Transcriptome Analysis of Two Rice Varieties Contrasting for Nitrogen Use Efficiency under Chronic N Starvation Reveals Differences in Chloroplast and Starch Metabolism-Related Genes

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatment
2.2. Plant Phenotyping and Enzyme Assay
2.3. RNA Extraction and Sequencing
2.4. RNA Sequencing Data Processing, Read Alignment, and Analysis
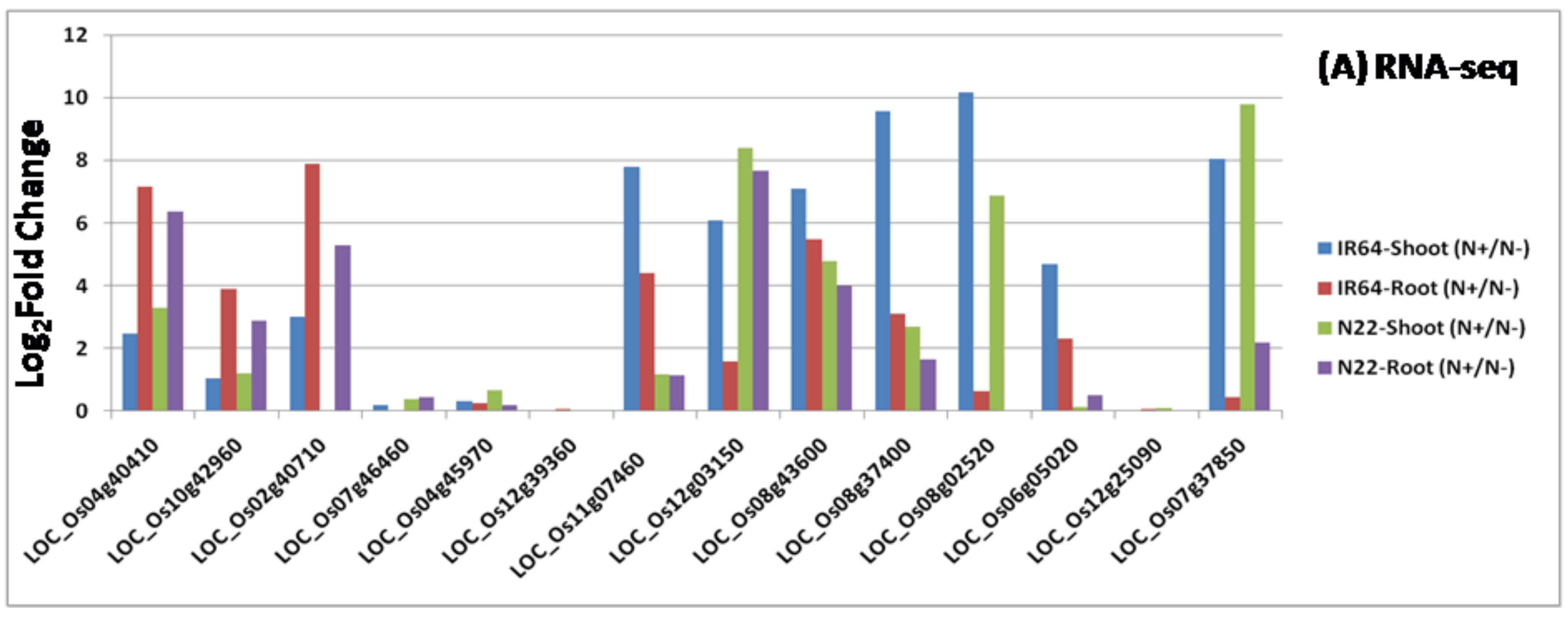
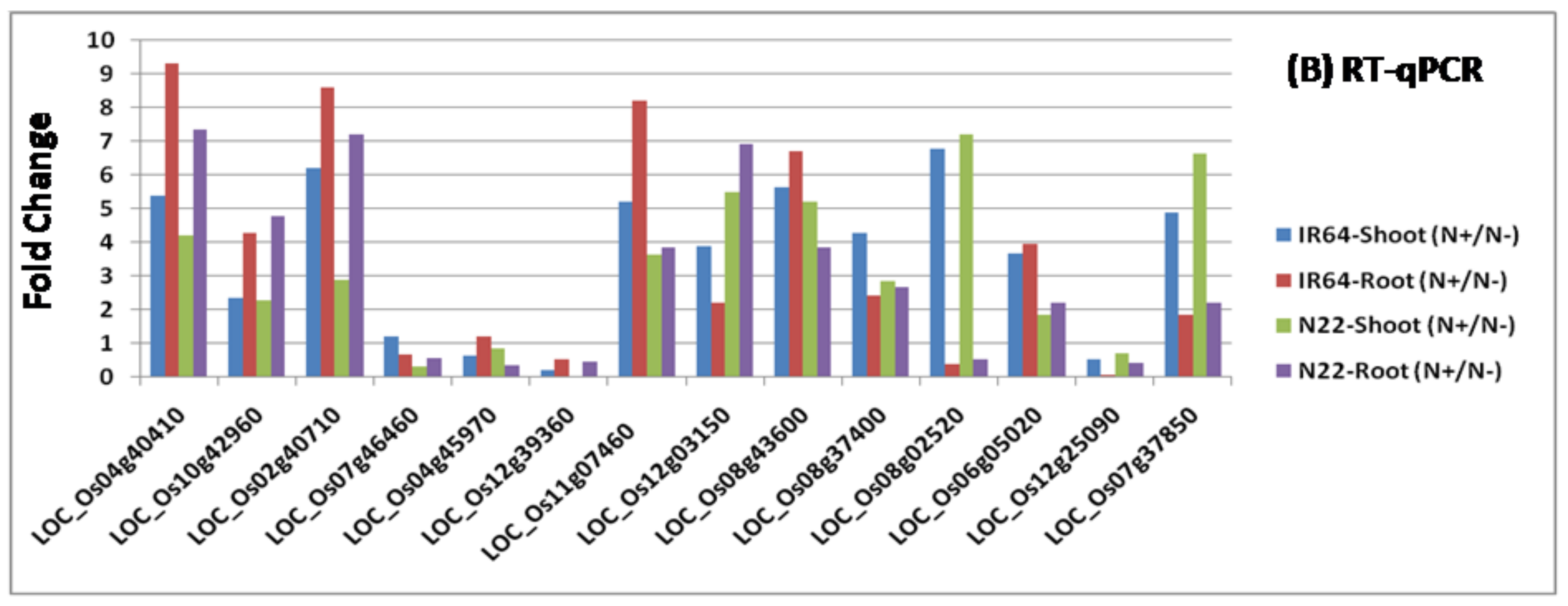
2.5. Validation of RNA Sequencing Results Using Real Time Reverse Transcription Polymerase Chain Reaction
2.6. Co-Localization of Differentially Expressed Genes Identified with Quantitative Trait Loci for Nitrogen Use Efficiency
3. Results
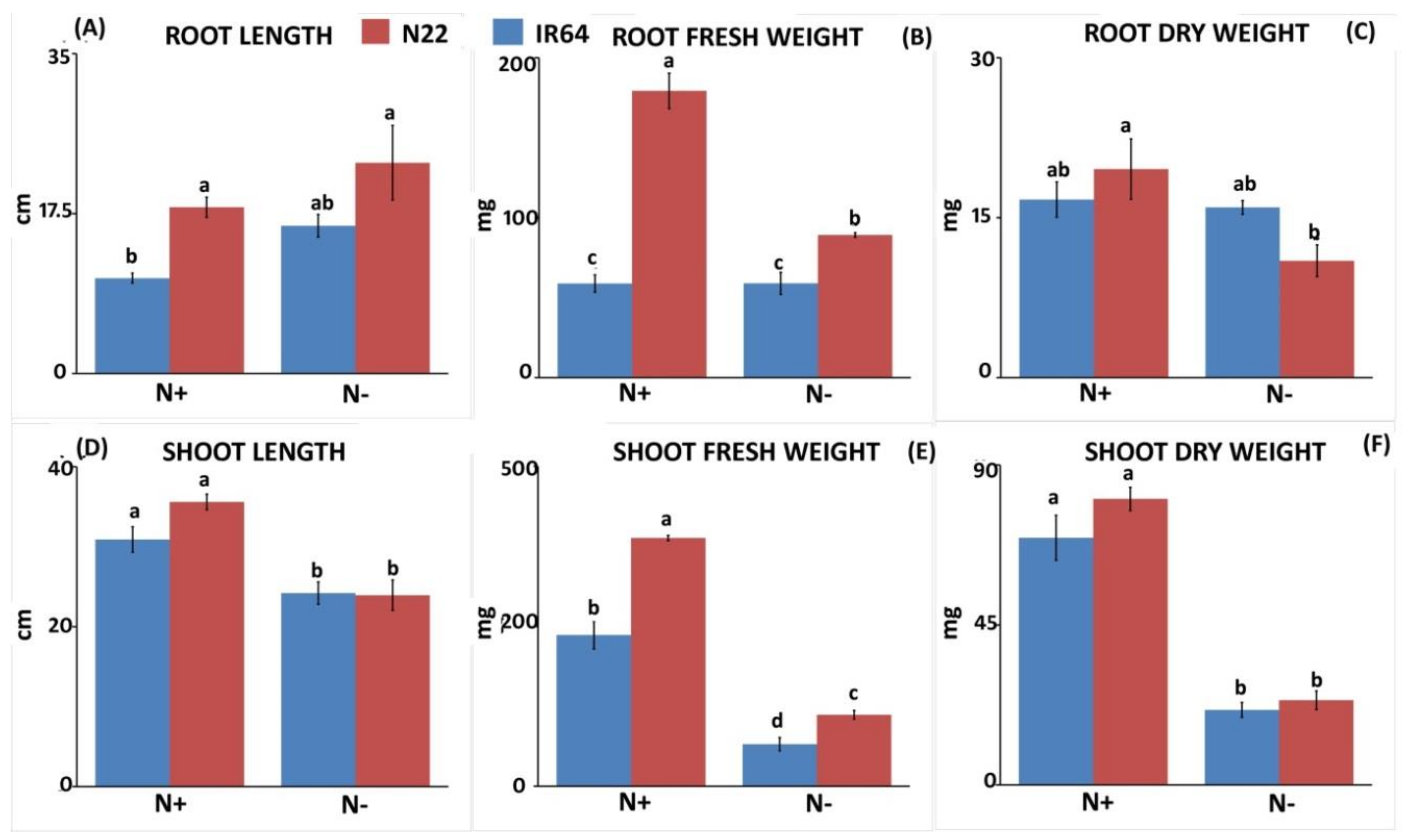
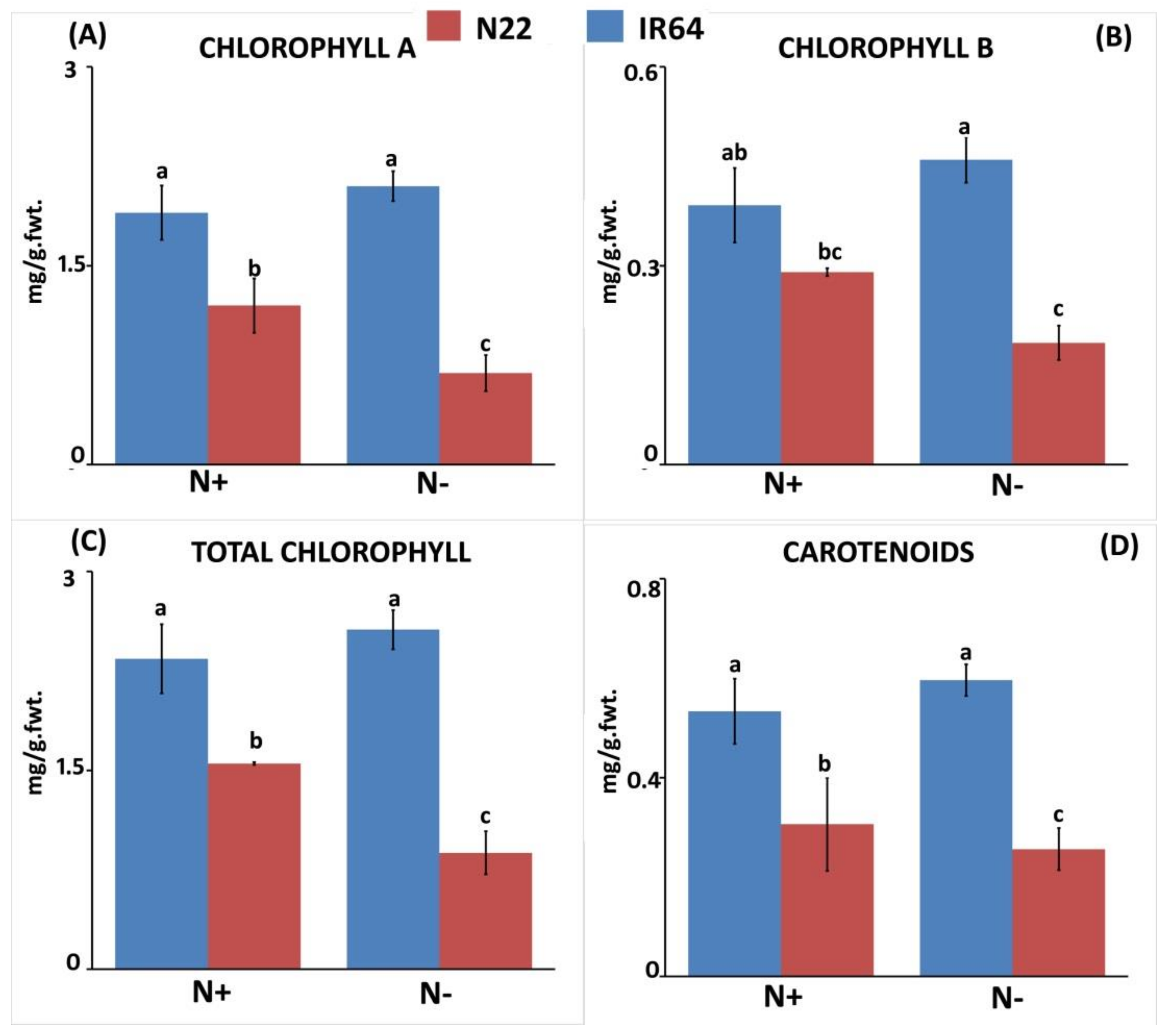
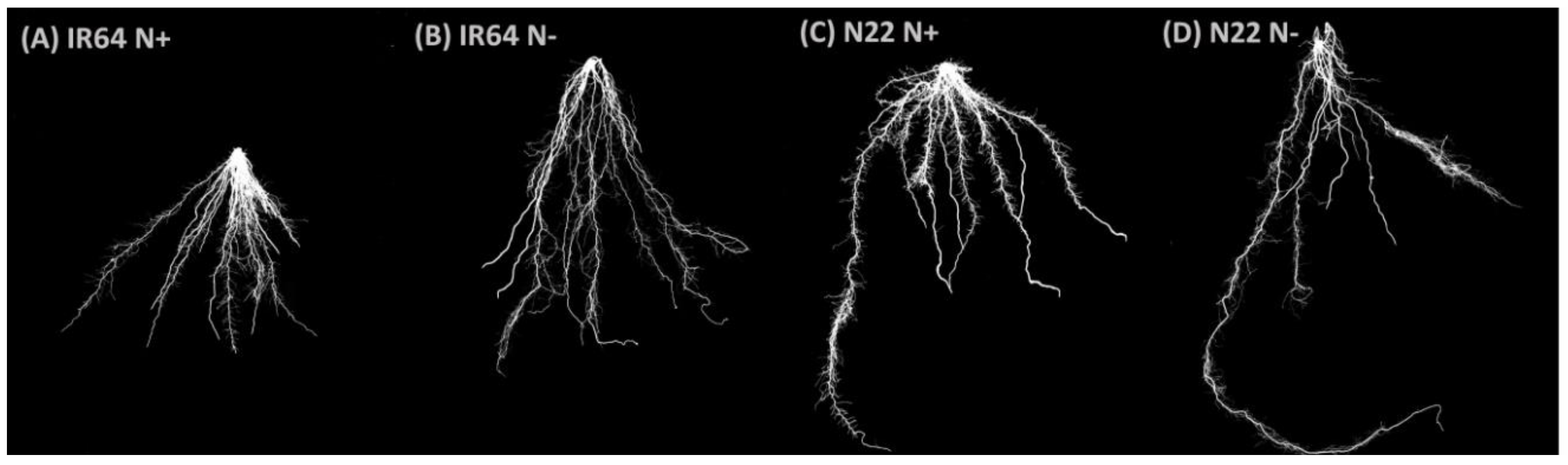
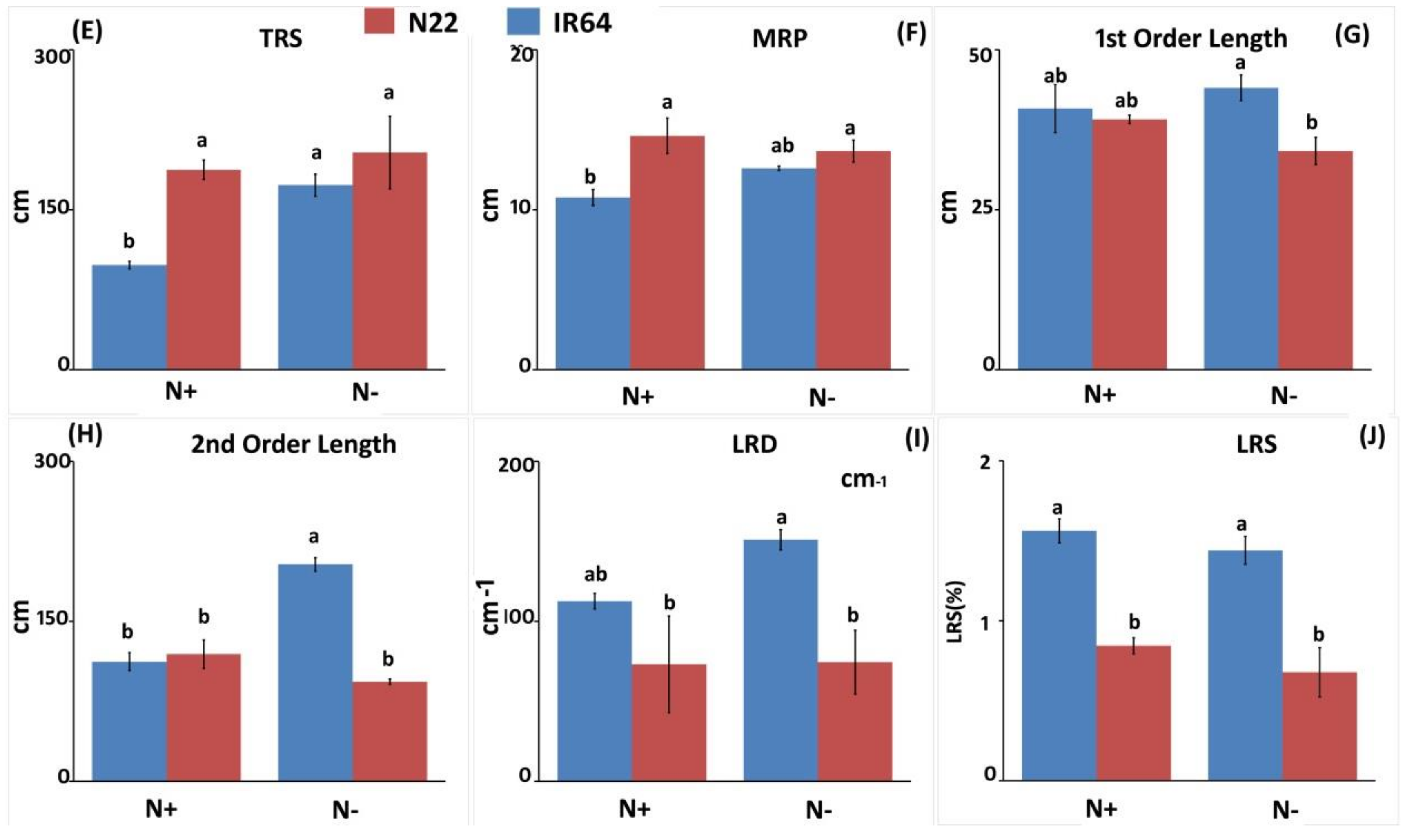
3.1. Plant Phenotyping and Enzyme Assay
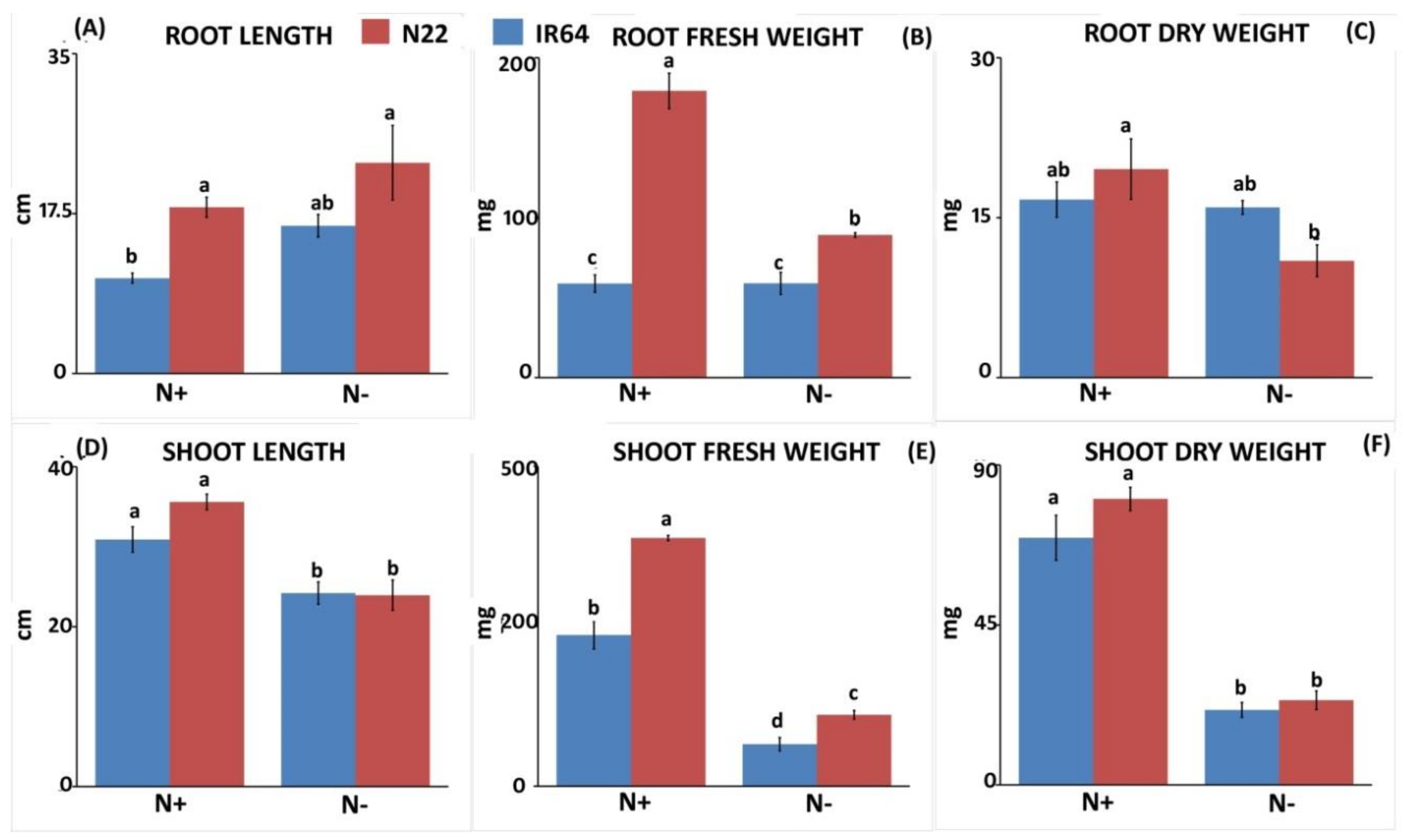
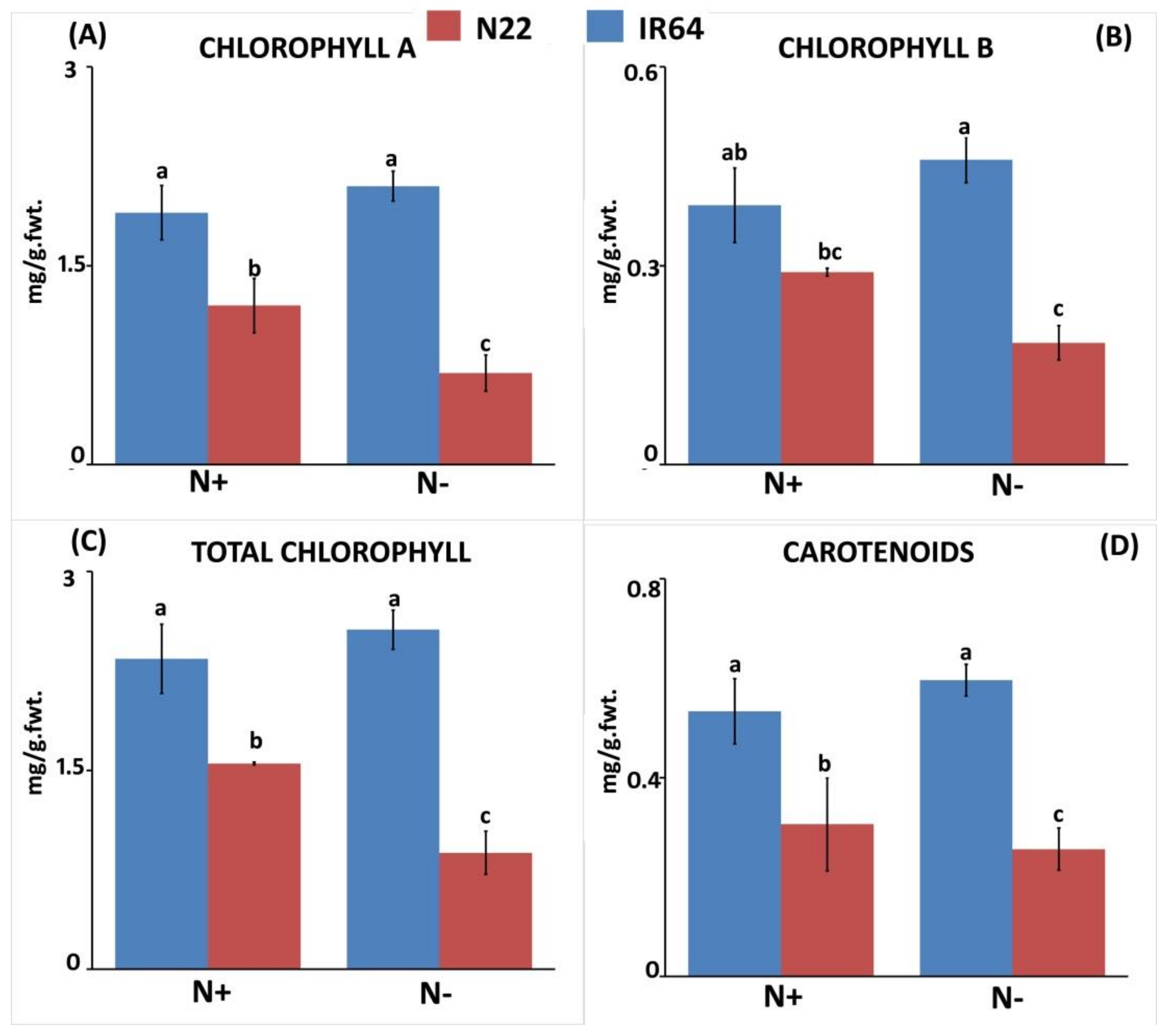
3.1.1. Effect of N Stress on Biomass and Chlorophyll Pigment
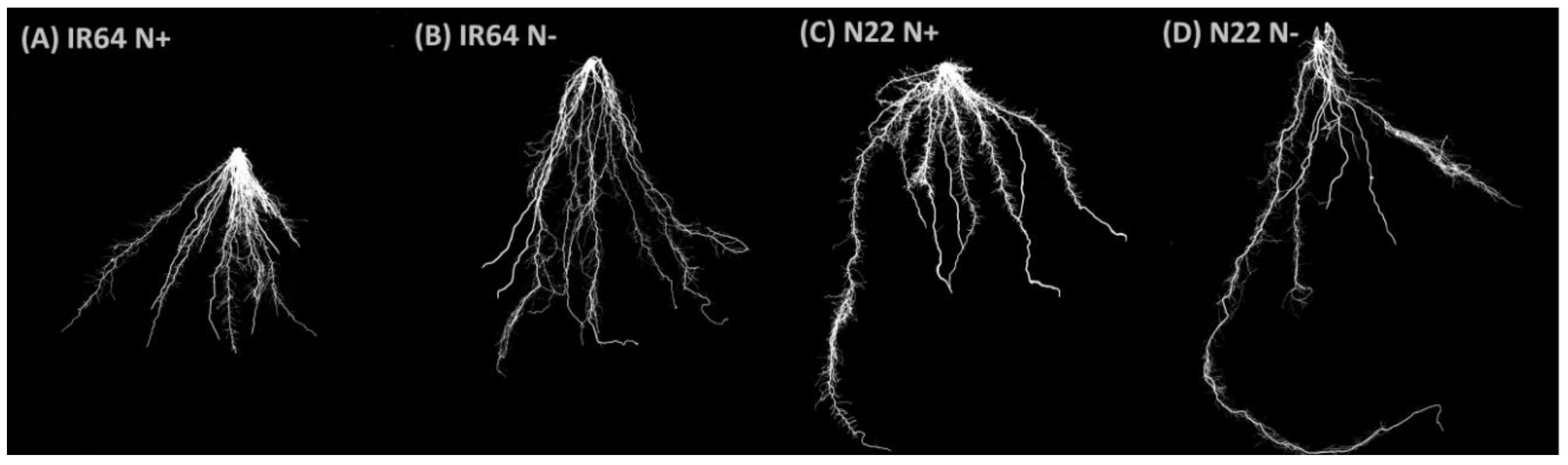
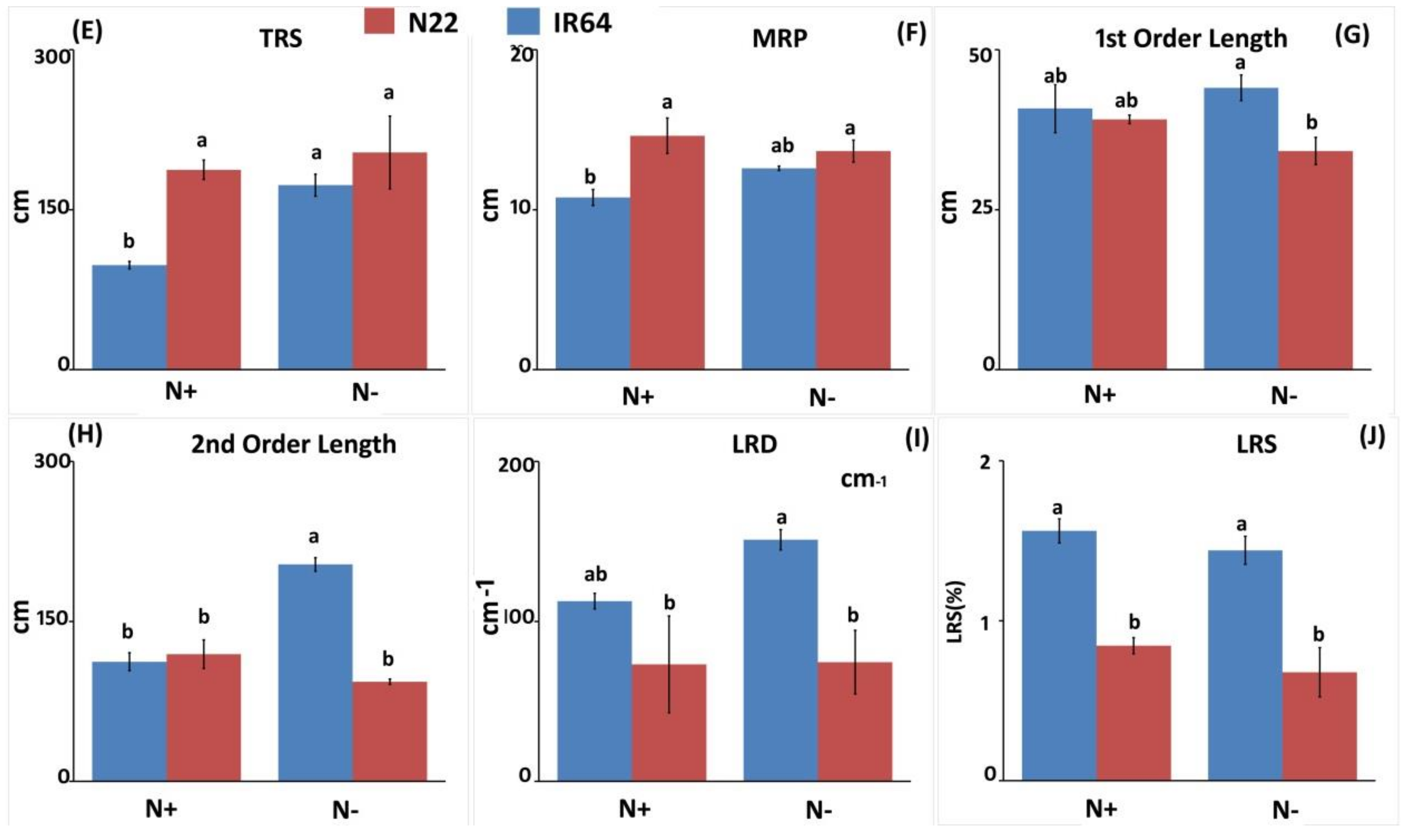
3.1.2. Effect of N Stress on Root System Architecture
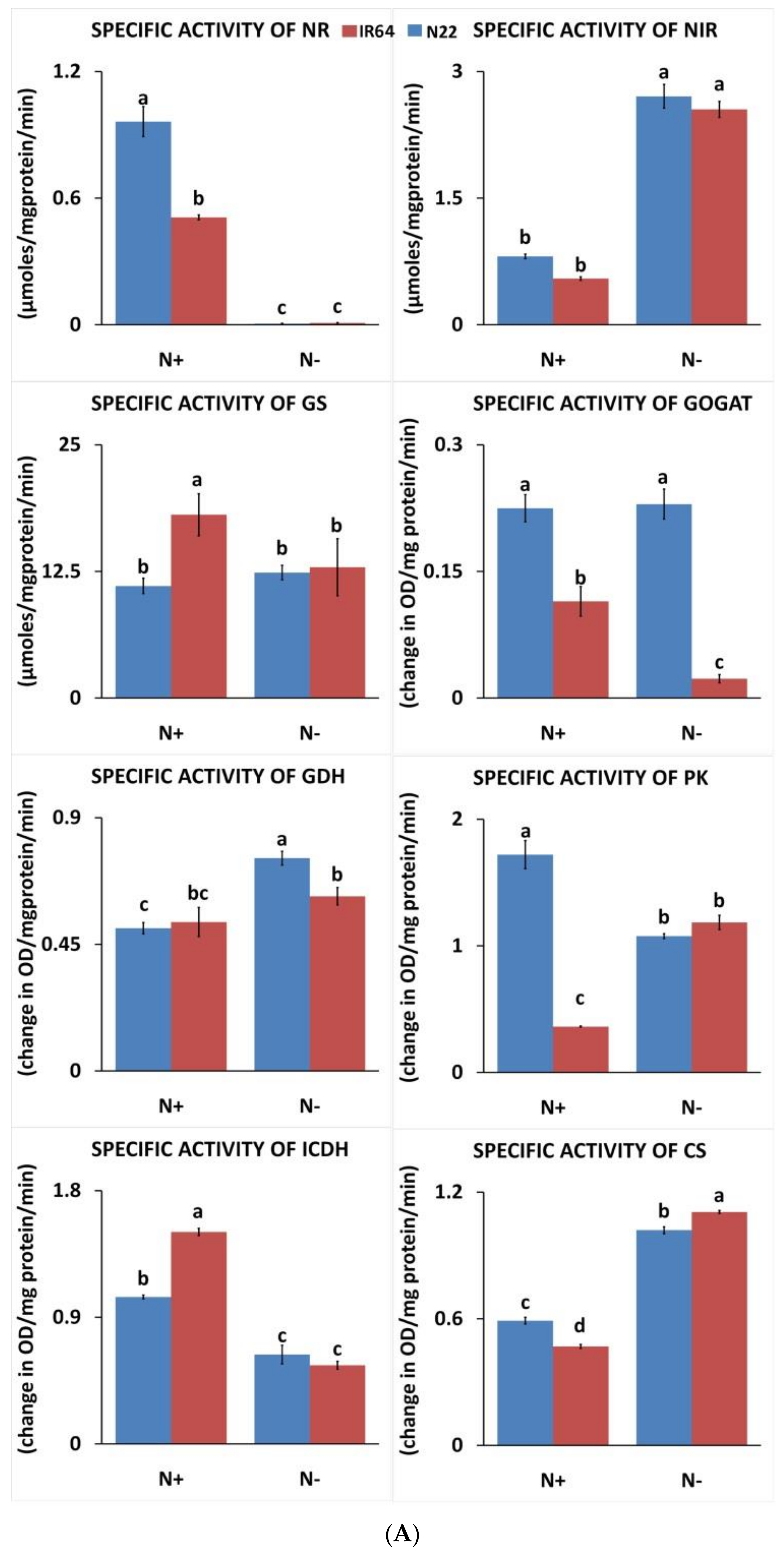
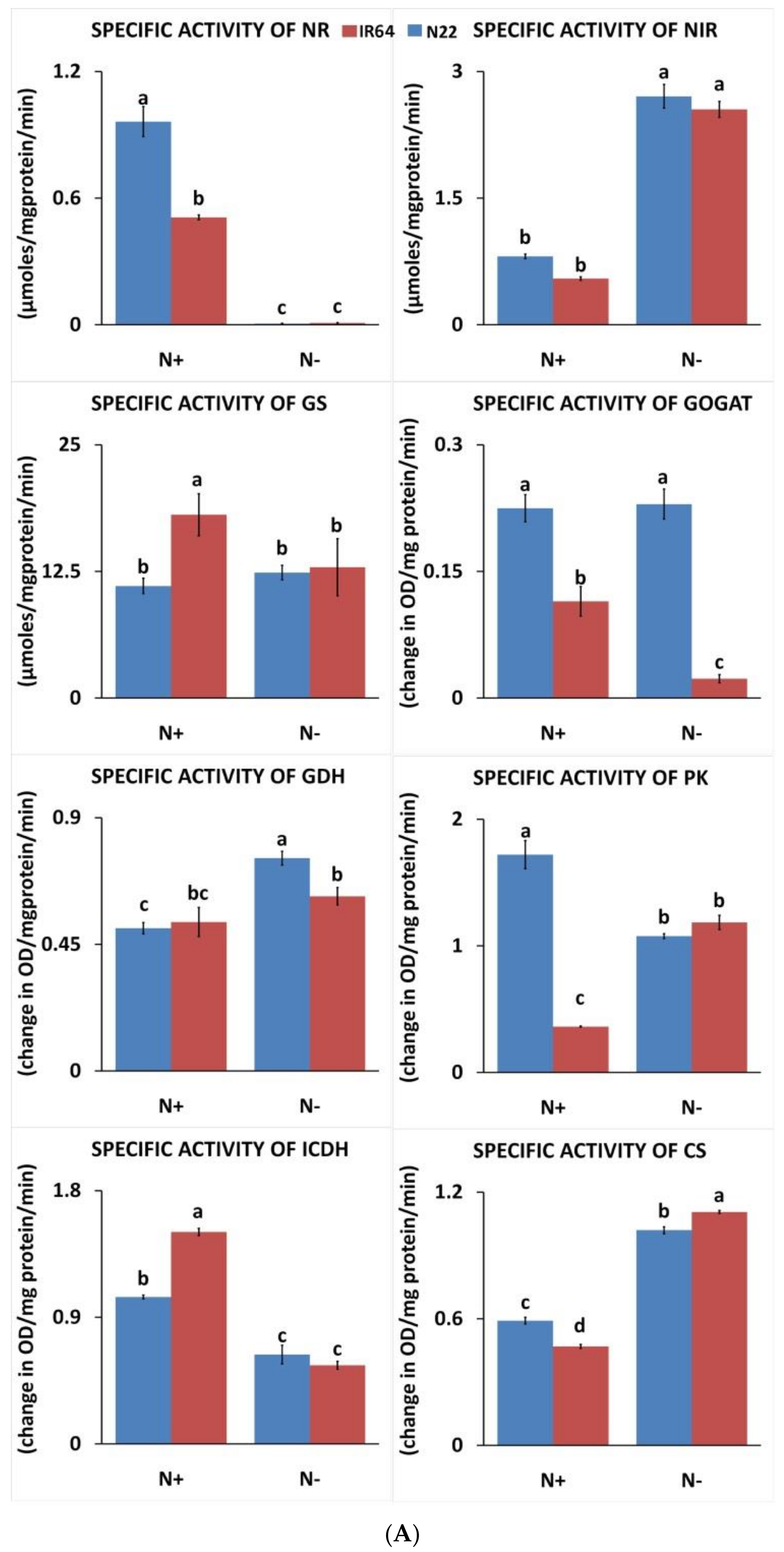
3.1.3. Effect of N Stress on Carbon and Nitrogen Metabolizing Enzymes
3.2. RNA Sequencing Data Analysis: Quality Control, Assembly, and Mapping
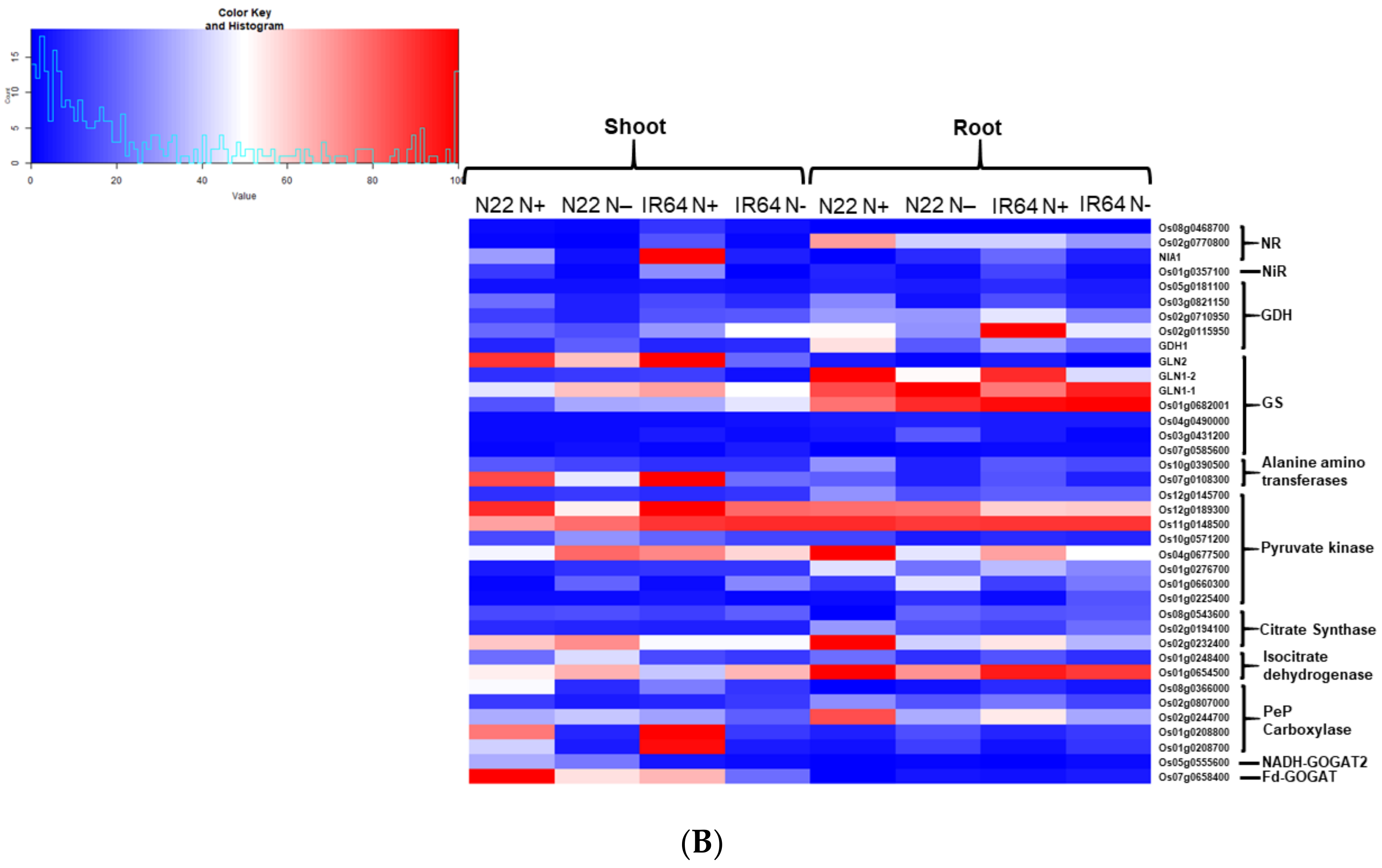
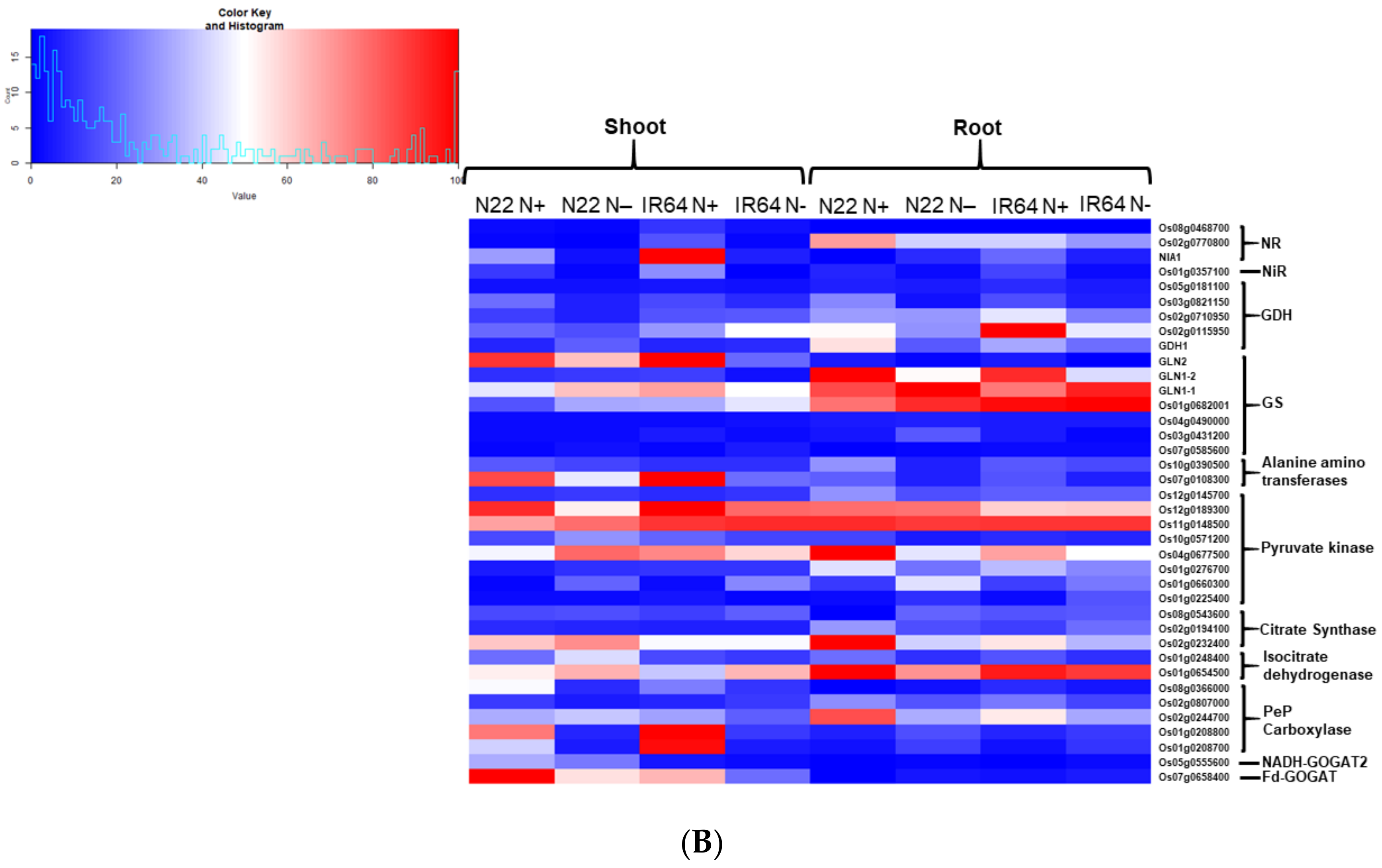
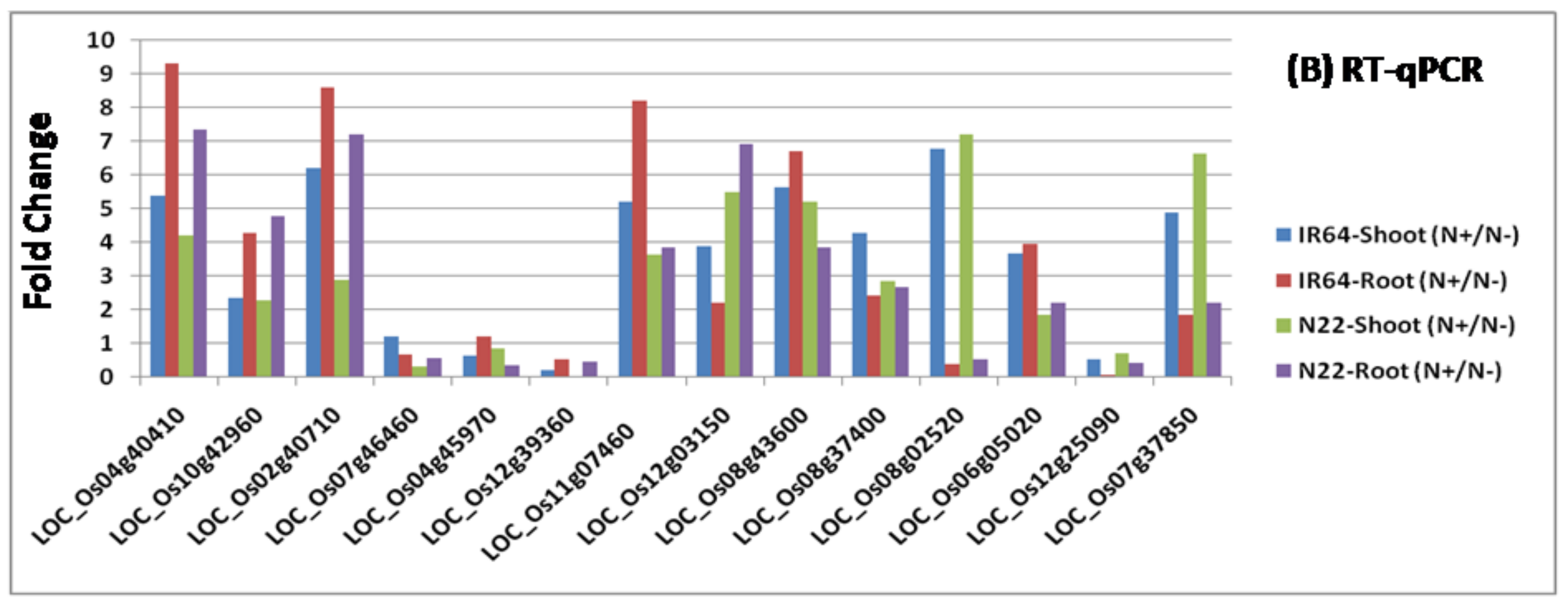
3.3. Identification of Differentially Expressed Genes and Their Validation
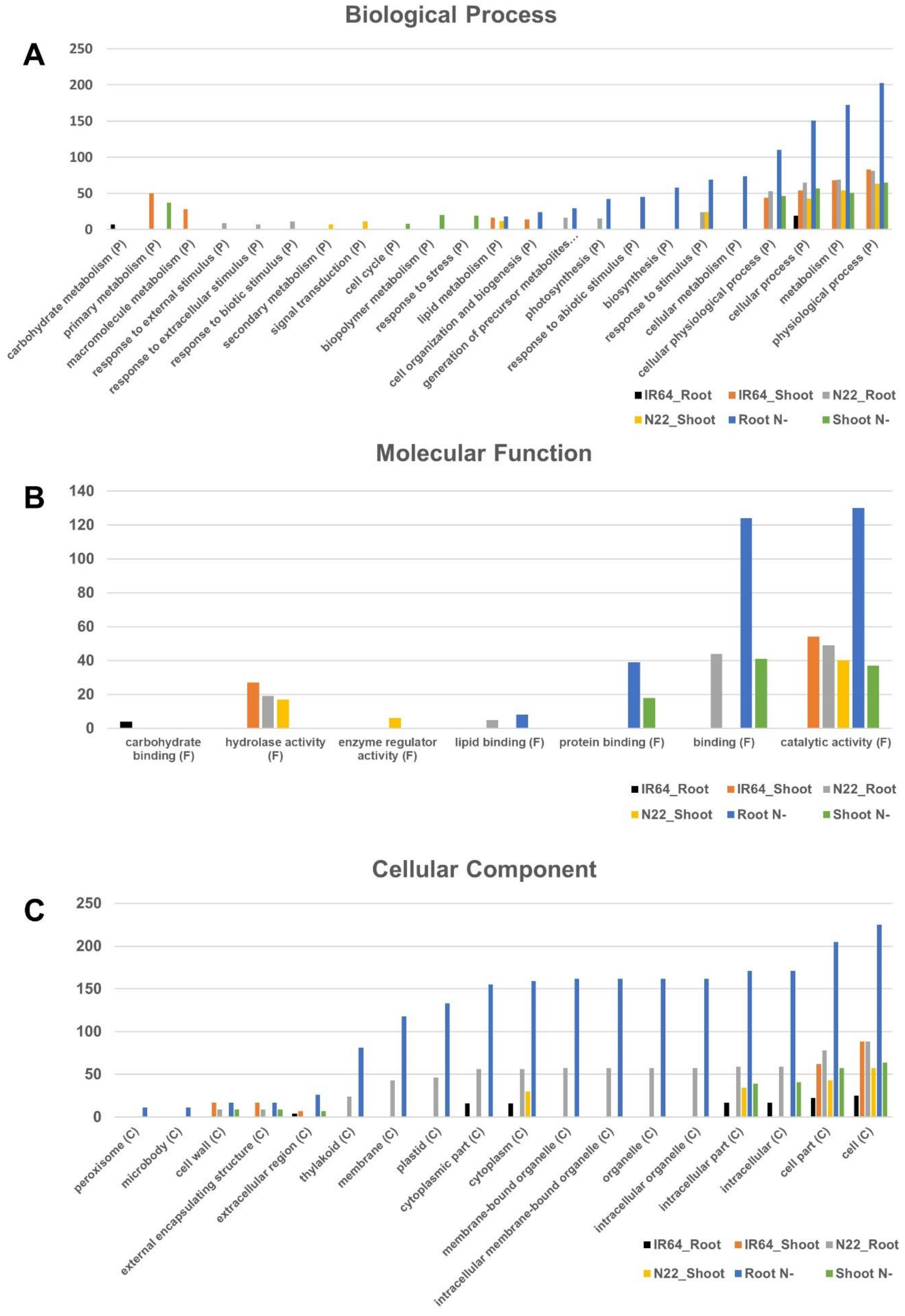
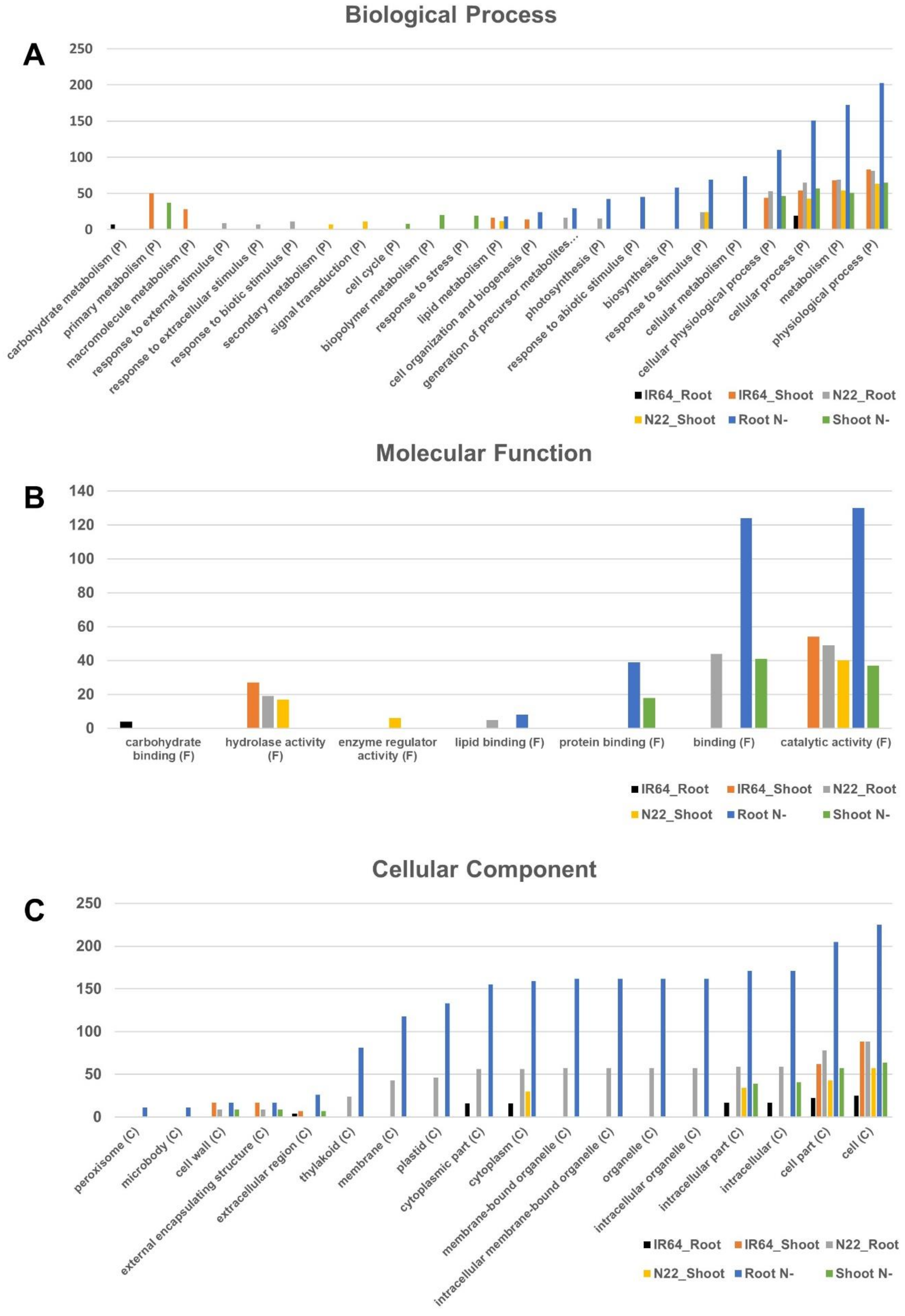
3.4. Gene Ontology Enrichment Analysis for Differentially Expressed Genes
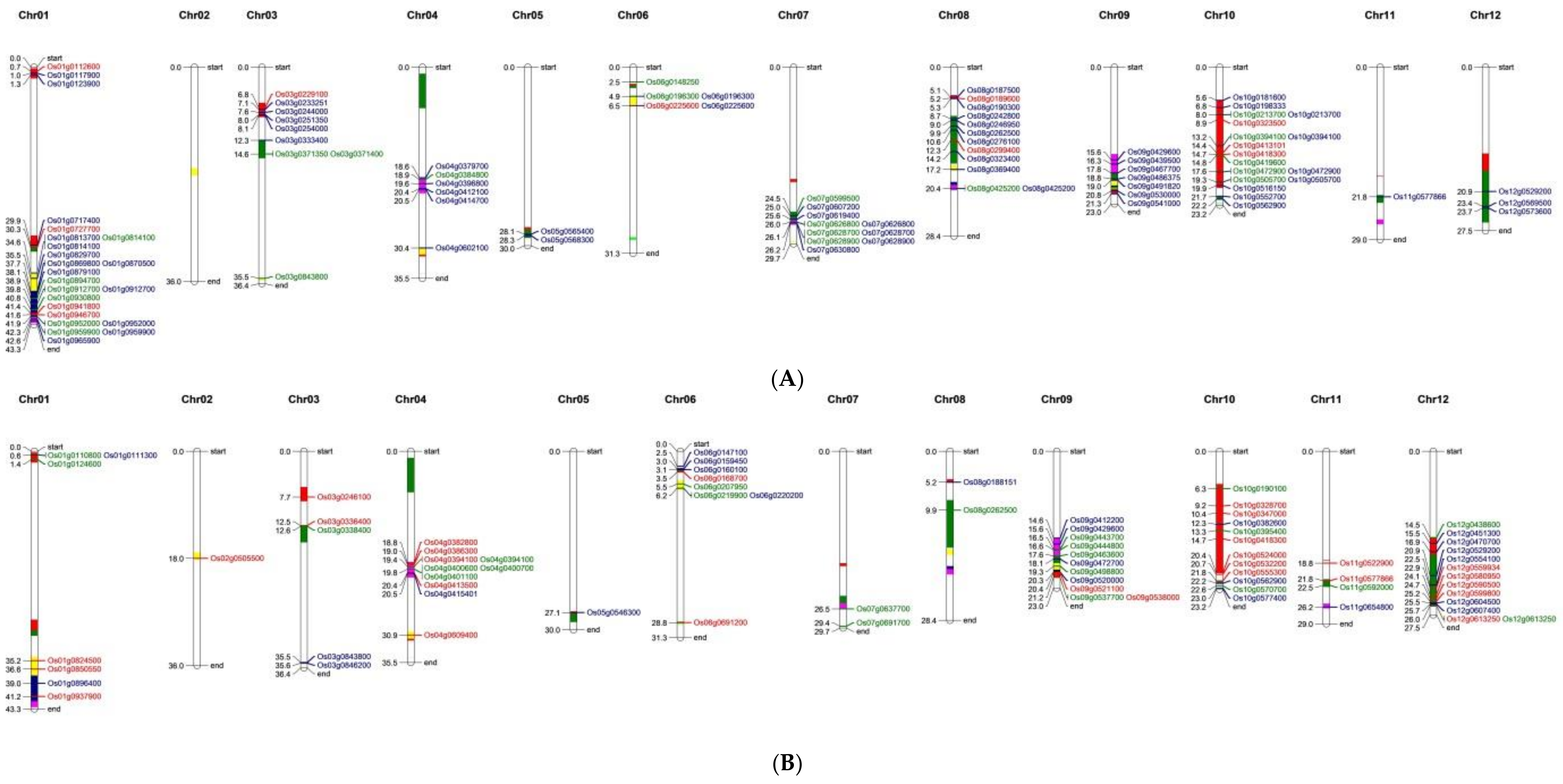
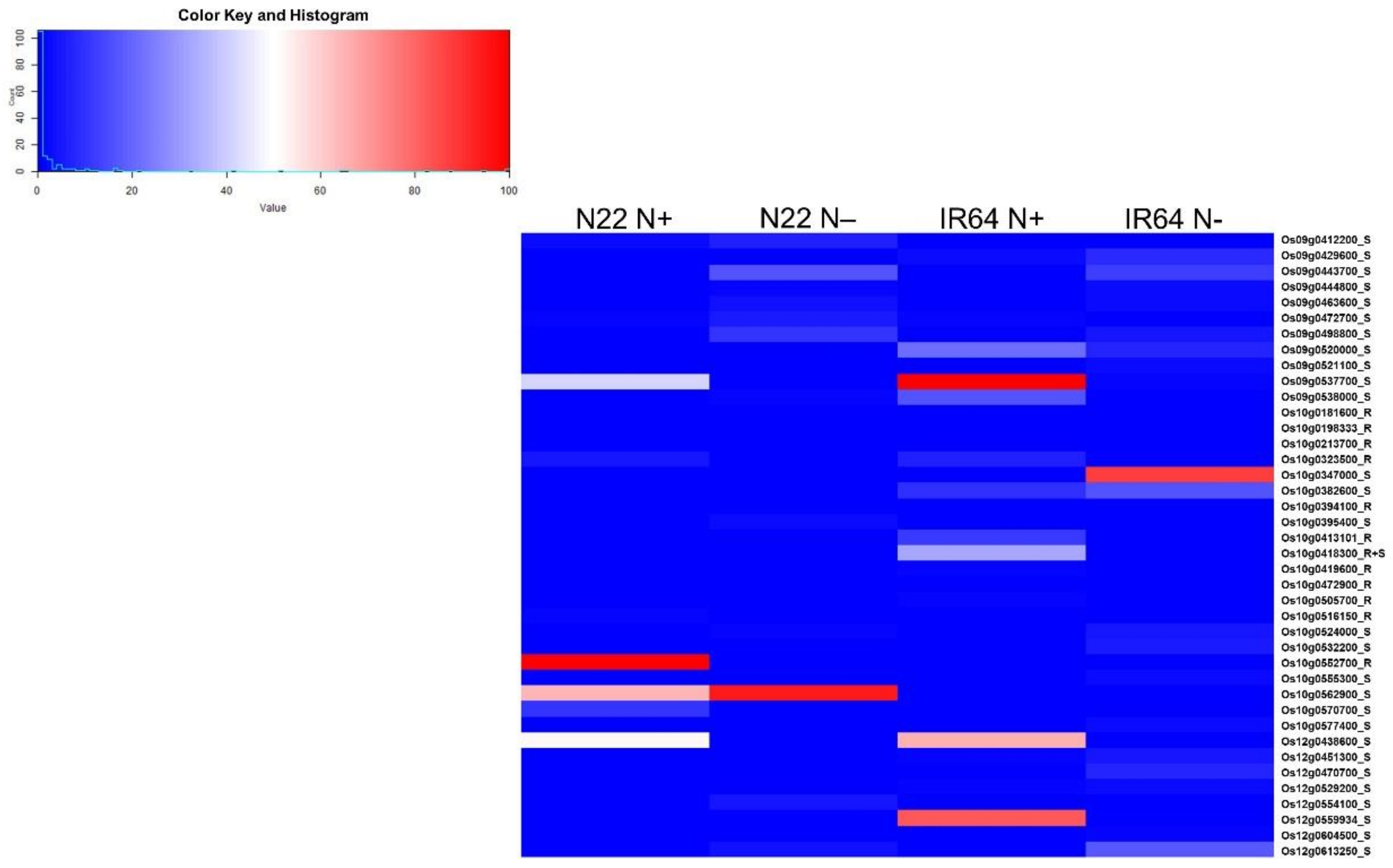
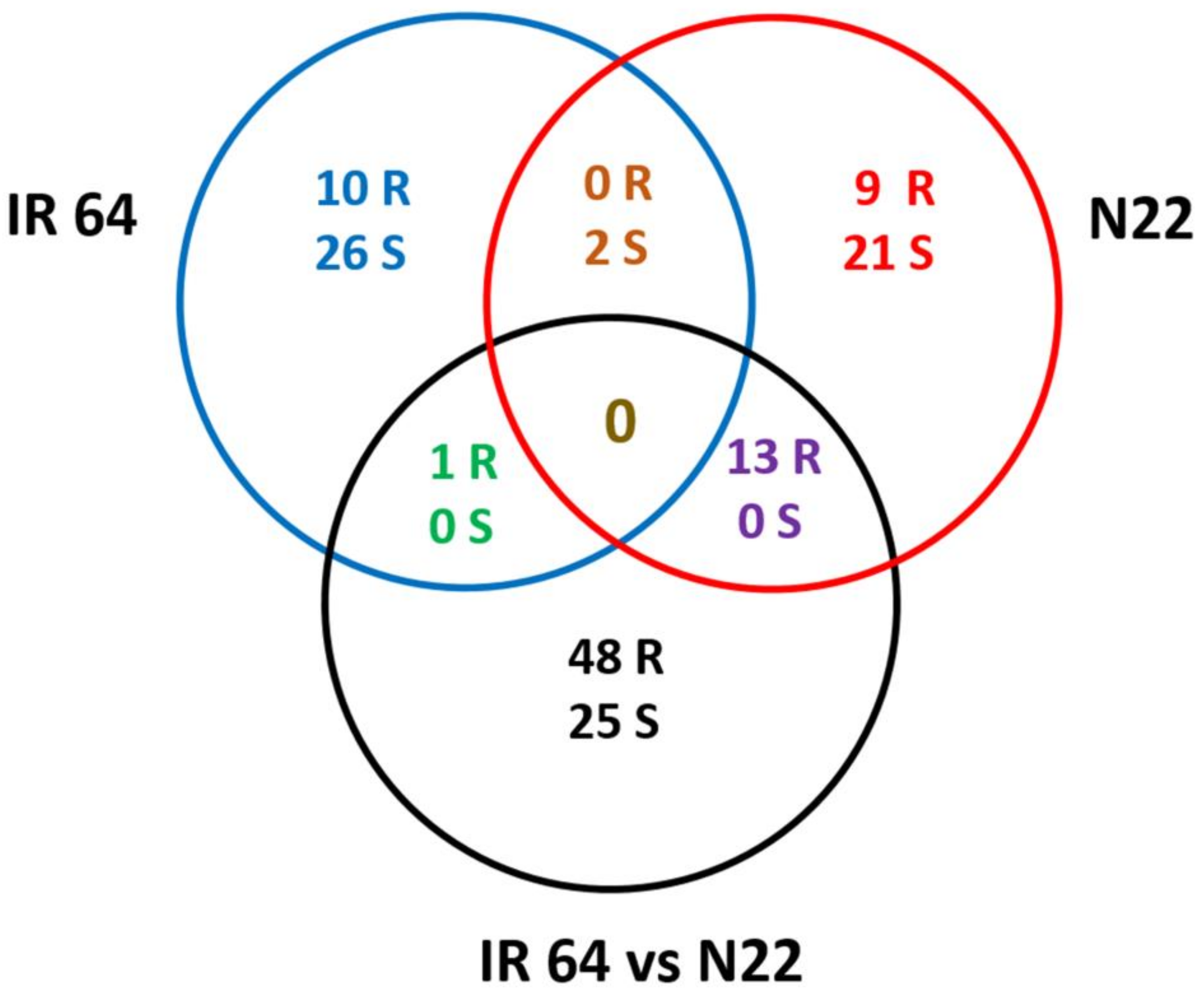
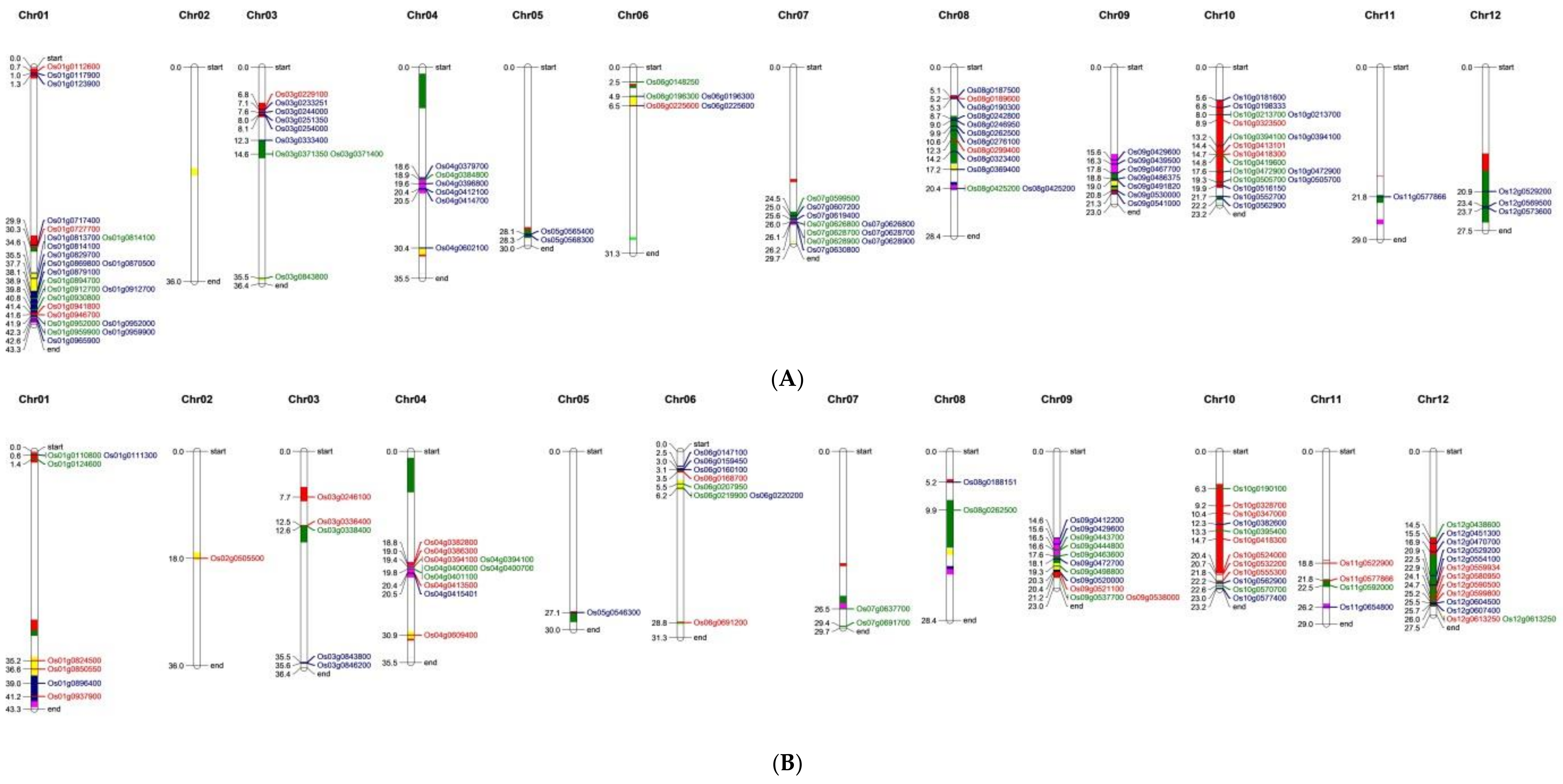
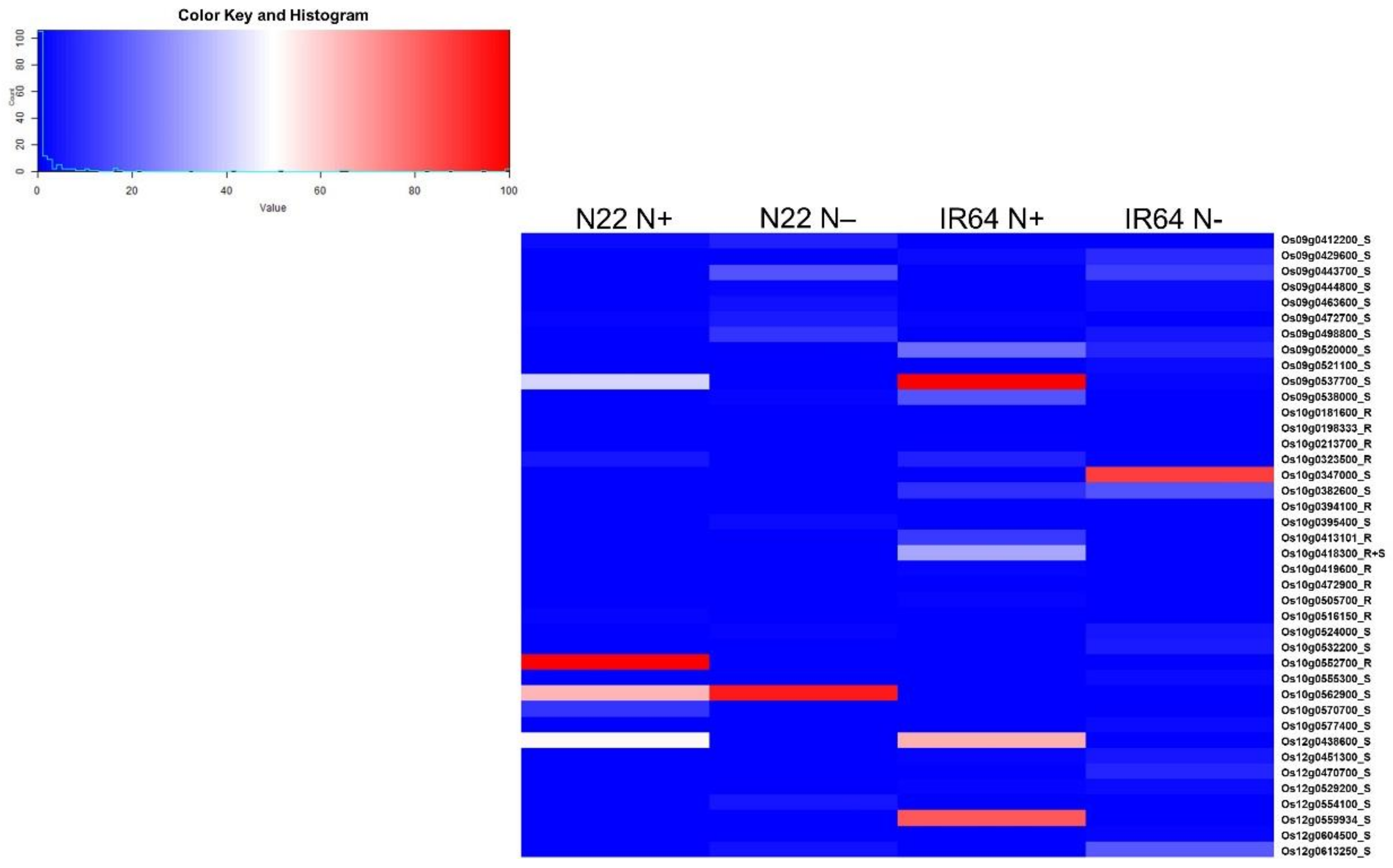
3.5. Nitrogen Use Efficiency Quantitative Trait Loci in Rice and Co-Localization of Differentially Expressed Genes to the Quantitative Trait Loci Regions
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Frink, C.R.; Waggoner, P.E.; Ausubel, J.H. Nitrogen fertilizer: Retrospect and prospect. Proc. Natl. Acad. Sci. USA 1999, 96, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Horchani, F.; Prévot, M.; Boscari, A.; Evangelisti, E.; Meilhoc, E.; Bruand, C.; Brouquisse, R. Both plant and bacterial nitrate reductases contribute to nitric oxide production in Medicago truncatula nitrogen-fixing nodules. Plant Physiol. 2011, 155, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Good, A.G.; Johnson, S.J.; De Pauw, M.; Carroll, R.T.; Savidov, N.; Vidmar, J.; Lu, Z.; Taylor, G.; Stroeher, V. Engineering nitrogen use efficiency with alanine aminotransferase. Botany 2007, 85, 252–262. [Google Scholar] [CrossRef]
- Raun, W.R.; Solie, J.B.; Johnson, G.V.; Stone, M.L.; Mullen, R.W.; Freeman, K.W.; Lukina, E.V. Improving nitrogen use efficiency in cereal grain production with optical sensing and variable rate application. Agron. J. 2002, 94, 815–820. [Google Scholar] [CrossRef]
- Raven, J.A.; Taylor, R. Macroalgal growth in nutrient-enriched estuaries: A biogeochemical and evolutionary perspective. Water Air Soil Pollut. Focus 2003, 3, 7–26. [Google Scholar] [CrossRef]
- Guo, J.H.; Liu, X.J.; Zhang, Y.; Shen, J.L.; Han, W.X.; Zhang, W.F.; Zhang, F.S. Significant acidification in major Chinese croplands. Science 2010, 327, 1008–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, N.M. Nitrate: Nutrient and signal for plant growth. Plant Cell 1995, 7, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Krapp, A.; David, L.C.; Chardin, C.; Girin, T.; Marmagne, A.; Leprince, A.S.; Daniel-Vedele, F. Nitrate transport and signalling in Arabidopsis. J. Exp. Bot. 2014, 65, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Kiba, T.; Krapp, A. Plant nitrogen acquisition under low availability: Regulation of uptake and root architecture. Plant Cell Physiol. 2016, 57, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.; Duan, F.; An, X.; Zhang, F.; von Wiren, N.; Yuan, L. Characterization of AMT-mediated high-affinity ammonium uptake in roots of maize (Zea mays L.). Plant Cell Physiol. 2013, 54, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Rawal, H.C.; Kumar, S.; Mithra, S.V.A.; Solanke, A.U. High quality unigenes and microsatellite markers from tissue specific transcriptome and development of a database in Clusterbean (Cyamopsis tetragonoloba, L. Taub). Genes 2017, 8, 313. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Wang, S.; Zhang, J.; Feng, Q.; Zhang, L.; Fan, D.; Li, X.; Yuan, D.; Han, B.; Zhang, Q. Expression profiles of 10,422 genes at early stage of low nitrogen stress in rice assayed using a cDNA microarray. Plant Mol. Biol. 2006, 60, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yoon, J.; Choi, H.; Fan, Y.; Chen, R.; An, G. Transcriptome analysis of nitrogen-starvation-responsive genes in rice. BMC Plant Biol. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.N.; Zhou, X.A.; Ai, H.S.; Wang, C.; Zhou, R.; Chen, S.L. Identification of genes associated with nitrogen-use efficiency by genome-wide transcriptional analysis of two soybean genotypes. BMC Genom. 2011, 12, 525. [Google Scholar] [CrossRef] [PubMed]
- Gelli, M.; Duo, Y.; Konda, A.R.; Zhang, C.; Holding, D.; Dweikat, I. Identification of differentially expressed genes between sorghum genotypes with contrasting nitrogen stress tolerance by genome-wide transcriptional profiling. BMC Genom. 2014, 15, 179. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xiang, F.; Zhong, M.; Zhou, L.; Liu, H.; Li, S.; Wang, X. Transcriptome and metabolite analysis identifies nitrogen utilization genes in tea plant (Camellia sinensis). Sci. Rep. 2017, 7, 1693. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.I.; Jiang, W.Z.; Chin, J.H.; Piao, Z.Z.; Cho, Y.G.; McCouch, S.R.; Koh, H.J. Identification of QTLs associated with physiological nitrogen use efficiency in rice. Mol. Cells 2007, 23, 72–79. [Google Scholar] [PubMed]
- Wei, D.; Cui, K.H.; Ye, G.Y.; Pan, J.F.; Xiang, J.; Huang, J.L.; Nie, L. QTL mapping for nitrogen-use efficiency and nitrogen-deficiency tolerance traits in rice. Plant Soil 2012, 359, 281–295. [Google Scholar] [CrossRef]
- Zhou, Y.; Tao, Y.; Tang, D.; Wang, J.; Zhong, J.; Wang, Y.; Liang, G. Identification of QTLs associated with nitrogen uptake and nitrogen use efficiency using high throughput genotyped CSSLs in rice (Oryza sativa L.). Front. Plant Sci. 2017, 8, 1166. [Google Scholar] [CrossRef] [PubMed]
- Pandit, A.; Rai, V.; Bal, S.; Sinha, S.; Kumar, V.; Chauhan, M.; Gautam, R.K.; Singh, R.; Sharma, P.C.; Singh, A.K.; et al. Combining QTL mapping and transcriptome profiling of bulked RILs for identification of functional polymorphism for salt tolerance genes in rice (Oryza sativa L.). Mol. Genet. Genom. 2013, 284, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, J.T.; Israelstam, G.F. A method for the extraction of chlorophyll from leaf tissue without maceration. Can. J. Bot. 1979, 57, 1332–1334. [Google Scholar] [CrossRef]
- Sinha, S.K.; Rani, M.; Bansal, N.; Gayatri; Venkatesh, K.; Mandal, P.K. Nitrate starvation induced changes in root system architecture, carbon: Nitrogen metabolism, and miRNA expression in nitrogen-responsive wheat genotypes. Appl. Biochem. Biotechnol. 2015, 177, 1299–1312. [Google Scholar] [CrossRef] [PubMed]
- Joy, K.W.; Hageman, R.H. The purification and properties of nitrite reductase from higher plants, and its dependence on ferredoxin. Biochem. J. 1966, 100, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Fieuw, S.; Muller-Rober, B.; Galvez, S.; Willmitzer, L. Cloning and expression analysis of the cytosolic NADP+-dependent isocitrate dehydrogenase from potato (implications for nitrogen metabolism). Plant Physiol. 1995, 107, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Srere, P.A. Enzyme concentrations in tissues. Science 1967, 158, 936–937. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Rice Genome Annotation Project. Available online: http://rice.plantbiology.msu.edu/ (accessed on 23 January 2018).
- Primer3Web. Available online: http://primer3.ut.ee/ (accessed on 12 December 2016).
- Voorrips, R. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Xing, Y.; Yan, H.; Xu, C.; Li, X.; Zhang, Q. QTLs for low nitrogen tolerance at seedling stage identified using a recombinant inbred line population derived from an elite rice hybrid. Theor. Appl. Genet. 2005, 112, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Senthilvel, S.; Vinod, K.K.; Malarvizhi, P.; Maheswaran, M. QTL and QTL x environment effects on agronomic and nitrogen acquisition traits in rice. J. Integr. Plant Biol. 2008, 50, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, K.; Kobayashi, N.; Ono, K.; Yano, M.; Ohsugi, R. Are contents of Rubisco, soluble protein and nitrogen in flag leaves of rice controlled by the same genetics? J. Exp. Bot. 2001, 52, 1827–1833. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Lu, Y.; Xie, W.; Zhu, T.; Lian, X. Transcriptome response to nitrogen starvation in rice. J. Biosci. 2012, 37, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Takehisa, H.; Sato, Y.; Antonio, B.A.; Nagamura, Y. Global transcriptome profile of rice root in response to essential macronutrient deficiency. Plant Signal. Behavi. 2013, 8, e24409. [Google Scholar] [CrossRef] [PubMed]
- Takehisa, H.; Sato, Y.; Antonio, B.A.; Nagamura, Y. Coexpression Network Analysis of Macronutrient Deficiency Response Genes in Rice. Rice 2015, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Drew, M.C. Comparison of the effects of a localised supply of phosphate, nitrate, ammonium and potassium on the growth of the seminal root system, and the shoot, in barley. New Phytol. 1975, 75, 479–490. [Google Scholar] [CrossRef]
- Gu, B.; Ju, X.; Chang, J.; Ge, Y.; Vitousek, P.M. Integrated reactive nitrogen budgets and future trends in China. Proc. Natl. Acad. Sci. USA 2015, 112, 8792–8797. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, D.; Shi, P.; Omasa, K. Estimating rice chlorophyll content and leaf nitrogen concentration with a digital still color camera under natural light. Plant Methods 2014, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Forde, B.G.; Lea, P.J. Glutamate in plants: Metabolism, regulation, and signalling. J. Exp. Bot. 2007, 58, 2339–2358. [Google Scholar] [CrossRef] [PubMed]
- Hirona, T.; Saito, Y.; Ushimaru, H.; Michiyama, H. The effect of amount of nitrogen fertilizer on starch metabolism in leaf sheaths of Japonica and Indica rice varieties during the heading period. Plant Prod. Sci. 2005, 8, 122–130. [Google Scholar] [CrossRef]
- Evans, J.R.; Terashima, I. Effects of nitrogen nutrition on electron transport components and photosynthesis in spinach. Aust. J. Plant Physiol. 1987, 14, 59–68. [Google Scholar] [CrossRef]
- Spreitzer, R.J.; Salvucci, M.E. Rubisco: Structure, regulatory interactions, and possibilities for a better enzyme. Ann. Rev. Plant Biol. 2002, 53, 449–479. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ren, B.; Ding, L.; Shen, Q.; Peng, S.; Guo, S. Does chloroplast size influence photosynthetic nitrogen use efficiency? PLoS ONE 2013, 8, e62036. [Google Scholar] [CrossRef] [PubMed]
- El-kereamy, A.; Guevara, D.; Bi, Y.-M.; Chen, Xi.; Rothstein, S.J. Exploring the molecular and metabolic factors contributing to the adaptation of maize seedlings to nitrate limitation. Front. Plant Sci. 2011, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Kim, B.K.; Kwon, S.J.; Jin, H.C.; Park, O.K. Arabidopsis GDSL lipase 2 plays a role in pathogen defense via negative regulation of auxin signaling. Biochem. Biophys. Res. Commun. 2009, 379, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, M.Á.; Forment, J.; Roldán, M.; Serrano, R.; Vicente, O. Overexpression of Arabidopsis thaliana LTL1, a salt-induced gene encoding a GDSL-motif lipase, increases salt tolerance in yeast and transgenic plants. Plant Cell Environ. 2006, 29, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.K.; Choi, H.W.; Hwang, I.S.; Kim, D.S.; Kim, N.H.; Choi, D.S.; Kim, Y.J.; Hwang, B.K. Function of a novel GDSL-type pepper lipase gene, CaGLIP1, in disease susceptibility and abiotic stress tolerance. Planta 2008, 227, 539–558. [Google Scholar] [CrossRef] [PubMed]
- Buchanan-Wollaston, V.; Page, T.; Harrison, E.; Breeze, E.; Lim, P.O.; Nam, H.G.; Lin, J.-F.; Wu, S.-H.; Swidzinski, J.; Ishizaki, K.; et al. Comparative transcriptome analysis reveals significant differences in gene expression and signalling pathways between developmental and dark/starvation-induced senescence in Arabidopsis. Plant J. 2005, 42, 567–585. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, M.; Sureshkumar, V.; Prakash, C.; Dixit, R.; Solanke, A.U.; Sharma, T.R.; Mohapatra, T.; Amitha Mithra, S.V. RiceMetaSys for salt and drought stress responsive genes in rice: A web interface for crop improvement. BMC Bioinform. 2017, 18, 432. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library | Number of Raw Reads | Number of Trimmed Reads | Number of Uniquely Mapped Reads |
|---|---|---|---|
| IAR | 66,536,324 | 50,191,956 | 37,645,788 |
| IAS | 70,824,528 | 55,136,806 | 41,531,371 |
| IDR | 71,789,678 | 65,991,386 | 12,203,558 |
| IDS | 59,320,262 | 54,086,464 | 14,888,256 |
| NAR | 85,535,858 | 78,152,624 | 40,574,926 |
| NAS | 80,808,216 | 74,622,520 | 13,694,430 |
| NDR | 68,228,978 | 64,247,540 | 40,574,926 |
| NDS | 72,947,448 | 67,688,471 | 50,455,258 |
| Comparisons | Number of Upregulated Genes | Number of Downregulated Genes | Total Number of DEGs | Roots + Shoots |
|---|---|---|---|---|
| IR64 root (N+/N−) | 43 | 40 | 83 | 209 |
| IR64 shoot (N+/N−) | 102 | 24 | 126 | |
| N22 root (N+/N−) | 86 | 6 | 92 | 188 |
| N22 shoot (N+/N−) | 66 | 30 | 96 | |
| IR64/N22 root (N+) | 151 | 267 | 418 | 732 |
| IR64/N22 shoot (N+) | 218 | 96 | 314 | |
| IR64/N22 root (N−) | 294 | 304 | 598 | 855 |
| IR64/N22 shoot (N−) | 138 | 119 | 257 | |
| IR64/N22 root (N-responsive) | 28 | 251 | 279 | 385 |
| IR64/N22 shoot (N-responsive) | 73 | 33 | 106 |
| Chromosome | IR 64 (1) | Nagina 22 (2) | IR 64 vs. Nagina 22 (3) |
|---|---|---|---|
| Root | |||
| 1 | 9 (1) | 12 (7) | 34 |
| 2 | 2 | 15 (9) | 26 |
| 3 | 3 (1) | 6 (1) | 34 |
| 4 | 4 (1 *) | 12 (6 + 1 *) | 20 |
| 5 | 3 | 4(4) | 22 |
| 6 | 6 (1 + 1 *) | 8 (5 + 1 *) | 21 |
| 7 | 4 (2) | 7 (5) | 30 |
| 8 | 2 | 6 (3) | 27 |
| 9 | 1 | 4 (3) | 18 |
| 10 | 5 | 10 (8) | 20 |
| 11 | 3 | 3 (1) | 13 |
| 12 | 1 | 2 (2) | 14 |
| Total | 43 (5 + 2 *) | 89 (52 + 2 *) | 279 |
| Shoot | |||
| 1 | 10 (1 **) | 10 (1 + 1 **) | 9 (1 **) |
| 2 | 13 (2) | 10 | 9 |
| 3 | 18 (2 + 1 *) | 8 (1 *) | 10 |
| 4 | 17 (2 + 1 *) | 11 (1 *) | 11 |
| 5 | 6 (2 + 1 *) | 4 (1 *) | 9 |
| 6 | 12 (1) | 6 (1) | 11 |
| 7 | 3 | 6 | 5 |
| 8 | 7 | 5 | 2 |
| 9 | 6 (1) | 9 | 10 |
| 10 | 10 (2 + 2 *) | 6 (2 *) | 9 |
| 11 | 4 (1 *) | 6 (1 *) | 12 |
| 12 | 10 (2 *) | 4 (2 *) | 9 |
| Total | 116 (12 + 8 * + 1 **) | 85 (2 + 8 * + 1 **) | 106 (1 **) |
| Grand total | 159 (22 + 13 *) | 174 (60 + 13 *) | 385 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, S.K.; Sevanthi V., A.M.; Chaudhary, S.; Tyagi, P.; Venkadesan, S.; Rani, M.; Mandal, P.K. Transcriptome Analysis of Two Rice Varieties Contrasting for Nitrogen Use Efficiency under Chronic N Starvation Reveals Differences in Chloroplast and Starch Metabolism-Related Genes. Genes 2018, 9, 206. https://doi.org/10.3390/genes9040206
Sinha SK, Sevanthi V. AM, Chaudhary S, Tyagi P, Venkadesan S, Rani M, Mandal PK. Transcriptome Analysis of Two Rice Varieties Contrasting for Nitrogen Use Efficiency under Chronic N Starvation Reveals Differences in Chloroplast and Starch Metabolism-Related Genes. Genes. 2018; 9(4):206. https://doi.org/10.3390/genes9040206
Chicago/Turabian StyleSinha, Subodh Kumar, Amitha Mithra Sevanthi V., Saurabh Chaudhary, Punit Tyagi, Sureshkumar Venkadesan, Manju Rani, and Pranab Kumar Mandal. 2018. "Transcriptome Analysis of Two Rice Varieties Contrasting for Nitrogen Use Efficiency under Chronic N Starvation Reveals Differences in Chloroplast and Starch Metabolism-Related Genes" Genes 9, no. 4: 206. https://doi.org/10.3390/genes9040206
APA StyleSinha, S. K., Sevanthi V., A. M., Chaudhary, S., Tyagi, P., Venkadesan, S., Rani, M., & Mandal, P. K. (2018). Transcriptome Analysis of Two Rice Varieties Contrasting for Nitrogen Use Efficiency under Chronic N Starvation Reveals Differences in Chloroplast and Starch Metabolism-Related Genes. Genes, 9(4), 206. https://doi.org/10.3390/genes9040206