Influence of Maternal Aging on Mitochondrial Heterogeneity, Inheritance, and Function in Oocytes and Preimplantation Embryos



{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Germline Mitochondrial DNA Bottleneck and Purifying Selection
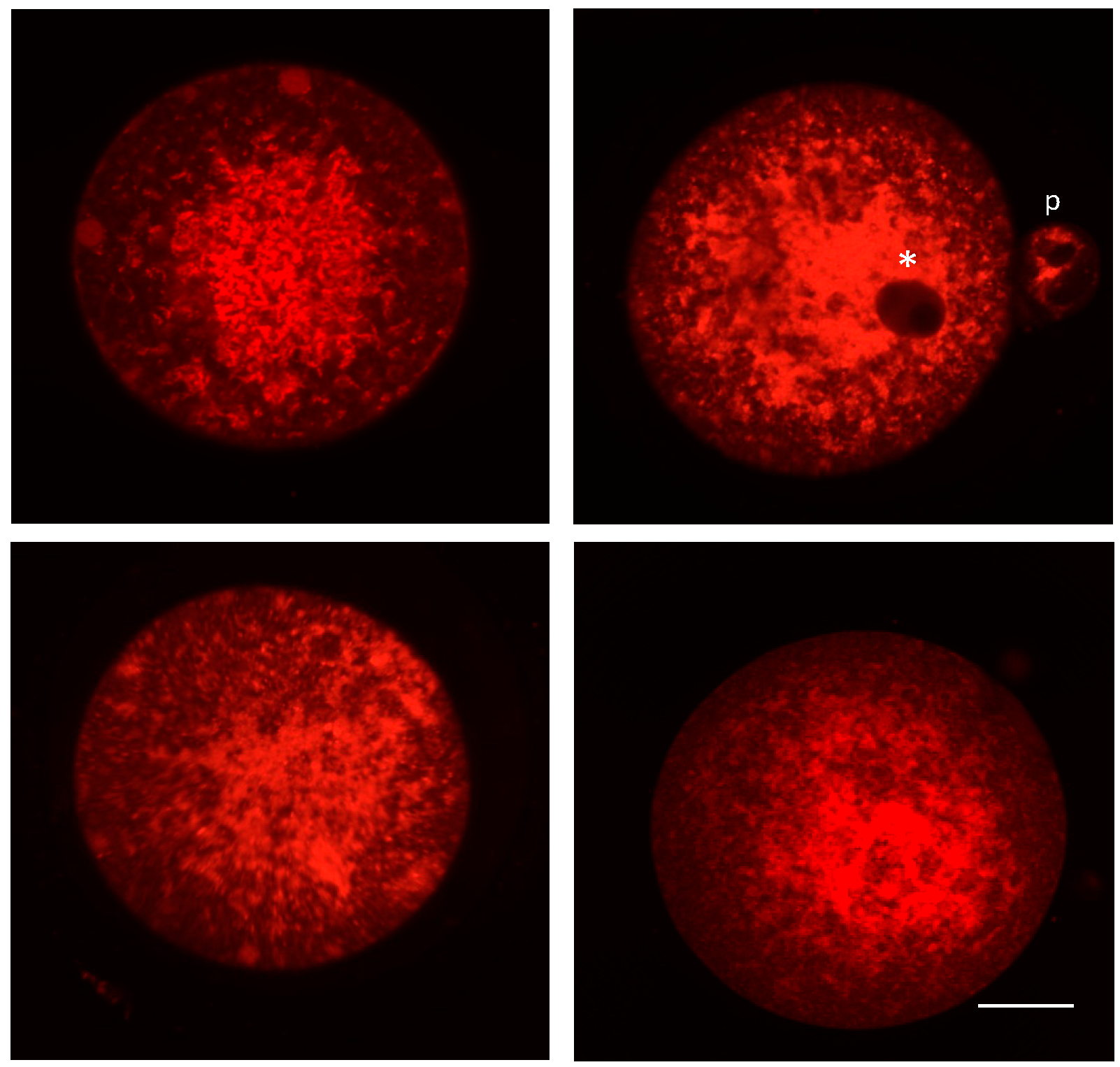
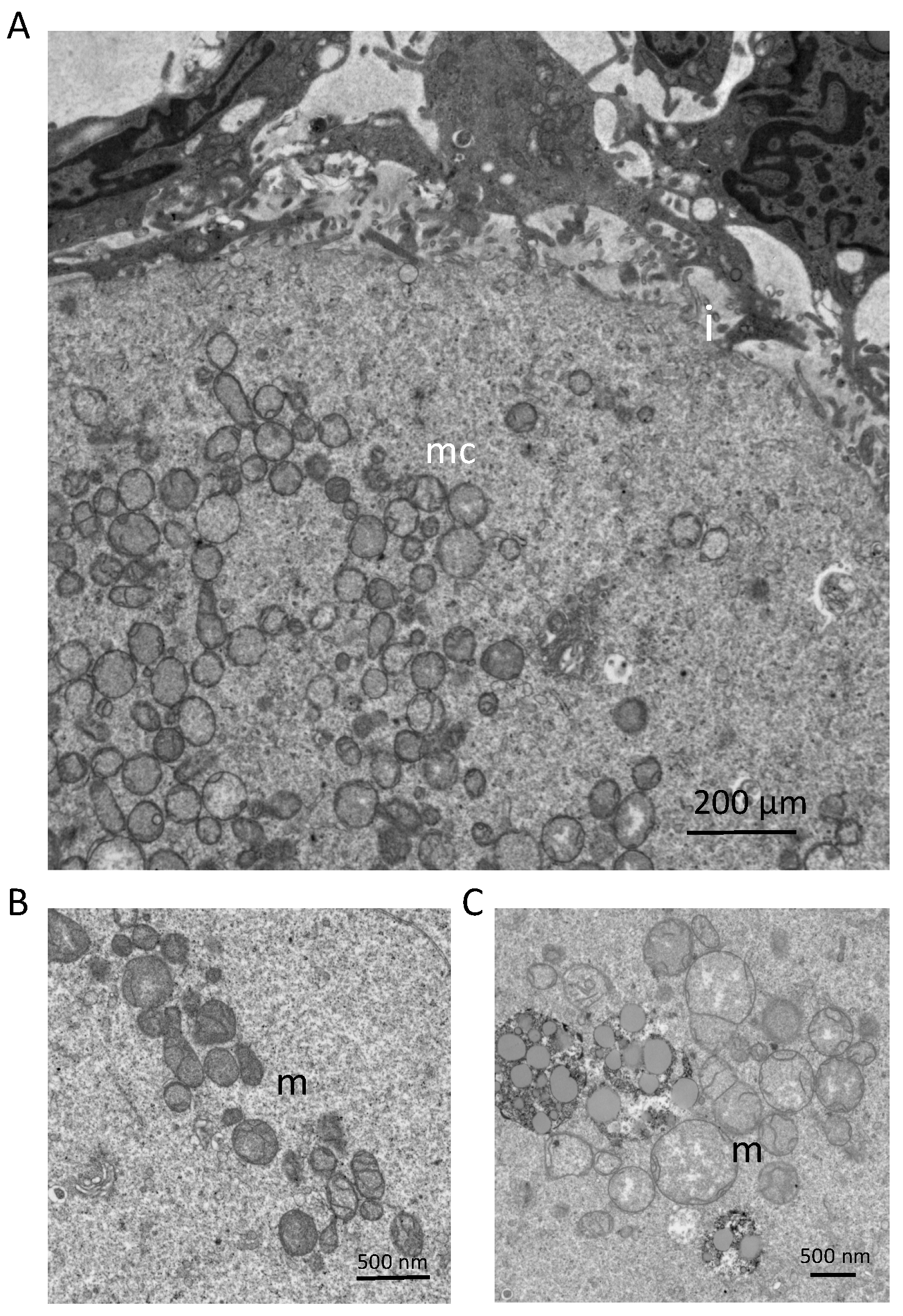
3. Mitochondrial Dynamics and Heterogeneity in Eggs and Embryos
4. Boosting Egg and Embryo Quality through Exogenous Mitochondrial Transfer
5. Mitochondrial Dynamics in Artificial Oocytes
6. Conclusions and Future Studies
Acknowledgments
Conflicts of Interest
References
- Rockstein, M.; Brandt, K.F. Enzyme changes in flight muscle correlated with aging and flight availability in the male housefly. Science 1963, 139, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-C.; Wei, Y.-H. Mitochondria and aging. Adv. Exp. Med. Biol. 2012, 942, 311–327. [Google Scholar] [PubMed]
- Bractic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. BioMed Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Freire, M.; de Cabo, R.; Bernier, M.; Sollott, S.J.; Fabbri, E.; Navas, P.; Ferrucci, L. Reconsidering the role of mitochondria in aging. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The mitochondrial basis of aging. Mol. Cell 2016, 61, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.-G. Mammalian mitochondria and aging: An update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Bittremieux, M.; Missiroli, S.; Morganti, C.; Patergnani, S.; Sbano, L.; Rimessi, A.; Kerkhofs, M.; Parys, J.B.; Bultynck, G.; et al. Endoplasmic reticulum-mitochondria communication through Ca2+ signaling: The importance of mitochondria-associated membranes (MAMs). Adv. Exp. Med. Biol. 2017, 997, 49–67. [Google Scholar] [PubMed]
- Suofu, Y.; Li, W.; Jean-Alphonse, F.G.; Jia, J.; Khattar, N.K.; Li, J.; Baranov, S.V.; Leronni, D.; Mihalik, A.C.; He, Y.; et al. Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc. Natl. Acad. Sci. USA 2017, 114, E7997–E8006. [Google Scholar] [CrossRef] [PubMed]
- Palmfeldt, J.; Bross, P. Proteomics of human mitochondria. Mitochondrion 2017, 33, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Kruse, R.; Hojlund, H. Mitochondrial phosphoproteomics of mammalian tissues. Mitochondrion 2017, 33, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, W. The mitochondrial acylome emerges: Proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Butow, R.A.; Avadhani, N.G. Mitochondrial signaling: The retrograde response. Mol. Cell 2004, 14, 1–15. [Google Scholar] [CrossRef]
- Guha, M.; Avadhani, N.G. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 2013, 13, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Jazwinski, S.M. The retrograde response: When mitochondrial quality control is not enough. Biochim. Biophys. Acta 2013, 1833, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Cagin, U.; Enriquez, J.A. The complex crosstalk between mitochondria and the nucleus: What goes on in between? Int. J. Biochem. Cell Biol. 2015, 63, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, C.A.; Newbold, J.E.; Potter, S.S. Maternal inheritance of mammalian mitochondrial DNA. Nature 1974, 251, 536–538. [Google Scholar] [CrossRef]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, H.; Hayashi, J.; Takahama, S.; Taya, C.; Lindahl, K.F.; Yonekawa, H. Elimination of paternal mitochondrial DNA in intraspecific crosses during early mouse embryogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 4542–4546. [Google Scholar] [CrossRef] [PubMed]
- Cummins, J. Mitochondrial DNA in mammalian reproduction. Rev. Reprod. 1998, 3, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Sutovsky, P.; Moreno, R.D.; Ramalho-Santos, J.; Dominko, T.; Simerly, C.; Schatten, G. Ubiquitinated sperm mitochondria, selective proteolysis, and the regulation of mitochondrial inheritance in mammalian embryos. Biol. Reprod. 2000, 63, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Sutovsky, P.; Van Leyen, K.; McCauley, T.; Day, B.N.; Sutovsky, M. Degradation of paternal mitochondria after fertilization: Implications for heteroplasmy, assisted reproductive technologies and mtDNA inheritance. Reprod. Biomed. Online 2004, 8, 24–33. [Google Scholar] [CrossRef]
- Al Rawi, S.; Louvet-Vallée, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011, 334, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Piko, L.; Matsumoto, L. Number of mitochondria and some properties of mitochondrial DNA in the mouse egg. Dev. Biol. 1976, 49, 1–10. [Google Scholar] [CrossRef]
- Motta, P.M.; Nottola, S.A.; Makabe, S.; Heyn, R. Mitochondrial morphology in human fetal and adult female germ cells. Hum. Reprod. 2000, 15 (Suppl. 2), 129–147. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.P.; Burton, G.J. Mitochondrial dysfunction in reproduction. Mitochondrion 2004, 4, 577–600. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K.; Coller, H.A.; Andre, P.C.; Li, X.C.; Hanekamp, J.S.; Thilly, W.G. Mitochondrial mutational spectra in human cells and tissues. Proc. Natl. Acad. Sci. USA 1997, 94, 13798–13803. [Google Scholar] [CrossRef] [PubMed]
- Faddy, M.J.; Gosden, R.G.; Gougeon, A.; Richardson, S.J.; Nelson, J.F. Accelerated disappearance of ovarian follicles in mid-life: Implications for forecasting menopause. Hum. Reprod. 1992, 7, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Szamatowicz, M.; Grochowski, D. Fertility and infertility in aging women. Gynecol. Endocrinol. 1998, 12, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Buckler, H. The menopause transition: Endocrine changes and clinical symptoms. J. Br. Menopause Soc. 2005, 11, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Oktem, O.; Oktay, K. The ovary: Anatomy and function throughout human life. Ann. N. Y. Acad. Sci. 2008, 1127, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Broekmans, F.J.; Soules, M.R.; Fauser, B.C. Ovarian aging: Mechanisms and clinical consequences. Endocr. Rev. 2009, 30, 465–493. [Google Scholar] [CrossRef] [PubMed]
- Tilly, J.L.; Sinclair, D.A. Germline energetics, aging and female infertility. Cell Metab. 2013, 17, 838–850. [Google Scholar] [CrossRef] [PubMed]
- Navot, D.; Bergh, P.A.; Williams, M.A.; Garrisi, G.J.; Guzman, I.; Sandler, B.; Grunfeld, L. Poor oocyte quality rather than implantation failure as a cause of age-related decline in female fertility. Lancet 1991, 337, 1375–1377. [Google Scholar] [CrossRef]
- Sauer, M.V.; Paulson, R.J.; Lobo, R.A. Pregnancy in women 50 or more years of age: Outcomes of 22 consecutively established pregnancies from oocyte donation. Fertil. Steril. 1995, 64, 111–115. [Google Scholar] [PubMed]
- Henderson, S.A.; Edwards, R.G. Chiasma frequency and maternal age in mammals. Nature 1968, 218, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Hook, E.B. Rates of chromosome abnormalities at different maternal ages. Obstet. Gynecol. 1981, 58, 282–285. [Google Scholar] [PubMed]
- Hassold, T.; Chiu, D. Maternal age-specific rates of numerical chromosome abnormalities with special reference to trisomy. Hum. Genet. 1985, 70, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, D.E.; Goodwin, P.; Klein, N.A.; Soules, M.R. Influence of maternal age on meiotic spindle assembly in oocytes from naturally cycling women. Hum. Reprod. 1996, 11, 2217–2222. [Google Scholar] [CrossRef] [PubMed]
- Tarín, J.J.; Pérez-Albalá, S.; Cano, A. Cellular and morphological traits of oocytes retrieved from aging mice after exogenous ovarian stimulation. Biol. Reprod. 2001, 65, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Eichenlaub-Ritter, U.; Vogt, E.; Yin, H.; Gosden, R. Spindles, mitochondria and redox potential in ageing oocytes. Reprod. Biomed. Online 2004, 8, 45–58. [Google Scholar] [CrossRef]
- Pan, H.; Ma, P.; Zhu, W.; Schultz, R.M. Age-associated increase in aneuploidy and changes in gene expression in mouse eggs. Dev. Biol. 2008, 316, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Hunt, P.A.; Hassold, T.J. Human female meiosis: What makes a good egg go bad? Trends Genet. 2008, 24, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Selesniemi, K.; Lee, H.-J.; Muhlhauser, A.; Tilly, J.L. Prevention of maternal aging-associated oocyte aneuploidy and meiotic spindle defects in mice by dietary and genetic strategies. Proc. Natl. Acad. Sci. USA 2011, 108, 12319–12324. [Google Scholar] [CrossRef] [PubMed]
- Merriman, J.A.; Jennings, P.C.; McLaughlin, E.A.; Jones, K.T. Effect of aging on superovulation efficiency, aneuploidy rates, and sister chromatid cohesion in mice aged up to 15 months. Biol. Reprod. 2012, 86, 49. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Cheng, J.; Hou, Y.; Zhu, S. The association between the oocyte pool and aneuploidy: A comparative study of the reproductive potential of young and aged mice. J. Assist. Reprod. Genet. 2014, 31, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Franasiak, J.M.; Forman, E.J.; Hong, K.H.; Werner, M.D.; Upham, K.M.; Treff, N.R.; Scott, R.J., Jr. The nature of aneuploidy with increasing age of the female partner: A review of 15,169 consecutive trophectoderm biopsies evaluated with comprehensive chromosomal screening. Fertil. Steril. 2014, 101, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Tarín, J.J.; Pérez-Albalá, S.; Cano, A. Oral antioxidants counteract the negative effects of female aging on oocyte quantity and quality in the mouse. Mol. Reprod. Dev. 2002, 61, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Selesniemi, K.; Lee, H.-J.; Tilly, J.L. Moderate caloric restriction initiated in rodents during adulthood sustains function of the female reproductive axis into advanced chronological age. Aging Cell 2008, 7, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Bentov, Y.; Yavorska, T.; Esfandiari, N.; Jurisicova, A.; Casper, R.F. The contribution of mitochondrial function to reproductive aging. J. Assist. Reprod. Genet. 2011, 28, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.C.; Tilly, J.L. Autologous germline mitochondrial energy transfer (AUGMENT) in human assisted reproduction. Semin. Reprod. Med. 2015, 33, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene 2002, 286, 81–89. [Google Scholar] [CrossRef]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yin, Y.; Ye, X.; Zeng, M.; Zhao, Q.; Keefe, D.L.; Liu, L. Resveratrol protects against age-associated infertility in mice. Hum. Reprod. 2013, 28, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Ben-Meir, A.; Burstein, E.; Borrego-Alvarez, A.; Chong, J.; Wong, E.; Yavorska, T.; Naranian, T.; Chi, M.; Wang, Y.; Bentov, Y.; et al. Coenzyme Q10 restores oocyte mitochondrial function and fertility during reproductive aging. Aging Cell 2015, 14, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; Li, S.; Lin, C.C. Effect of resveratrol and pterostilbene on aging and longevity. Biofactors 2018, 44, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Camacho, J.D.; Bernier, M.; López-Lluch, G.; Navas, P. Coenzyme Q10 supplementation in aging and disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Keefe, D.L.; Niven-Fairchild, T.; Powell, S.; Buradagunta, S. Mitochondrial deoxyribonucleic acid deletions in oocytes and reproductive aging in women. Fertil. Steril. 1995, 64, 577–583. [Google Scholar] [CrossRef]
- Hsieh, R.H.; Tsai, N.M.; Chang, S.J.; Wei, Y.H.; Tzeng, C.R. Multiple rearrangements of mitochondrial DNA in unfertilized human oocytes. Fertil. Steril. 2002, 77, 1012–1017. [Google Scholar] [CrossRef]
- Chan, C.C.; Liu, V.W.; Lau, E.Y.; Yeung, W.S.; Ng, E.H.; Ho, P.C. Mitochondrial DNA content and 4977 bp deletion in unfertilized oocytes. Mol. Hum. Reprod. 2005, 11, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Ashley, M.V.; Laipis, P.J.; Hauswirth, W.W. Rapid segregation of heteroplasmic bovine mitochondria. Nucl. Acids Res. 1989, 17, 7325–7331. [Google Scholar] [CrossRef] [PubMed]
- Jenuth, J.P.; Peterson, A.C.; Fu, K.; Shoubridge, E.A. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat. Genet. 1996, 14, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.P.; de Boer, K. The bottleneck: Mitochondrial imperatives in oogenesis and ovarian follicular fate. Mol. Cell. Endocrinol. 1998, 145, 81–88. [Google Scholar] [CrossRef]
- Stewart, J.B.; Freyer, C.; Elson, J.L.; Wredenberg, A.; Cansu, Z.; Trifunovic, A.; Larsson, N.-G. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 2008, 6, e10. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Teoli, D.; Shoubridge, E.A. The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat. Genet. 2008, 40, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Cree, L.M.; Samuels, D.C.; de Sousa Lopes, S.C.; Rajasimha, H.K.; Wonnapinij, P.; Mann, J.R.; Dahl, H.H.; Chinnery, P.F. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat. Genet. 2008, 40, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Rebolledo-Jaramillo, B.; Su, M.S.-W.; Stoler, N.; McElhoe, J.A.; Dickins, B.; Blankenberg, D.; Korneliussen, T.S.; Chiaromonte, F.; Nielsen, R.; Holland, M.M.; et al. Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 2014, 111, 15474–15479. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Larsson, N.-G. Keeping mtDNA in shape between generations. PLoS Genet. 2014, 10, e1004670. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Floros, V.I.; Pyle, A.; Dietmann, S.; Wei, W.; Tang, W.C.; Irie, N.; Payne, B.; Capalbo, A.; Noli, L.; Coxhead, J.; et al. Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. Nat. Cell Biol. 2018, 20, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Shitara, H.; Horii, T.; Nagao, Y.; Imai, H.; Abe, K.; Hara, T.; Hayashi, J.; Yonekawa, H. The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat. Genet. 2007, 39, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Samuels, D.C.; Wonnapinij, P.; Cree, L.M.; Chinnery, P.F. Reassessing evidence for a postnatal mitochondrial genetic bottleneck. Nat. Genet. 2010, 42, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Shoubridge, E.A. Reply: Reassessing evidence for a postnatal mitochondrial genetic bottleneck. Nat. Genet. 2010, 42, 472–473. [Google Scholar] [CrossRef]
- Lee, H.S.; Ma, H.; Juanes, R.C.; Tachibana, M.; Sparman, M.; Woodward, J.; Ramsey, C.; Xu, J.; Kang, E.J.; Amato, P.; et al. Rapid mitochondrial DNA segregation in primate preimplantation embryos precedes somatic and germline bottleneck. Cell Rep. 2012, 1, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.M.; Stewart, J.B.; Hagström, E.; Brené, S.; Mourier, A.; Coppotelli, G.; Freyer, C.; Lagouge, M.; Hoffer, B.J.; Olson, L.; et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 2013, 501, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.M.; Coppotelli, G.; Hoffer, B.J.; Olson, L. Maternally transmitted mitochondrial DNA mutations can reduce lifespan. Sci. Rep. 2014, 4, 6569. [Google Scholar] [CrossRef] [PubMed]
- Van Blerkom, J. Mitochondria in human oogenesis and preimplantation embryogenesis: Engines of metabolism, ionic regulation and developmental competence. Reproduction 2004, 128, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Van Blerkom, J.; Davis, P.; Mathwig, V.; Alexander, S. Domains of high-polarized and low-polarized mitochondria may occur in mouse and human oocytes and early embryos. Hum. Reprod. 2002, 17, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Van Blerkom, J.; Davis, P.; Alexander, S. Inner mitochondrial membrane potential (ΔΨm), cytoplasmic ATP content and free Ca2+ levels in metaphase II mouse oocytes. Hum. Reprod. 2003, 18, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Morimoto, N.; Yamanaka, M.; Matsumoto, H.; Yamochi, T.; Goto, H.; Inoue, M.; Nakaoka, Y.; Shibahara, H.; Morimoto, Y. Quantitative and qualitative changes of mitochondria in human preimplantation embryos. J. Assist. Reprod. Genet. 2017, 34, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.J.; Zernicka-Goetz, M. The acquisition of cell fate in mouse development: How do cells first become heterogeneous? Curr. Top. Dev. Biol. 2016, 117, 671–695. [Google Scholar] [PubMed]
- Mihajlovic, A.I.; Bruce, A.W. The first cell-fate decision of mouse preimplantation embryo development: Integrating cell position and polarity. Open Biol. 2017, 7, 170210. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Partin, J. Evidence for mitochondrial control of neuronal polarity. J. Neurosci. Res. 1999, 56, 8–20. [Google Scholar] [CrossRef]
- Fu, D.; Mitra, K.; Sengupta, P.; Jarnik, M.; Lippincott-Schwartz, J.; Arias, I.M. Coordinated elevation of mitochondrial oxidative phosphorylation and autophagy help drive hepatocyte polarity. Proc. Natl. Acad. Sci. USA 2013, 110, 7288–7293. [Google Scholar] [CrossRef] [PubMed]
- Diaz, G.; Setzu, H.; Zucca, A.; Isola, R.; Diana, A.; Murru, R.; Sogos, V.; Gremo, F. Subcellular heterogeneity of mitochondrial membrane potential: Relationship with organelle distribution and intercellular contacts in normal, hypoxic and apoptotic cells. J. Cell Sci. 1999, 112, 1077–1084. [Google Scholar] [PubMed]
- Reynier, P.; May-Panloup, P.; Chrétien, M.F.; Morgan, C.J.; Jean, M.; Savagner, F.; Barrière, P.; Malthièry, Y. Mitochondrial DNA content affects the fertilizability of human oocytes. Mol. Hum. Reprod. 2001, 7, 425–429. [Google Scholar] [CrossRef] [PubMed]
- May-Panloup, P.; Chrétien, M.F.; Jacques, C.; Vasseur, C.; Malthièry, Y.; Reynier, P. Low oocyte mitochondrial DNA content in ovarian insufficiency. Hum. Reprod. 2005, 20, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.A.; El Shourbagy, S.; St John, J.C. Mitochondrial content reflects oocyte variability and fertilization outcome. Fertil. Steril. 2006, 85, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Duran, H.E.; Simsek-Duran, F.; Oehninger, S.C.; Jones, H.W.; Castora, F.J. The association of reproductive senescence with mitochondrial quantity, function, and DNA integrity in human oocytes at different stages of maturation. Fertil. Steril. 2011, 96, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Murakoshi, Y.; Sueoka, K.; Takahashi, K.; Sato, S.; Sakurai, T.; Tajima, H.; Yoshimura, Y. Embryo developmental capability and pregnancy outcome are related to the mitochondrial DNA copy number and ooplasmic volume. J. Assist. Reprod. Genet. 2013, 30, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Simsek-Duran, F.; Li, F.; Ford, W.; Swanson, R.J.; Jones H.W., Jr.; Castora, F.J. Age-associated metabolic and morphologic changes in mitochondria of individual mouse and hamster oocytes. PLoS ONE 2013, 8, e64955. [Google Scholar] [CrossRef] [PubMed]
- Larrson, N.; Wang, J.; Wilhemsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.; Clayton, D. Mitochondria transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Thundathil, J.; Filion, F.; Smith, L.C. Molecular control of mitochondrial function in preimplantation mouse embryos. Mol. Reprod. Dev. 2005, 71, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Boudoures, A.L.; Saben, J.; Drury, A.; Scheaffer, S.; Modi, Z.; Zhang, W.; Moley, K.H. Obesity-exposed oocytes accumulate and transmit damaged mitochondria due to an inability to activate mitophagy. Dev. Biol. 2017, 426, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Kuma, A.; Mizushima, N. The role of autophagy during the oocyte-to-embryo transition. Autophagy 2008, 4, 1076–1078. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.K.; Shin, H.; Lim, H.J. Rapamycin influences the efficiency of in vitro fertilization and development in the mouse: A role for autophagic activation. Asian-Australas. J. Anim. Sci. 2016, 29, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; Spath, K.; Alfarawati, S.; Kaper, F.; Craig, A.; Michel, C.E.; Kokocinski, F.; Cohen, J.; Munné, S.; Wells, D. Altered levels of mitochondrial DNA are associated with female age, aneuploidy, and provide an independent measure of embryonic implantation potential. PLoS Genet. 2015, 11, e1005241. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, V.A.; Ludaway, T.; Russ, R.B.; Fields, E.J.; Koczor, C.; Lewis, W. Reproductive aging is associated with decreased mitochondrial abundance and altered structure in murine oocytes. J. Assist. Reprod. Genet. 2012, 29, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.; McCaffrey, C.; Grifo, J.; Morales, A.; Perloe, M.; Munné, S.; Wells, D.; Fragouli, E. Mitochondrial DNA quantification as a tool for embryo viability assessment: Retrospective analysis of data from single euploid transfers. Hum. Reprod. 2017, 32, 1282–1292. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; McCaffrey, C.; Ravichandran, K.; Spath, K.; Grifo, J.A.; Munné, S.; Wells, D. Clinical implications of mitochondrial DNA quantification on pregnancy outcomes: A blinded prospective non-selection study. Hum. Reprod. 2017, 32, 2340–2347. [Google Scholar] [CrossRef] [PubMed]
- Leese, H.J. Quiet please, do not disturb: A hypothesis of embryo metabolism and viability. Bioessays 2002, 9, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Scott, R.; Schimmel, T.; Levron, J.; Willadsen, S. Birth of an infant after transfer of anucleate donor oocyte cytoplasm into recipient eggs. Lancet 1997, 350, 186–187. [Google Scholar] [CrossRef]
- Cohen, J.; Scott, R.; Alikani, M.; Schimmel, T.; Munné, S.; Levron, J.; Wu, L.; Brenner, C.A.; Warner, C.; Willadsen, S. Ooplasmic transfer in mature human oocytes. Mol. Hum. Reprod. 1998, 4, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Lazendorf, S.E.; Mayer, J.F.; Toner, J.; Oehninger, S.; Saffan, D.S.; Muasher, S. Pregnancy following transfer of ooplasm from cryopreserved-thawed donor oocytes into recipient oocytes. Fertil. Steril. 1999, 71, 575–577. [Google Scholar] [CrossRef]
- Huang, C.C.; Cheng, T.C.; Chang, H.H.; Chang, C.C.; Chen, C.I.; Liu, J.; Lee, M.S. Birth after the injection of sperm and the cytoplasm of tripronucleate zygotes into metaphase II oocytes in patients with repeated implantation failure after assisted fertilization procedures. Fertil. Steril. 1999, 74, 702–706. [Google Scholar] [CrossRef]
- Dale, B.; Wilding, M.; Botta, G.; Rasile, M.; Marino, M.; Di Matteo, L.; De Placido, G.; Izzo, A. Pregnancy after cytoplasmic transfer in a couple suffering from idiopathic infertility: Case report. Hum. Reprod. 2001, 16, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Barritt, J.A.; Brenner, C.A.; Malter, H.E.; Cohen, J. Mitochondria in human offspring derived from ooplasmic transplantation. Hum. Reprod. 2001, 16, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.A.; Barritt, J.A.; Willadsen, S.; Cohen, J. Mitochondrial DNA heteroplasmy after human ooplasmic transplantation. Fertil. Steril. 2000, 74, 573–578. [Google Scholar] [CrossRef]
- Barritt, J.; Willadsen, S.; Brenner, C.; Cohen, J. Cytoplasmic transfer in assisted reproduction. Hum. Reprod. Update 2001, 7, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Zoon, K.C. Human cells used in therapy involving the transfer of genetic material by means other than the union of gamete nuclei. Available online: https://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ucm2007205.htm (accessed on 20 May 2018).
- El Shourbagy, S.H.; Spikings, E.C.; Freitas, M.; St John, J.C. Mitochondria directly influence fertilisation outcome in the pig. Reproduction 2006, 131, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.C.; Chen, M.J.; Ho, J.Y.P.; Guu, H.F.; Ho, E.S. Mitochondrial transfer can enhance the murine embryo development. J. Assist. Reprod. Genet. 2007, 24, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.H.; El Shmoury, M.; Szeptycki, J.; dela Cruz, D.B.; Lux, C.; Verjee, S.; Burgess, C.M.; Cohn, G.M.; Casper, R.F. The AUGMENTSM treatment: Physician reported outcomes of the initial global patient experience. JFIV Reprod. Med. Genet. 2015, 3, 154. [Google Scholar] [CrossRef]
- Oktay, K.; Baltaci, V.; Sonmezer, M.; Turan, V.; Unsal, E.; Baltaci, A.; Aktuna, S.; Moy, F. Oogonial precursor cell-derived autologous mitochondria injection (AMI) to improve outcomes in women with multiple IVF failures due to low oocyte quality: A clinical translation. Reprod. Sci. 2015, 22, 1612–1617. [Google Scholar] [CrossRef] [PubMed]
- Cagnone, G.L.; Tsai, T.S.; Makanji, Y.; Matthews, P.; Gould, J.; Bonkowski, M.S.; Elgass, K.D.; Wong, A.S.; Wu, L.E.; St John, J.C. Restoration of normal embryogenesis by mitochondrial supplementation in pig oocytes exhibiting mitochondrial DNA deficiency. Sci. Rep. 2016, 6, 23229. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-B.; Hao, J.-X.; Meng, T.-G.; Guo, L.; Dong, M.-Z.; Fan, L.-H.; Ouyang, Y.-C.; Wang, G.; Sun, Q.-Y.; Ou, X.-H.; et al. Transfer of autologous mitochondria from adipose tissue-derived stem cells rescues oocyte quality and infertility in aged mice. Aging 2017, 9, 2480–2488. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, H.; Takahashi, T.; Abe, H.; Nakano, H.; Nakajima, O.; Nagase, S. Poor embryo development in post-ovulatory in vivo-aged mouse oocytes is associated with mitochondrial dysfunction, but mitochondrial transfer from somatic cells is not sufficient for rejuvenation. Hum. Reprod. 2016, 31, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wen, B.; Zhao, H.; Ouyang, N.; Ou, S.; Wang, W.; Han, J.; Yang, D. Embryo development after mitochondrial supplementation from induced pluripotent stem cells. J. Assist. Reprod. Genet. 2017, 34, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Cherry, A.B.C.; Daley, G.Q. Reprogramming and cellular identity for regenerative medicine. Cell 2012, 148, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. Japanese woman is first recipient of next-generation stem cells. Nature 2014. [Google Scholar] [CrossRef]
- Cyranoski, D. Stem cells cruise to clinic: Japanese study of induced pluripotent stem cells aims to demonstrate safety in humans. Nature 2015, 494, 413. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous induced stem-cell–derived retinal cells for macular degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. Japanese man is first to receive ‘reprogrammed’ stem cells from another person. Nature 2017. [Google Scholar] [CrossRef]
- Hayashi, K.; Ohta, H.; Kurimoto, K.; Aramaki, S.; Saitou, M. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 2011, 146, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Ogushi, S.; Kurimoto, K.; Shimamoto, S.; Ohta, H.; Saitou, M. Offspring from oocytes derived from in vitro primordial germ cell-like cells in mice. Science 2012, 338, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, M.; Wang, X.; Fu, R.; Wan, H.; Xie, M.; Liu, M.; Guo, X.; Zheng, Y.; Feng, G.; et al. Complete meiosis from embryonic stem cell-derived germ cells in vitro. Cell Stem Cell 2016, 18, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Hikabe, O.; Hamazaki, N.; Nagamatsu, G.; Obata, Y.; Hirao, Y.; Hamada, N.; Shimamoto, S.; Imamura, T.; Nakashima, K.; Saitou, M.; et al. Reconstitution in vitro of the entire cycle of the mouse female germ line. Nature 2016, 539, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Panula, S.; Medrano, J.V.; Kee, K.; Bergström, R.; Nguyen, H.N.; Byers, B.; Wilson, K.D.; Wu, J.C.; Simon, C.; Hovatta, O.; et al. Human germ cell differentiation from fetal- and adult-derived induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Irie, N.; Weinberger, L.; Tang, W.W.C.; Kobayashi, T.; Viukov, S.; Manor, Y.S.; Dietman, S.; Hanna, J.H.; Surani, M.A. SOX17 is a critical specifier of human primordial germ cell fate. Cell 2015, 160, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. Rudimentary egg and sperm cells made from stem cells. Nature 2014. [Google Scholar] [CrossRef]
- Cyranoski, D. Mouse eggs made from skin cells in a dish. Nature 2016, 538, 301. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.G.; Daley, G.Q.; Adashi, E.Y. Disruptive reproductive technologies. Sci. Transl. Med. 2017, 9, eaag2959. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.E.J.; Gilchrist, R.B. Are human oocytes from stem cells next? Nat. Biotechnol. 2016, 34, 1247–1248. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Wang, X.; Tippner-Hedges, R.; Ma, H.; Folmes, C.; Gutierrez, N.M.; Lee, Y.; Van Dyken, C.; Ahmed, R.; Li, Y.; et al. Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human iPSCs. Cell Stem Cell 2016, 18, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Zheng, X.; Ma, L.; Kutty, G.; Gogineni, E.; Sun, Q.; Sherman, B.T.; Hu, X.; Jones, K.; Raley, C.; et al. A benchmark study on error assessment and quality control of CCS reads derived from the PacBio RS. J. Data Min. Genom. Proteom. 2013, 4, 16008. [Google Scholar] [CrossRef] [PubMed]
- Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Rev. Genet. 2018, 19, 269–285. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woods, D.C.; Khrapko, K.; Tilly, J.L. Influence of Maternal Aging on Mitochondrial Heterogeneity, Inheritance, and Function in Oocytes and Preimplantation Embryos. Genes 2018, 9, 265. https://doi.org/10.3390/genes9050265
Woods DC, Khrapko K, Tilly JL. Influence of Maternal Aging on Mitochondrial Heterogeneity, Inheritance, and Function in Oocytes and Preimplantation Embryos. Genes. 2018; 9(5):265. https://doi.org/10.3390/genes9050265
Chicago/Turabian StyleWoods, Dori C., Konstantin Khrapko, and Jonathan L. Tilly. 2018. "Influence of Maternal Aging on Mitochondrial Heterogeneity, Inheritance, and Function in Oocytes and Preimplantation Embryos" Genes 9, no. 5: 265. https://doi.org/10.3390/genes9050265
APA StyleWoods, D. C., Khrapko, K., & Tilly, J. L. (2018). Influence of Maternal Aging on Mitochondrial Heterogeneity, Inheritance, and Function in Oocytes and Preimplantation Embryos. Genes, 9(5), 265. https://doi.org/10.3390/genes9050265