The Oncojanus Paradigm of Respiratory Complex I



Abstract
1. Introduction
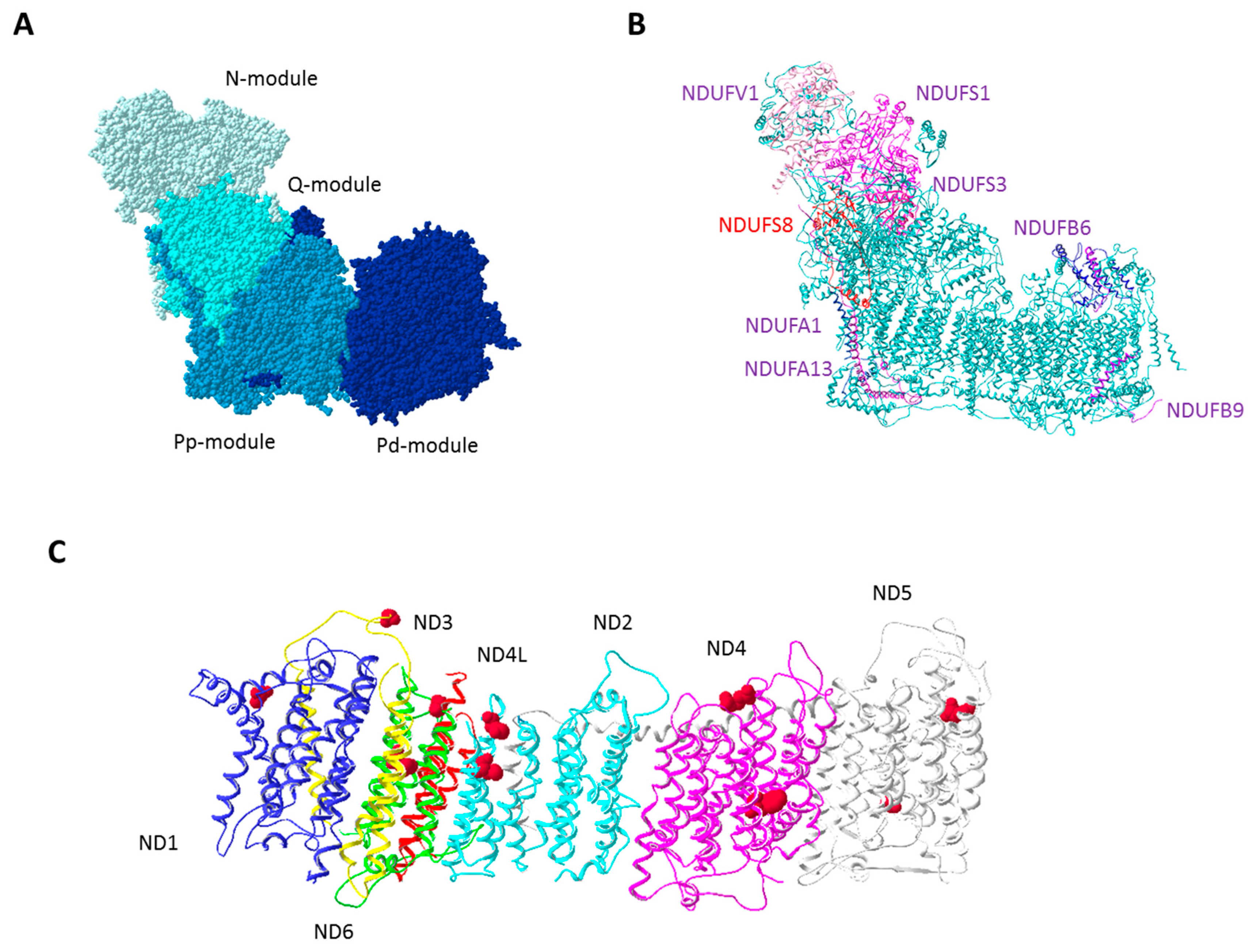
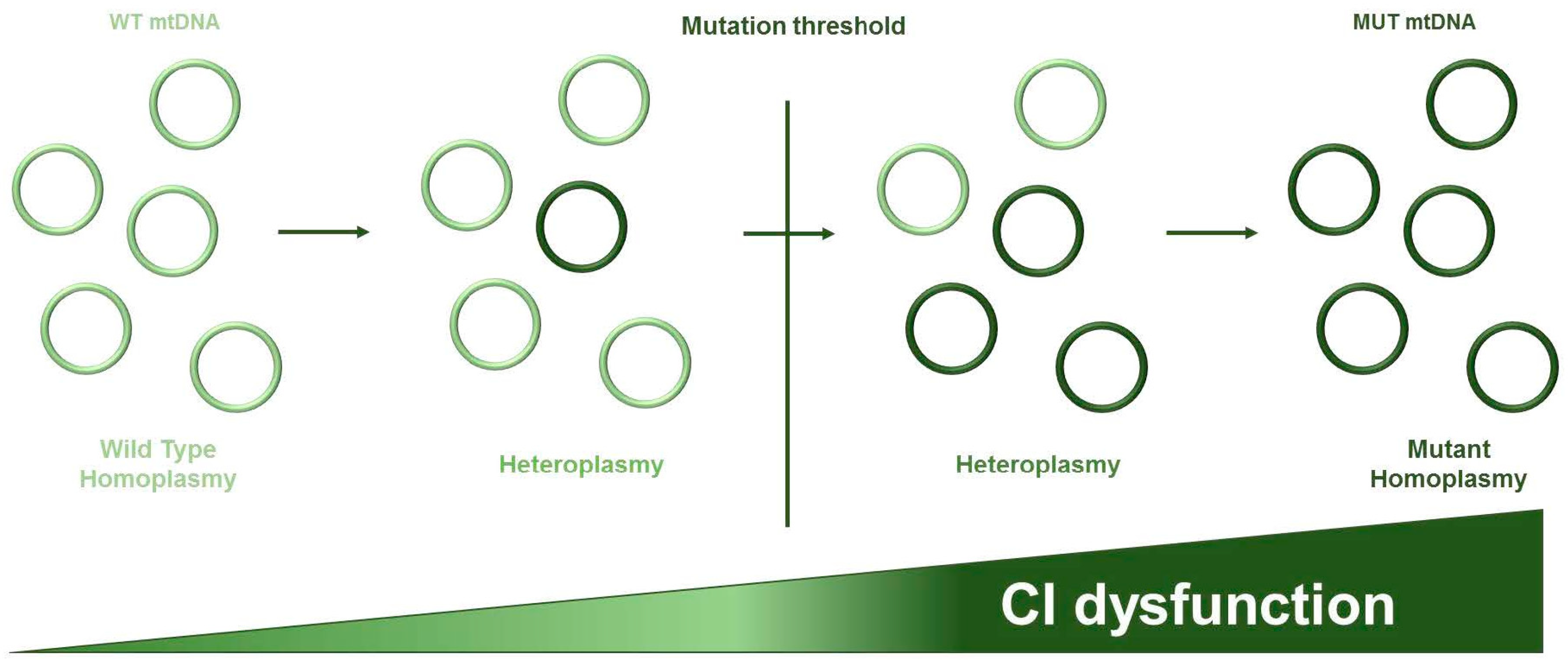
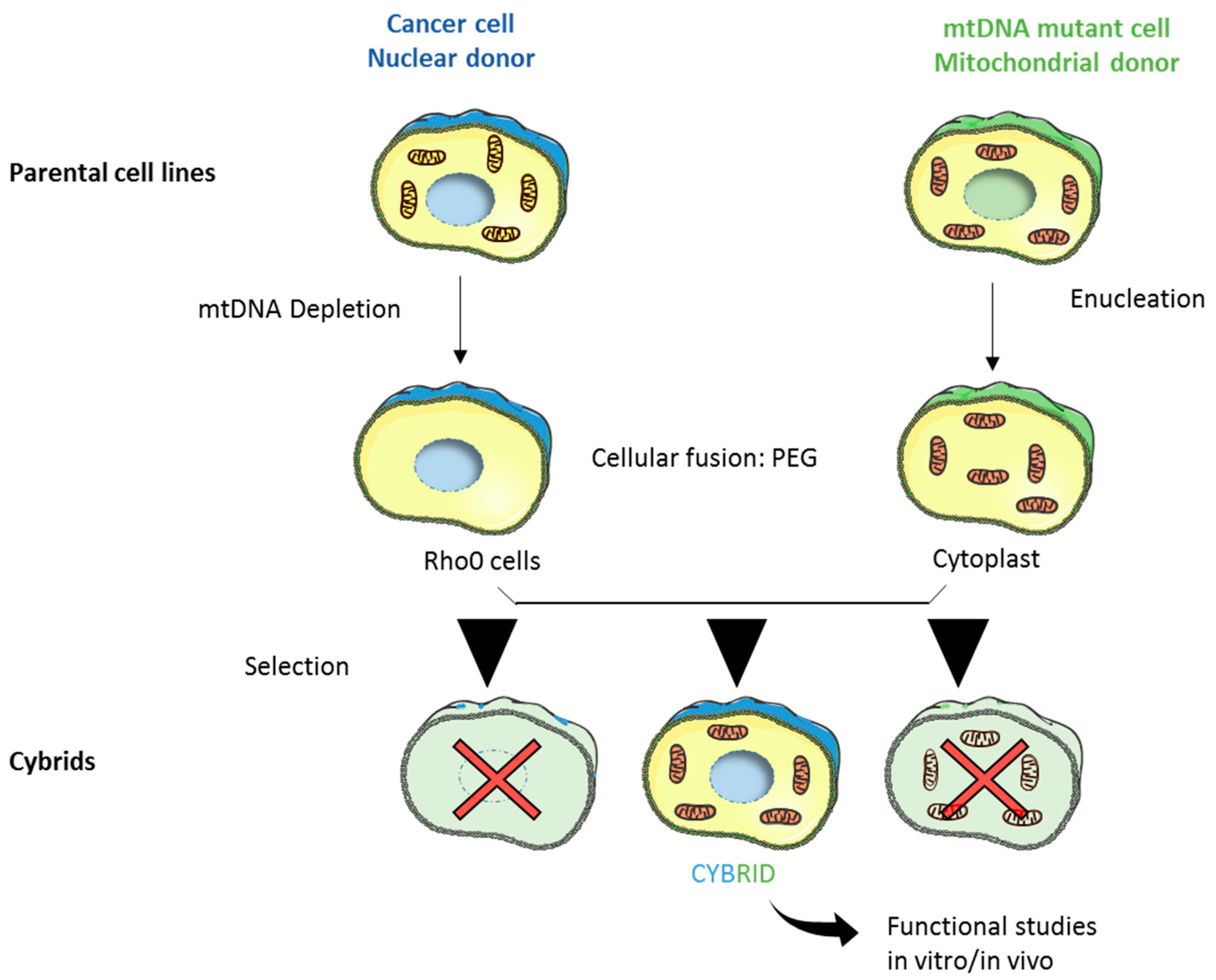
2. Mitochondrial DNA Features and Genetics
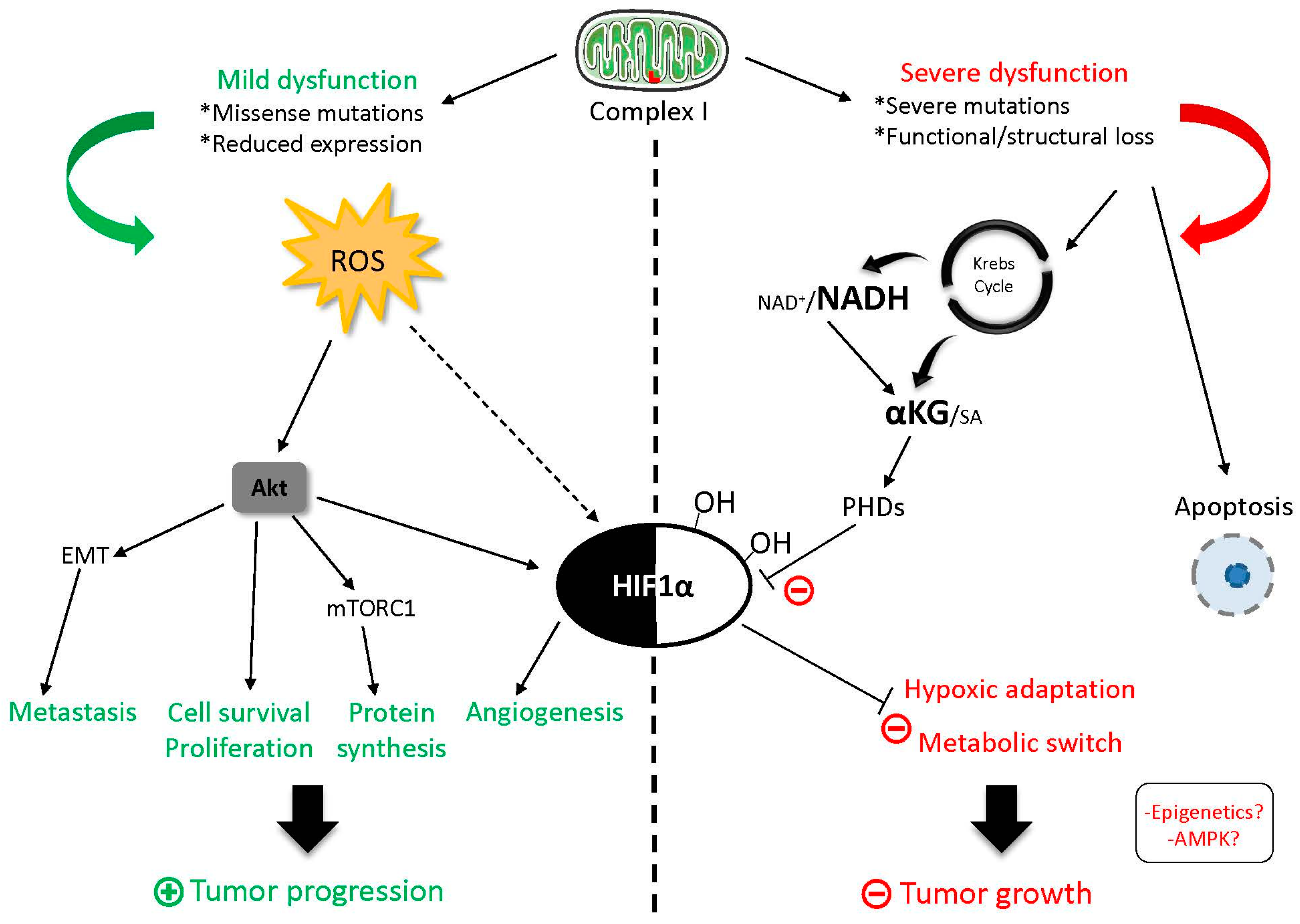
3. Relevance of mtDNA Mutations in Complex I Genes in Cancer Progression, Metastasis Formation, and Chemoresistance
4. Emerging Role of nDNA-Encoded Subunits and Assembly Factors of Complex I in Cancer
5. Complex I as a Target for Anti-Cancer Therapy?
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Milenkovic, D.; Blaza, J.N.; Larsson, N.-G.; Hirst, J. The enigma of the respiratory chain supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Caballero, L.; Guerrero-Castillo, S.; Nijtmans, L. Unraveling the complexity of mitochondrial complex I assembly: A dynamic process. Biochim. Biophys. Acta 2016. [Google Scholar] [CrossRef] [PubMed]
- Hofhaus, G.; Weiss, H.; Leonard, K. Electron microscopic analysis of the peripheral and membrane parts of mitochondrial NADH dehydrogenase (complex I). J. Mol. Biol. 1991, 221, 1027–1043. [Google Scholar] [CrossRef]
- Ragan, C.I.; Hatefi, Y. Isolation of the iron–sulfur-containing polypeptides of NADH:Oxidoreductase ubiquinone. Methods Enzymol. 1986, 126, 360–369. [Google Scholar] [PubMed]
- Vinogradov, A.D.; Grivennikova, V.G. Oxidation of NADH and ROS production by respiratory complex I. Biochim. Biophys. Acta 2016, 1857, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Iommarini, L.; Calvaruso, M.A.; Kurelac, I.; Gasparre, G.; Porcelli, A.M. Complex I impairment in mitochondrial diseases and cancer: Parallel roads leading to different outcomes. Int. J. Biochem. Cell Biol. 2013, 45, 47–63. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Blandini, F.; Nappi, G.; Greenamyre, J.T. Quantitative study of mitochondrial complex I in platelets of parkinsonian patients. Mov. Disord. Off. J. Mov. Disord. Soc. 1998, 13, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Giordano, L.; Deceglie, S.; d’Adamo, P.; Valentino, M.L.; La Morgia, C.; Fracasso, F.; Roberti, M.; Cappellari, M.; Petrosillo, G.; Ciaravolo, S.; et al. Cigarette toxicity triggers Leber’s hereditary optic neuropathy by affecting mtDNA copy number, oxidative phosphorylation and ROS detoxification pathways. Cell Death Dis. 2015, 6, e2021. [Google Scholar] [CrossRef] [PubMed]
- Ghelli, A.; Porcelli, A.M.; Zanna, C.; Vidoni, S.; Mattioli, S.; Barbieri, A.; Iommarini, L.; Pala, M.; Achilli, A.; Torroni, A.; et al. The background of mitochondrial DNA haplogroup J increases the sensitivity of Leber’s hereditary optic neuropathy cells to 2,5-hexanedione toxicity. PLoS ONE 2009, 4, e7922. [Google Scholar] [CrossRef] [PubMed]
- Rodenburg, R.J. Mitochondrial complex I-linked disease. Biochim. Biophys. Acta 2016, 1857, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Giannoccaro, M.P.; La Morgia, C.; Rizzo, G.; Carelli, V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 346–363. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Li, Y.; Zhu, H.; Lengauer, C.; Willson, J.K.; Markowitz, S.D.; Trush, M.A.; Kinzler, K.W.; Vogelstein, B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 1998, 20, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Fiedorczuk, K.; Letts, J.A.; Degliesposti, G.; Kaszuba, K.; Skehel, M.; Sazanov, L.A. Atomic structure of the entire mammalian mitochondrial complex I. Nature 2016, 538, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Bestwick, M.L.; Shadel, G.S. Accessorizing the human mitochondrial transcription machinery. Trends Biochem. Sci. 2013, 38, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.-P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Iommarini, L.; Porcelli, A.M.; Lang, M.; Ferri, G.G.; Kurelac, I.; Zuntini, R.; Mariani, E.; Pennisi, L.F.; Pasquini, E.; et al. An inherited mitochondrial DNA disruptive mutation shifts to homoplasmy in oncocytic tumor cells. Hum. Mutat. 2009, 30, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Grandhi, S.; Bosworth, C.; Maddox, W.; Sensiba, C.; Akhavanfard, S.; Ni, Y.; LaFramboise, T. Heteroplasmic shifts in tumor mitochondrial genomes reveal tissue-specific signals of relaxed and positive selection. Hum. Mol. Genet. 2017, 26, 2912–2922. [Google Scholar] [CrossRef] [PubMed]
- Coller, H.A.; Khrapko, K.; Bodyak, N.D.; Nekhaeva, E.; Herrero-Jimenez, P.; Thilly, W.G. High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat. Genet. 2001, 28, 147–150. [Google Scholar] [CrossRef] [PubMed]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Frank, M. Prevalence of neoplasms in definite and probable mitochondrial disorders. Mitochondrion 2016, 29, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Canter, J.A.; Kallianpur, A.R.; Parl, F.F.; Millikan, R.C. Mitochondrial DNA G10398A polymorphism and invasive breast cancer in African-American women. Cancer Res. 2005, 65, 8028–8033. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.K.; Leal, S.M.; Covarrubias, D.; Liu, A.; Wong, L.J.C. Mitochondrial genetic background modifies breast cancer risk. Cancer Res. 2007, 67, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Darvishi, K.; Sharma, S.; Bhat, A.K.; Rai, E.; Bamezai, R.N.K. Mitochondrial DNA G10398A polymorphism imparts maternal haplogroup N a risk for breast and esophageal cancer. Cancer Lett. 2007, 249, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Krawczyk, T.; Zdrozny, M.; Lubiński, J.; Arnold, R.S.; Kukwa, W.; Scińska, A.; Golik, P.; Bartnik, E.; Petros, J.A. Mitochondrial NADH-dehydrogenase subunit 3 (ND3) polymorphism (A10398G) and sporadic breast cancer in Poland. Breast Cancer Res. Treat. 2010, 121, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Mims, M.P.; Hayes, T.G.; Zheng, S.; Leal, S.M.; Frolov, A.; Ittmann, M.M.; Wheeler, T.M.; Prchal, J.T. Mitochondrial DNA G10398A polymorphism and invasive breast cancer in African-American women. Cancer Res. 2006, 66, 1880; author reply 1880–1881. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Soudry, E.; Mukhopadhyay, N.; Shao, C.; Yee, J.; Lam, S.; Lam, W.; Zhang, W.; Gazdar, A.F.; Fisher, P.B.; et al. Mitochondrial DNA mutations in respiratory complex-I in never-smoker lung cancer patients contribute to lung cancer progression and associated with EGFR gene mutation. J. Cell. Physiol. 2012, 227, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhou, S.; Chang, S.S.; McFate, T.; Verma, A.; Califano, J.A. Mitochondrial mutations contribute to HIF1α accumulation via increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Kachhap, S.; Sun, W.; Wu, G.; Chuang, A.; Poeta, L.; Grumbine, L.; Mithani, S.K.; Chatterjee, A.; Koch, W.; et al. Frequency and phenotypic implications of mitochondrial DNA mutations in human squamous cell cancers of the head and neck. Proc. Natl. Acad. Sci. USA 2007, 104, 7540–7545. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, W.; Li, H.; Yu, Y.; Tao, J.; Huang, S.; Zeng, Z. Nonsense and missense mutation of mitochondrial ND6 gene promotes cell migration and invasion in human lung adenocarcinoma. BMC Cancer 2015, 15, 346. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Bermúdez, A.; Vallejo, C.G.; Vicente-Blanco, R.J.; Gallardo, M.E.; Fernández-Moreno, M.Á.; Quintanilla, M.; Garesse, R. Enhanced tumorigenicity by mitochondrial DNA mild mutations. Oncotarget 2015, 6, 13628–13643. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; La Morgia, C.; Iommarini, L.; Carroccia, R.; Mattiazzi, M.; Sangiorgi, S.; Farne’, S.; Maresca, A.; Foscarini, B.; Lanzi, L.; et al. Mitochondrial optic neuropathies: How two genomes may kill the same cell type? Biosci. Rep. 2007, 27, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Iommarini, L.; Kurelac, I.; Capristo, M.; Calvaruso, M.A.; Giorgio, V.; Bergamini, C.; Ghelli, A.; Nanni, P.; De Giovanni, C.; Carelli, V.; et al. Different mtDNA mutations modify tumor progression in dependence of the degree of respiratory complex I impairment. Hum. Mol. Genet. 2014, 23, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Fang, H.; Liu, J.; Vartak, R.; Deng, J.; Bai, Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum. Mol. Genet. 2011, 20, 4605–4616. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Kurelac, I.; Capristo, M.; Iommarini, L.; Ghelli, A.; Ceccarelli, C.; Nicoletti, G.; Nanni, P.; De Giovanni, C.; Scotlandi, K.; et al. A mutation threshold distinguishes the antitumorigenic effects of the mitochondrial gene MTND1, an oncojanus function. Cancer Res. 2011, 71, 6220–6229. [Google Scholar] [CrossRef] [PubMed]
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-canonical mechanisms regulating hypoxia-inducible factor 1 α in cancer. Front. Oncol. 2017, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Iommarini, L.; Kurelac, I.; Calvaruso, M.A.; Capristo, M.; Lollini, P.L.; Nanni, P.; Bergamini, C.; Nicoletti, G.; Giovanni, C.D.; et al. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab. 2013, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- De Luise, M.; Girolimetti, G.; Okere, B.; Porcelli, A.M.; Kurelac, I.; Gasparre, G. Molecular and metabolic features of oncocytomas: Seeking the blueprints of indolent cancers. Biochim. Biophys. Acta 2017, 1858, 591–601. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.; LaFramboise, T. Mutational patterns in the breast cancer mitochondrial genome, with clinical correlates. Carcinogenesis 2014, 35, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.I. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, O.; Shimizu, A.; Yokota, M.; Sugiyama, A.; Nakada, K.; Miyoshi, H.; Itami, M.; Ohira, M.; Nagase, H.; Takenaga, K.; et al. Specific mitochondrial DNA mutation in mice regulates diabetes and lymphoma development. Proc. Natl. Acad. Sci. USA 2012, 109, 10528–10533. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, O.; Yamanashi, H.; Taketo, M.M.; Nakada, K.; Hayashi, J.-I. A specific nuclear DNA background is required for high frequency lymphoma development in transmitochondrial mice with G13997A mtDNA. PLoS ONE 2015, 10, e0118561. [Google Scholar] [CrossRef] [PubMed]
- Kulawiec, M.; Owens, K.M.; Singh, K.K. mtDNA G10398A variant in African-American women with breast cancer provides resistance to apoptosis and promotes metastasis in mice. J. Hum. Genet. 2009, 54, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, H.; Hattori, K.; Wada, R.; Ishikawa, K.; Fukuda, S.; Takenaga, K.; Nakada, K.; Hayashi, J. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS ONE 2011, 6, e23401. [Google Scholar] [CrossRef] [PubMed]
- Koshikawa, N.; Akimoto, M.; Hayashi, J.I.; Nagase, H.; Takenaga, K. Association of predicted pathogenic mutations in mitochondrial ND genes with distant metastasis in NSCLC and colon cancer. Sci. Rep. 2017, 7, 15535. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Kenny, T.C.; Hart, P.; Ragazzi, M.; Sersinghe, M.; Chipuk, J.; Sagar, M.A.K.; Eliceiri, K.W.; LaFramboise, T.; Grandhi, S.; Santos, J.; et al. Selected mitochondrial DNA landscapes activate the SIRT3 axis of the UPRmtto promote metastasis. Oncogene 2017, 36, 4393–4404. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, S.; Miyato, Y.; Shidara, Y.; Asoh, S.; Tokunaga, A.; Tajiri, T.; Ohta, S. Mutations in the mitochondrial genome confer resistance of cancer cells to anticancer drugs. Cancer Sci. 2009, 100, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Jones, A.W.E.; Fassone, E.; Sweeney, M.G.; Lebiedzinska, M.; Suski, J.M.; Wieckowski, M.R.; Tajeddine, N.; Hargreaves, I.P.; Yasukawa, T.; et al. PGC-1β mediates adaptive chemoresistance associated with mitochondrial DNA mutations. Oncogene 2013, 32, 2592–2600. [Google Scholar] [CrossRef] [PubMed]
- Girolimetti, G.; Guerra, F.; Iommarini, L.; Kurelac, I.; Vergara, D.; Maffia, M.; Vidone, M.; Amato, L.B.; Leone, G.; Dusi, S.; et al. Platinum-induced mitochondrial DNA mutations confer lower sensitivity to paclitaxel by impairing tubulin cytoskeletal organization. Hum. Mol. Genet. 2017, 26, 2961–2974. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Perrone, A.M.; Kurelac, I.; Santini, D.; Ceccarelli, C.; Cricca, M.; Zamagni, C.; De Iaco, P.; Gasparre, G. Mitochondrial DNA mutation in serous ovarian cancer: Implications for mitochondria-coded genes in chemoresistance. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, e373–e378. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, J.; Poss, M.; Brüggemann, M.; Gromes, A.; Schmidt, D.; Ellinger, N.; Tolkach, Y.; Dietrich, D.; Kristiansen, G.; Müller, S.C. Systematic expression analysis of mitochondrial complex I identifies NDUFS1 as a biomarker in clear-cell renal-cell carcinoma. Clin. Genitourin. Cancer 2017, 15, e551–e562. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Cheng, X.; Fu, Z.; Zhou, C.; Lu, W.; Xie, X. Reduced expression of NDUFS3 and its clinical significance in serous ovarian cancer. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2013, 23, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Mamelak, A.J.; Kowalski, J.; Murphy, K.; Yadava, N.; Zahurak, M.; Kouba, D.J.; Howell, B.G.; Tzu, J.; Cummins, D.L.; Liégeois, N.J.; et al. Downregulation of NDUFA1 and other oxidative phosphorylation-related genes is a consistent feature of basal cell carcinoma. Exp. Dermatol. 2005, 14, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.E.; Braun, M.; Syring, I.; Klümper, N.; Schmidt, D.; Perner, S.; Hauser, S.; Müller, S.C.; Ellinger, J. NDUFA4 expression in clear cell renal cell carcinoma is predictive for cancer-specific survival. Am. J. Cancer Res. 2015, 5, 2816–2822. [Google Scholar] [PubMed]
- Narimatsu, T.; Matsuura, K.; Nakada, C.; Tsukamoto, Y.; Hijiya, N.; Kai, T.; Inoue, T.; Uchida, T.; Nomura, T.; Sato, F.; et al. Downregulation of NDUFB6 due to 9p24.1-p13.3 loss is implicated in metastatic clear cell renal cell carcinoma. Cancer Med. 2015, 4, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Poulos, R.C.; Thoms, J.A.I.; Shah, A.; Beck, D.; Pimanda, J.E.; Wong, J.W.H. Systematic screening of promoter regions pinpoints functional cis-regulatory mutations in a cutaneous melanoma genome. Mol. Cancer Res. 2015, 13, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Li, L.D.; Sun, H.F.; Liu, X.X.; Gao, S.P.; Jiang, H.L.; Hu, X.; Jin, W. Down-regulation of NDUFB9 promotes breast cancer cell proliferation, metastasis by mediating mitochondrial metabolism. PLoS ONE 2015, 10, e0144441. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhou, A.; Lu, H.; Chen, Y.; Huang, G.; Yue, X.; Zhao, P.; Wu, Y. Suppression of mitochondrial complex I influences cell metastatic properties. PLoS ONE 2013, 8, e61677. [Google Scholar] [CrossRef] [PubMed]
- Santidrian, A.F.; Matsuno-Yagi, A.; Ritland, M.; Seo, B.B.; LeBoeuf, S.E.; Gay, L.J.; Yagi, T.; Felding-Habermann, B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J. Clin. Investig. 2013, 123, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Nallar, S.C.; Kalvakolanu, D.V. GRIM-19: A master regulator of cytokine induced tumor suppression, metastasis and energy metabolism. Cytokine Growth Factor Rev. 2017, 33, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Angell, J.E.; Lindner, D.J.; Shapiro, P.S.; Hofmann, E.R.; Kalvakolanu, D.V. Identification of GRIM-19, a novel cell death-regulatory gene induced by the interferon-β and retinoic acid combination, using a genetic approach. J. Biol. Chem. 2000, 275, 33416–33426. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, I.M.; Carroll, J.; Shannon, R.J.; Runswick, M.J.; Walker, J.E.; Hirst, J. GRIM-19, a cell death regulatory gene product, is a subunit of bovine mitochondrial NADH: Ubiquinone oxidoreductase (complex I). J. Biol. Chem. 2001, 276, 38345–38348. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Lu, H.; Hao, A.; Ng, D.C.H.; Ponniah, S.; Guo, K.; Lufei, C.; Zeng, Q.; Cao, X. GRIM-19, a cell death regulatory protein, is essential for assembly and function of mitochondrial complex I. Mol. Cell. Biol. 2004, 24, 8447–8456. [Google Scholar] [CrossRef] [PubMed]
- Kalakonda, S.; Nallar, S.C.; Jaber, S.; Keay, S.K.; Rorke, E.; Munivenkatappa, R.; Lindner, D.J.; Fiskum, G.M.; Kalvakolanu, D.V. Monoallelic loss of tumor suppressor GRIM-19 promotes tumorigenesis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, E4213–E4222. [Google Scholar] [CrossRef] [PubMed]
- Lufei, C.; Ma, J.; Huang, G.; Zhang, T.; Novotny-Diermayr, V.; Ong, C.T.; Cao, X. GRIM-19, a death-regulatory gene product, suppresses Stat3 activity via functional interaction. EMBO J. 2003, 22, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Kalakonda, S.; Nallar, S.C.; Lindner, D.J.; Hu, J.; Reddy, S.P.; Kalvakolanu, D.V. Tumor-suppressive activity of the cell death activator GRIM-19 on a constitutively active signal transducer and activator of transcription 3. Cancer Res. 2007, 67, 6212–6220. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ren, W.; Zhao, Y.; Fu, Z.; Ji, Y.; Zhu, Y.; Qin, C. Downregulation of GRIM-19 is associated with hyperactivation of p-STAT3 in hepatocellular carcinoma. Med. Oncol. Northwood Lond. Engl. 2012, 29, 3046–3054. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, M.; Wei, Y.; Feng, D.; Peng, C.; Weng, H.; Ma, Y.; Bao, L.; Nallar, S.; Kalakonda, S.; et al. Down-regulation of GRIM-19 expression is associated with hyperactivation of STAT3-induced gene expression and tumor growth in human cervical cancers. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2009, 29, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.B.; Luo, X.L.; Liu, S.Y.; Tao, D.D.; Gong, J.P.; Hu, J.B. Correlations of GRIM-19 and its target gene product STAT3 to malignancy of human colorectal carcinoma. Ai Zheng 2007, 26, 683–687. (In Chinese) [Google Scholar] [PubMed]
- Alchanati, I.; Nallar, S.C.; Sun, P.; Gao, L.; Hu, J.; Stein, A.; Yakirevich, E.; Konforty, D.; Alroy, I.; Zhao, X.; et al. A proteomic analysis reveals the loss of expression of the cell death regulatory gene GRIM-19 in human renal cell carcinomas. Oncogene 2006, 25, 7138–7147. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chao, L.; Rong, G.; Wang, C.; Ma, R.; Wang, X. Down-regulation of GRIM-19 is associated with STAT3 overexpression in breast carcinomas. Hum. Pathol. 2013, 44, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Máximo, V.; Botelho, T.; Capela, J.; Soares, P.; Lima, J.; Taveira, A.; Amaro, T.; Barbosa, A.P.; Preto, A.; Harach, H.R.; et al. Somatic and germline mutation in GRIM-19, a dual function gene involved in mitochondrial metabolism and cell death, is linked to mitochondrion-rich (Hurthle cell) tumours of the thyroid. Br. J. Cancer 2005, 92, 1892–1898. [Google Scholar] [CrossRef] [PubMed]
- Suhane, S.; Berel, D.; Ramanujan, V.K. Biomarker signatures of mitochondrial NDUFS3 in invasive breast carcinoma. Biochem. Biophys. Res. Commun. 2011, 412, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-Y.; Chang, Y.-C.; Yang, C.-J.; Huang, M.-S.; Hsiao, M. The opposite prognostic effect of NDUFS1 and NDUFS8 in lung cancer reflects the oncojanus role of mitochondrial complex I. Sci. Rep. 2016, 6, 31357. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, A.M.; Ghelli, A.; Ceccarelli, C.; Lang, M.; Cenacchi, G.; Capristo, M.; Pennisi, L.F.; Morra, I.; Ciccarelli, E.; Melcarne, A.; et al. The genetic and metabolic signature of oncocytic transformation implicates HIF1α destabilization. Hum. Mol. Genet. 2010, 19, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Porcelli, A.M.; Bonora, E.; Pennisi, L.F.; Toller, M.; Iommarini, L.; Ghelli, A.; Moretti, M.; Betts, C.M.; Martinelli, G.N.; et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 9001–9006. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Hervouet, E.; de Laplanche, E.; Demont, J.; Pennisi, L.F.; Colombel, M.; Mège-Lechevallier, F.; Scoazec, J.Y.; Bonora, E.; Smeets, R.; et al. Clonal expansion of mutated mitochondrial DNA is associated with tumor formation and complex I deficiency in the benign renal oncocytoma. Hum. Mol. Genet. 2008, 17, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E.; Porcelli, A.M.; Gasparre, G.; Biondi, A.; Ghelli, A.; Carelli, V.; Baracca, A.; Tallini, G.; Martinuzzi, A.; Lenaz, G.; et al. Defective oxidative phosphorylation in thyroid oncocytic carcinoma is associated with pathogenic mitochondrial DNA mutations affecting complexes I and III. Cancer Res. 2006, 66, 6087–6096. [Google Scholar] [CrossRef] [PubMed]
- Palorini, R.; Simonetto, T.; Cirulli, C.; Chiaradonna, F. Mitochondrial complex I inhibitors and forced oxidative phosphorylation synergize in inducing cancer cell death. Int. J. Cell Biol. 2013, 2013, 243876. [Google Scholar] [CrossRef] [PubMed]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J.A. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006, 29, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Dowling, R.J.O.; Niraula, S.; Stambolic, V.; Goodwin, P.J. Metformin in cancer: Translational challenges. J. Mol. Endocrinol. 2012, 48, R31–R43. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, P.; Heisler, I.; Unterschemmann, K.; Haerter, M.; Beck, H.; Greschat, S.; Ehrmann, A.; Summer, H.; Flamme, I.; Oehme, F.; et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013, 2, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Bastian, A.; Matsuzaki, S.; Humphries, K.M.; Pharaoh, G.A.; Doshi, A.; Zaware, N.; Gangjee, A.; Ihnat, M.A. AG311, a small molecule inhibitor of complex I and hypoxia-induced HIF-1α stabilization. Cancer Lett. 2017, 388, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Schöckel, L.; Glasauer, A.; Basit, F.; Bitschar, K.; Truong, H.; Erdmann, G.; Algire, C.; Hägebarth, A.; Willems, P.H.; Kopitz, C.; et al. Targeting mitochondrial complex I using BAY 87-2243 reduces melanoma tumor growth. Cancer Metab. 2015, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Storozhuk, Y.; Hopmans, S.N.; Sanli, T.; Barron, C.; Tsiani, E.; Cutz, J.-C.; Pond, G.; Wright, J.; Singh, G.; Tsakiridis, T. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br. J. Cancer 2013, 108, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Prior, S.; Kim, A.; Yoshihara, T.; Tobita, S.; Takeuchi, T.; Higuchi, M. Mitochondrial respiratory function induces endogenous hypoxia. PLoS ONE 2014, 9, e88911. [Google Scholar] [CrossRef] [PubMed]
- Brunmair, B.; Lest, A.; Staniek, K.; Gras, F.; Scharf, N.; Roden, M.; Nohl, H.; Waldhäusl, W.; Fürnsinn, C. Fenofibrate impairs rat mitochondrial function by inhibition of respiratory complex I. J. Pharmacol. Exp. Ther. 2004, 311, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Wilk, A.; Wyczechowska, D.; Zapata, A.; Dean, M.; Mullinax, J.; Marrero, L.; Parsons, C.; Peruzzi, F.; Culicchia, F.; Ochoa, A.; et al. Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Mol. Cell. Biol. 2015, 35, 182–198. [Google Scholar] [CrossRef] [PubMed]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Vander Heiden, M.G. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, T.A.; Farias, L.C.; Santos, E.S.; de Carvalho Fraga, C.A.; Orsini, L.A.; de Freitas Teles, L.; Feltenberger, J.D.; de Jesus, S.F.; de Souza, M.G.; Santos, S.H.S.; et al. Metformin increases PDH and suppresses HIF-1α under hypoxic conditions and induces cell death in oral squamous cell carcinoma. Oncotarget 2016, 7, 55057–55068. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ren, L.; Liu, C.; Xia, T.; Zha, X.; Wang, S. Phenformin induces cell cycle change, apoptosis, and mesenchymal–epithelial transition and regulates the AMPK/mTOR/p70S6K and MAPK/ERK pathways in breast cancer cells. PLoS ONE 2015, 10, e0131207. [Google Scholar] [CrossRef] [PubMed]
- Appleyard, M.V.C.L.; Murray, K.E.; Coates, P.J.; Wullschleger, S.; Bray, S.E.; Kernohan, N.M.; Fleming, S.; Alessi, D.R.; Thompson, A.M. Phenformin as prophylaxis and therapy in breast cancer xenografts. Br. J. Cancer 2012, 106, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Narise, K.; Okuda, K.; Enomoto, Y.; Hirayama, T.; Nagasawa, H. Optimization of biguanide derivatives as selective antitumor agents blocking adaptive stress responses in the tumor microenvironment. Drug Des. Devel. Ther. 2014, 8, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Bastian, A.; Thorpe, J.E.; Disch, B.C.; Bailey-Downs, L.C.; Gangjee, A.; Devambatla, R.K.V.; Henthorn, J.; Humphries, K.M.; Vadvalkar, S.S.; Ihnat, M.A. A small molecule with anticancer and antimetastatic activities induces rapid mitochondrial-associated necrosis in breast cancer. J. Pharmacol. Exp. Ther. 2015, 353, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Akatsuka, A.; Kojima, N.; Okamura, M.; Dan, S.; Yamori, T. A novel thiophene-3-carboxamide analog of annonaceous acetogenin exhibits antitumor activity via inhibition of mitochondrial complex I. Pharmacol. Res. Perspect. 2016, 4, e00246. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, X.; Zhao, M.; Wang, Y.; Cheng, X.; Wang, D.; Xu, Y.; Du, Z.; Yu, X. Celastrol targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent cytotoxicity in tumor cells. BMC Cancer 2011, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.B.; Liu, Y.; Coothankandaswamy, V.; Mahdi, F.; Jekabsons, M.B.; Gerwick, W.H.; Valeriote, F.A.; Zhou, Y.D.; Nagle, D.G. Kalkitoxin inhibits angiogenesis, disrupts cellular hypoxic signaling, and blocks mitochondrial electron transport in tumor cells. Mar. Drugs 2015, 13, 1552–1568. [Google Scholar] [CrossRef] [PubMed]
- Jeso, V.; Yang, C.; Cameron, M.D.; Cleveland, J.L.; Micalizio, G.C. Synthesis and SAR of Lehualide B: A marine-derived natural product with potent anti-multiple myeloma activity. ACS Chem. Biol. 2013, 8, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Mechanism of Action | Cellular Model | References |
|---|---|---|---|
| Rotenone | Induces cell death under starvation in vitro Increases ROS production in vitro | Breast cancer cells, bovine heart mitochondria | [87,88] |
| Piericidin A | Induces cell death under starvation in vitro Increases ROS production in vitro | Breast, pancreatic and lung cancer cells, bovine heart mitochondria | [87] |
| Capsaicin | Induces cell death under starvation in vitro | Breast cancer cells | [87] |
| Metformin | Induces cell death under starvation in vitro Promotes AMPK phosphorylation Induces HIF1α destabilization under hypoxic conditions | Colon, lung, breast, cervical, osteosarcoma, oral carcinoma cancer cells | [94,96,100,101,102] |
| Phenformin | Induces cell death under starvation in vitro Promotes AMPK phosphorylation Induces HIF1α destabilization under hypoxic conditions | Colon and breast cancer cells and xenografts | [94,103,104,105] |
| BAY 87-2243 | Induces cell death under starvation in vitro Decreases tumor growth in vivo Promotes AMPK phosphorylation Induces HIF1α destabilization under hypoxic conditionsIncreases intracellular ROS levels | Lung cancer and melanoma cells and xenografts | [92,95] |
| AG311 | Induces cell death under starvation in vitro Decreases tumor growth in vivo Promotes AMPK phosphorylation Induces HIF1α destabilization under hypoxic conditions Induces mitochondrial superoxide production | Breast cancer cells and xenografts | [93,106] |
| Fenofibrate | Induces cell death in vitro | Glioblastoma cells | [99] |
| JCI-20679 | Induces cell death in vitro | A panel of 39 human cancer cell lines | [107] |
| Celastrol | Promotes ROS production and apoptosis in vitro | Lung cancer and hepatocellular carcinoma cells | [108] |
| Kalkitoxin | Induces cell death in vitro Induces HIF1α destabilization under hypoxic conditions | Neuroblastoma, breast and colon cancer cells | [109] |
| Lehualide B | Induces cell death in vitro | Multiple myeloma cells | [110] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leone, G.; Abla, H.; Gasparre, G.; Porcelli, A.M.; Iommarini, L. The Oncojanus Paradigm of Respiratory Complex I. Genes 2018, 9, 243. https://doi.org/10.3390/genes9050243
Leone G, Abla H, Gasparre G, Porcelli AM, Iommarini L. The Oncojanus Paradigm of Respiratory Complex I. Genes. 2018; 9(5):243. https://doi.org/10.3390/genes9050243
Chicago/Turabian StyleLeone, Giulia, Houda Abla, Giuseppe Gasparre, Anna Maria Porcelli, and Luisa Iommarini. 2018. "The Oncojanus Paradigm of Respiratory Complex I" Genes 9, no. 5: 243. https://doi.org/10.3390/genes9050243
APA StyleLeone, G., Abla, H., Gasparre, G., Porcelli, A. M., & Iommarini, L. (2018). The Oncojanus Paradigm of Respiratory Complex I. Genes, 9(5), 243. https://doi.org/10.3390/genes9050243