the β-type carbonic anhydrase gene bca by RamA in Corynebacterium glutamicum. PLoS ONE 2016, 11, e0154382. [Google Scholar] [CrossRef] [PubMed]







| Phase | µ, h−1 | YX/S, g g−1 | qS, g g−1 h−1 | YP/S, mol mol−1 | ||
|---|---|---|---|---|---|---|
| Lactate | Succinate | Acetate | ||||
| aerobic | 0.40 ± 0.01 | 0.52 ± 0.04 | 0.77 ± 0.06 | 0.03 ± 0.01 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| microaerobic | 0.21 ± 0.00 | 0.29 ± 0.02 | 0.72 ± 0.05 | 0.49 ± 0.03 | 0.22 ± 0.02 | 0.31 ± 0.01 |
| anaerobic | 0.09 ± 0.01 | 0.16 ± 0.01 | 0.56 ± 0.07 | 1.39 ± 0.05 | 0.37 ± 0.01 | 0.13 ± 0.02 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lange, J.; Münch, E.; Müller, J.; Busche, T.; Kalinowski, J.; Takors, R.; Blombach, B. Deciphering the Adaptation of Corynebacterium glutamicum in Transition from Aerobiosis via Microaerobiosis to Anaerobiosis. Genes 2018, 9, 297. https://doi.org/10.3390/genes9060297
Lange J, Münch E, Müller J, Busche T, Kalinowski J, Takors R, Blombach B. Deciphering the Adaptation of Corynebacterium glutamicum in Transition from Aerobiosis via Microaerobiosis to Anaerobiosis. Genes. 2018; 9(6):297. https://doi.org/10.3390/genes9060297
Chicago/Turabian StyleLange, Julian, Eugenia Münch, Jan Müller, Tobias Busche, Jörn Kalinowski, Ralf Takors, and Bastian Blombach. 2018. "Deciphering the Adaptation of Corynebacterium glutamicum in Transition from Aerobiosis via Microaerobiosis to Anaerobiosis" Genes 9, no. 6: 297. https://doi.org/10.3390/genes9060297
APA StyleLange, J., Münch, E., Müller, J., Busche, T., Kalinowski, J., Takors, R., & Blombach, B. (2018). Deciphering the Adaptation of Corynebacterium glutamicum in Transition from Aerobiosis via Microaerobiosis to Anaerobiosis. Genes, 9(6), 297. https://doi.org/10.3390/genes9060297