TRAP1 Regulation of Cancer Metabolism: Dual Role as Oncogene or Tumor Suppressor

 , and
, and
Abstract
1. Introduction
2. TRAP1 Background
3. TRAP1 Regulation of Energetic Metabolism
4. TRAP1 as an Oncogene
5. TRAP1 as a Tumor Suppressor
6. Conclusions and Future Perspectives
Acknowledgments
Conflicts of interest
References
- Coller, H.A. Is cancer a metabolic disease? Am. J. Pathol. 2014, 184, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Pratx, G. Imaging metabolic heterogeneity in cancer. Mol. Cancer 2016, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and mitochondrial respiration: A neglected connection. Cancer Res. 2015, 75, 3685–3686. [Google Scholar] [CrossRef] [PubMed]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant KRas-driven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell. Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.H.; Jung, W.H.; Koo, J.S. Metabolic phenotypes in triple-negative breast cancer. Tumor Biol. 2013, 34, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, M.R.; Matassa, D.S.; Sisinni, L.; Lettini, G.; Landriscina, M.; Esposito, F. TRAP1 revisited: Novel localizations and functions of a ‘next-generation’ biomarker. Int. J. Oncol. 2014, 45, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Song, H.Y.; Dunbar, J.D.; Zhang, Y.X.; Guo, D.; Donner, D.B. Identification of a protein with homology to Hsp90 that binds the Type 1 Tumor Necrosis Factor Receptor. J. Biol. Chem. 1995, 270, 3574–3581. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.F.; Chen, Y.; Dai, K.; Chen, P.L.; Riley, D.J.; Lee, W.H. A new member of the Hsp90 family of molecular chaperones interacts with the retinoblastoma protein during mitosis and after heat shock. Mol. Cell Biol. 1996, 16, 4691–4699. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013, 17, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Tsutsumi, S.; Muhlebach, G.; Sourbier, C.; Lee, M.J.; Lee, S.; Vartholomaiou, E.; Tatokoro, M.; Beebe, K.; Miyajima, N.; et al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc. Natl. Acad. Sci. USA 2013, 110, E1604–E1612. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Angelin, A.; Lisanti, S.; Kossenkov, A.V.; Speicher, K.D.; Wang, H.; Powers, J.F.; Tischler, A.S.; Pacak, K.; Fliedner, S.; et al. Landscape of the mitochondrial Hsp90 metabolome in tumors. Nat. Commun. 2013, 4, 2139. [Google Scholar] [CrossRef] [PubMed]
- Matassa, D.S.; Amoroso, M.R.; Agliarulo, I.; Maddalena, F.; Sisinni, L.; Paladino, S.; Romano, S.; Romano, M.F.; Sagar, V.; Loreni, F.; et al. Translational control in the stress adaptive response of cancer cells: A novel role for the heat shock protein TRAP1. Cell Death Dis. 2013, 4, e851. [Google Scholar] [CrossRef] [PubMed]
- Matassa, D.S.; Agliarulo, I.; Amoroso, M.R.; Maddalena, F.; Sepe, L.; Ferrari, M.C.; Sagar, V.; D’Amico, S.; Loreni, F.; Paolella, G.; Landriscina, M.; et al. TRAP1-dependent regulation of p70S6K is involved in the attenuation of protein synthesis and cell migration: Relevance in human colorectal tumors. Mol. Oncol. 2014, 8, 1482–1494. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, M.R.; Matassa, D.S.; Laudiero, G.; Egorova, A.V.; Polishchuk, R.S.; Maddalena, F.; Piscazzi, A.; Paladino, S.; Sarnataro, D.; Garbi, C.; et al. TRAP1 and the proteasome regulatory particle TBP7/Rpt3 interact in the endoplasmic reticulum and control cellular ubiquitination of specific mitochondrial proteins. Cell Death Differ. 2012, 19, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Neckers, L.; Picard, D. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends Cell Biol. 2014, 24, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C.; Stein, G.S.; Lian, J.B.; Languino, L.R. TRAP-1, the mitochondrial Hsp90. Biochim. Biophys. Acta 2012, 1823, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Lettini, G.; Maddalena, F.; Sisinni, L.; Condelli, V.; Matassa, D.S.; Costi, M.P.; Simoni, D.; Esposito, F.; Landriscina, M. TRAP1: a viable therapeutic target for future cancer treatments? Expert Opin. Ther. Targets 2017, 21, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Aust, S.; Bachmayr-Heyda, A.; Pateisky, P.; Tong, D.; Darb-Esfahani, S.; Denkert, C.; Chekerov, R.; Sehouli, J.; Mahner, S.; van Gorp, T.; et al. Role of TRAP1 and estrogen receptor α in patients with ovarian cancer—A study of the OVCAD consortium. Mol. Cancer 2012, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, M.A.; Guzzo, G.; Morandi, A.; Perra, A.; Menegon, S.; Masgras, I.; Trevisan, E.; Angioni, M.M.; Fornari, F.; Quagliata, L.; et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget 2016, 7, 32375–32393. [Google Scholar] [CrossRef] [PubMed]
- Matassa, D.S.; Amoroso, M.R.; Lu, H.; Avolio, R.; Arzeni, D.; Procaccini, C.; Faicchia, D.; Maddalena, F.; Simeon, V.; Agliarulo, I.; et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. 2016, 23, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Caino, M.C.; Chae, Y.C.; Vaira, V.; Ferrero, S.; Nosotti, M.; Martin, N.M.; Weeraratna, A.; O’Connell, M.; Jernigan, D.; Fatatis, A.; et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Investig. 2013, 123, 2907–2920. [Google Scholar] [CrossRef] [PubMed]
- Agliarulo, I.; Matassa, D.S.; Amoroso, M.R.; Maddalena, F.; Sisinni, L.; Sepe, L.; Ferrari, M.C.; Arzeni, D.; Avolio, R.; Paolella, G.; et al. TRAP1 controls cell migration of cancer cells in metabolic stress conditions: Correlations with AKT/p70S6K pathways. Biochim. Biophys. Acta 2015, 1853, 2570–2579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, J.; Huang, Z.; Wei, P.; Liu, Y.; Hao, J.; Zhao, L.; Zhang, F.; Tu, Y.; Wei, T. Aberrantly upregulated TRAP1 is required for tumorigenesis of breast cancer. Oncotarget 2015, 6, 44495–44508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Lu, Y.; Yu, D.; Zhang, D.; Hu, W. TRAP1 provides protection against myocardial ischemia-reperfusion injury by ameliorating mitochondrial dysfunction. Cell Physiol. Biochem. 2015, 36, 2072–2082. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, S.; Tavecchio, M.; Chae, Y.C.; Liu, Q.; Brice, A.K.; Thakur, M.L.; Languino, L.R.; Altieri, D.C. Deletion of the mitochondrial chaperone TRAP-1 uncovers global reprogramming of metabolic networks. Cell Rep. 2014, 8, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Maddalena, F.; Sisinni, L.; Lettini, G.; Condelli, V.; Matassa, D.S.; Piscazzi, A.; Amoroso, M.R.; La Torre, G.; Esposito, F.; Landriscina, M. Resistance to Paclitxel in breast carcinoma cells requires a quality control of mitochondrial antiapoptotic proteins by TRAP1. Mol. Oncol. 2013, 7, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Costantino, E.; Maddalena, F.; Calise, S.; Piscazzi, A.; Tirino, V.; Fersini, A.; Ambrosi, A.; Neri, V.; Esposito, F.; Landriscina, M. TRAP1, a novel mitochondrial chaperone responsible for multi-drug resistance and protection from apoptosis in human colorectal carcinoma cells. Cancer Lett. 2009, 279, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Pak, M.G.; Koh, H.J.; Roh, M.S. Clinicopathologic significance of TRAP1 expression in colorectal cancer: A large scale study of human colorectal adenocarcinoma tissues. Diagn. Pathol. 2017, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Maddalena, F.; Simeon, V.; Vita, G.; Bochicchio, A.; Possidente, L.; Sisinni, L.; Lettini, G.; Condelli, V.; Matassa, D.S.; Li Bergolis, V.; et al. TRAP1 protein signature predicts outcome in human metastatic colorectal carcinoma. Oncotarget 2017, 8, 21229–21240. [Google Scholar] [CrossRef] [PubMed]
- Siegelin, M.D.; Plescia, J.; Raskett, C.M.; Gilbert, C.A.; Ross, A.H.; Altieri, D.C. Global targeting of subcellular heat shock protein-90 networks for therapy of glioblastoma. Mol. Cancer Ther. 2010, 9, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- Im, C.N.; Seo, J.S. Overexpression of tumor necrosis factor receptor-associated protein 1 (TRAP1), leads to mitochondrial aberrations in mouse fibroblast NIH/3T3 cells. BMB Rep. 2014, 47, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.; MacLeod, K.; Williams, A.R.; Cameron, D.A.; Smyth, J.F.; Langdon, S.P. Estrogen-regulated gene expression predicts response to endocrine therapy in patients with ovarian cancer. Gynecol. Oncol. 2007, 106, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, M.R.; Matassa, D.S.; Agliarulo, I.; Avolio, R.; Lu, H.; Sisinni, L.; Lettini, G.; Gabra, H.; Landriscina, M.; Esposito, F. TRAP1 downregulation in human ovarian cancer enhances invasion and epithelial-mesenchymal transition. Cell Death Dis. 2016, 7, e2522. [Google Scholar] [CrossRef] [PubMed]
- Leav, I.; Plescia, J.; Goel, H.L.; Li, J.; Jiang, Z.; Cohen, R.J.; Languino, L.R.; Altieri, D.C. Cytoprotective mitochondrial chaperone TRAP-1 as a novel molecular target in localized and metastatic prostate cancer. Am. J. Pathol. 2010, 176, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, S.; Garlick, D.S.; Bryant, K.G.; Tavecchio, M.; Mills, G.B.; Lu, Y.; Kossenkov, A.V.; Showe, L.C.; Languino, L.R.; Altieri, D.C. Transgenic expression of the mitochondrial chaperone TNFR-associated protein 1 (TRAP1) accelerates prostate cancer development. J. Biol. Chem. 2016, 291, 25247–25254. [Google Scholar] [CrossRef] [PubMed]
- Condelli, V.; Piscazzi, A.; Sisinni, L.; Matassa, D.S.; Maddalena, F.; Lettini, G.; Simeon, V.; Palladino, G.; Amoroso, M.R.; Trino, S.; et al. TRAP1 is involved in BRAF regulation and downstream attenuation of ERK phosphorylation and cell-cycle progression: A novel target for BRAF-mutated colorectal tumors. Cancer Res. 2014, 74, 6693–6704. [Google Scholar] [CrossRef] [PubMed]
- Condelli, V.; Maddalena, F.; Sisinni, L.; Lettini, G.; Matassa, D.S.; Piscazzi, A.; Palladino, G.; Amoroso, M.R.; Esposito, F.; Landriscina, M. Targeting TRAP1 as a downstream effector of BRAF cytoprotective pathway: a novel strategy for human BRAF-driven colorectal carcinoma. Oncotarget 2015, 6, 22298–22309. [Google Scholar] [CrossRef] [PubMed]
- Lettini, G.; Sisinni, L.; Condelli, V.; Matassa, D.S.; Simeon, V.; Maddalena, F.; Gemei, M.; Lopes, E.; Vita, G.; del Vecchio, L.; et al. TRAP1 regulates stemness through Wnt/β-catenin pathway in human colorectal carcinoma. Cell Death Differ. 2016, 23, 1792–1803. [Google Scholar] [CrossRef] [PubMed]
- Sisinni, L.; Maddalena, F.; Lettini, G.; Condelli, V.; Matassa, D.S.; Esposito, F.; Landriscina, M. TRAP1 role in endoplasmic reticulum stress protection favors resistance to anthracyclins in breast carcinoma cells. Int. J. Oncol. 2014, 44, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Sisinni, L.; Maddalena, F.; Condelli, V.; Pannone, G.; Simeon, V.; Li Bergolis, V.; Lopes, E.; Piscazzi, A.; Matassa, D.S.; Mazzoccoli, C.; et al. TRAP1 controls cell cycle G2-M transition through the regulation of CDK1 and MAD2 expression/ubiquitination. J. Pathol. 2017, 243, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hu, J.; Agorreta, J.; Cesario, A.; Zhang, Y.; Harris, A.L.; Gatter, K.; Pezzella, F. Tumor necrosis factor receptor-associated protein 1 (TRAP1) regulates genes involved in cell cycle and metastases. Cancer Lett. 2010, 296, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Masgras, I.; Ciscato, F.; Brunati, A.M.; Tibaldi, E.; Indraccolo, S.; Curtarello, M.; Chiara, F.; Cannino, G.; Papaleo, E.; Lambrughi, M.; et al. Absence of neurofibromin induces an oncogenic metabolic switch via mitochondrial ERK-mediated phosphorylation of the chaperone TRAP1. Cell Rep. 2017, 18, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Montesano Gesualdi, N.; Chirico, G.; Pirozzi, G.; Costantino, E.; Landriscina, M.; Esposito, F. Tumor necrosis factor-associated protein 1 (TRAP-1) protects cells from oxidative stress and apoptosis. Stress 2007, 10, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Guzzo, G.; Sciacovelli, M.; Bernardi, P.; Rasola, A. Inhibition of succinate dehydrogenase by the mitochondrial chaperone TRAP1 has anti-oxidant and anti-apoptotic effects on tumor cells. Oncotarget 2014, 5, 11897–11908. [Google Scholar] [CrossRef] [PubMed]
- Tochhawng, L.; Deng, S.; Pervaiz, S.; Yap, C.T. Redox regulation of cancer cell migration and invasion. Mitochondrion 2013, 13, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, A.J.; Macleod, K.G.; Burns, D.J.; Smyth, J.F.; Langdon, S.P. Estrogen receptor-α mediates gene expression changes and growth response in ovarian cancer cells exposed to estrogen. Endocr. Relat. Cancer 2005, 12, 851–866. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Amoroso, M.R.; Matassa, D.S.; Agliarulo, I.; Avolio, R.; Maddalena, F.; Condelli, V.; Landriscina, M.; Esposito, F. Stress-adaptive response in ovarian cancer drug resistance: Role of TRAP1 in oxidative metabolism-driven inflammation. Adv. Protein Chem. Struct. Biol. 2017, 108, 163–198. [Google Scholar] [PubMed]


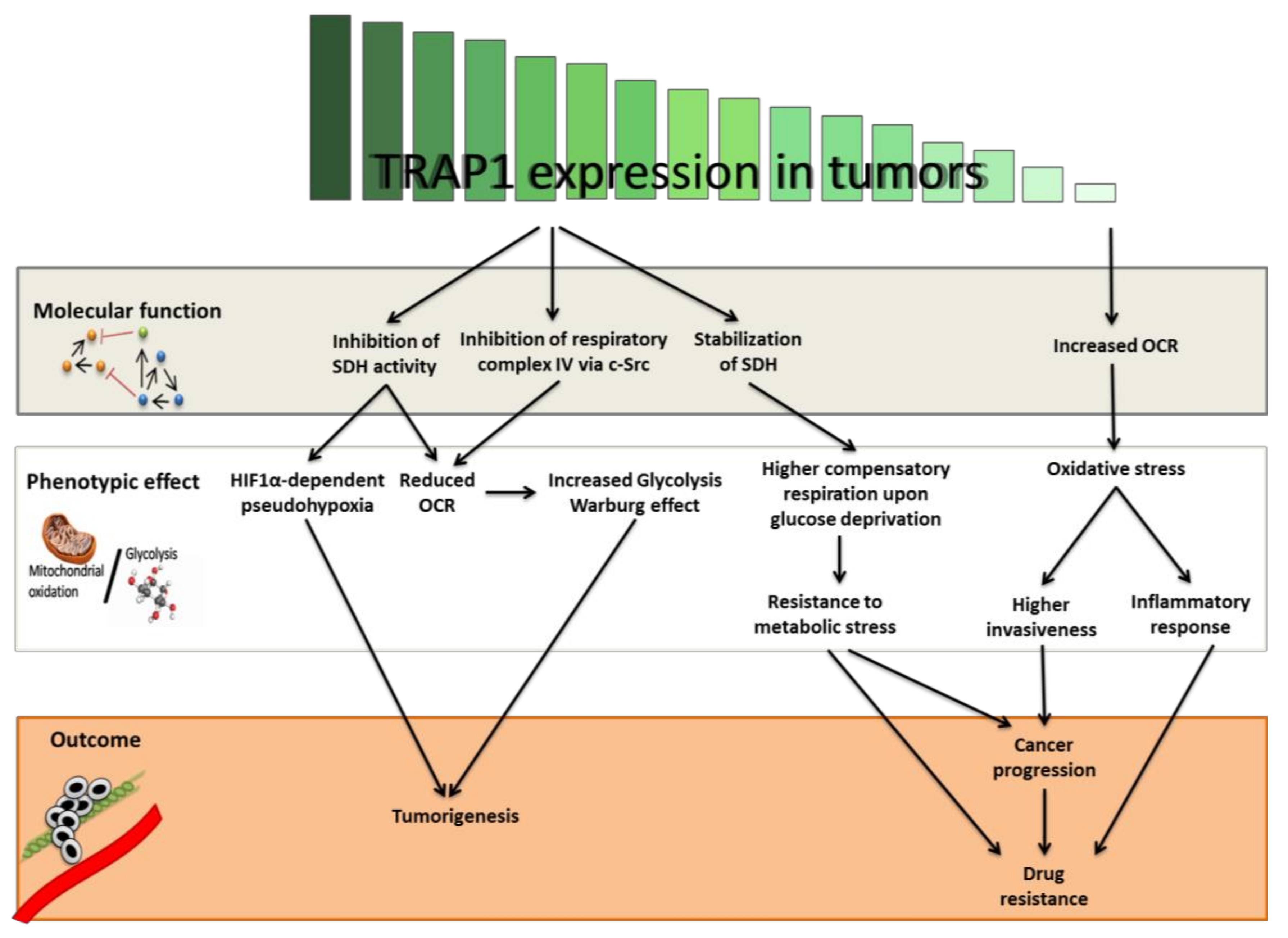{kind=link}
| Tumor Type | TRAP1 Expression | References |
|---|---|---|
| Bladder Cancer | Inversely correlated with tumor stage | Yoshida et al. [16] |
| Breast Cancer | High in tumor cells compared to surrounding normal tissue | Zhang et al. [29] Maddalena et al. [32] |
| Clear Cell Renal Cell Carcinoma | Inversely correlated with tumor stage | Yoshida et al. [16] |
| Cervical Cancer | Inversely correlated with tumor stage | Yoshida et al. [16] |
| Colorectal Carcinoma | High in tumor cells compared to surrounding healthy mucosa, increased in advanced pathological stage, positively correlated with worse survival, upregulated at the transition between low- and high-grade adenomas | Costantino et al. [33], Pak et al. [34] Maddalena et al. [35] |
| Glioblastoma | High in tumor cells compared to adjacent normal astrocytes | Siegelin et al. [36] |
| Lung Cancer | High in tumor cells compared to adjacent normal bronchial mucosa | Im et al. [37] |
| Ovarian Cancer | Inversely correlated with stage and grade, positively correlated with better survival, reduced in peritoneal deposit distant from primary site | Aust et al. [24] Matassa et al. [26] Walker et al. [38] Amoroso et al. [39] |
| Prostate Cancer | High in both localized and metastatic prostate cancers compared to normal prostate or benign prostatic hyperplasia | Leav et al. [40] Lisanti et al. [41] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matassa, D.S.; Agliarulo, I.; Avolio, R.; Landriscina, M.; Esposito, F. TRAP1 Regulation of Cancer Metabolism: Dual Role as Oncogene or Tumor Suppressor. Genes 2018, 9, 195. https://doi.org/10.3390/genes9040195
Matassa DS, Agliarulo I, Avolio R, Landriscina M, Esposito F. TRAP1 Regulation of Cancer Metabolism: Dual Role as Oncogene or Tumor Suppressor. Genes. 2018; 9(4):195. https://doi.org/10.3390/genes9040195
Chicago/Turabian StyleMatassa, Danilo Swann, Ilenia Agliarulo, Rosario Avolio, Matteo Landriscina, and Franca Esposito. 2018. "TRAP1 Regulation of Cancer Metabolism: Dual Role as Oncogene or Tumor Suppressor" Genes 9, no. 4: 195. https://doi.org/10.3390/genes9040195
APA StyleMatassa, D. S., Agliarulo, I., Avolio, R., Landriscina, M., & Esposito, F. (2018). TRAP1 Regulation of Cancer Metabolism: Dual Role as Oncogene or Tumor Suppressor. Genes, 9(4), 195. https://doi.org/10.3390/genes9040195