Is There Still Any Role for Oxidative Stress in Mitochondrial DNA-Dependent Aging?



{kind=link}
{kind=link}
Abstract
1. Introduction: The Mitochondrial Theory of Aging and Recent Controversial Findings
2. Sources of Reactive Oxygen Species: Mitochondria versus NAD(P)H Oxidase
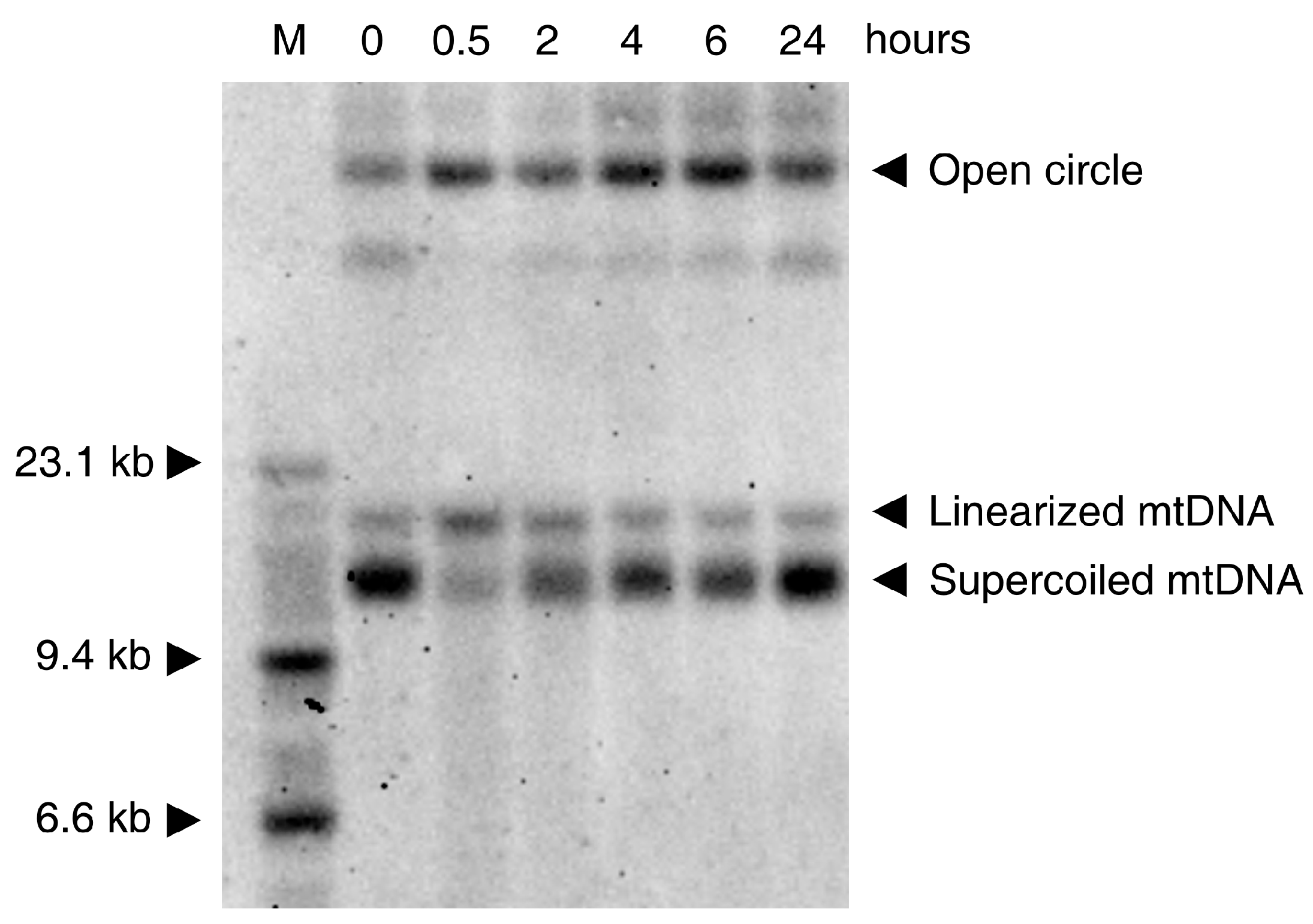
3. Effects of ROS on Mitochondrial DNA
4. Somatic Mitochondrial DNA Mutations in Aging: Free Radical Related Mutagenesis versus POLG Errors?
5. Somatic Mitochondrial DNA Deletions
6. Somatic Mitochondrial DNA Mutations in Neurodegenerative Diseases
7. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Alexeyev, M.F. Is there more to aging than mitochondrial DNA and reactive oxygen species? FEBS J. 2009, 276, 5768–5787. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, K.; Trifunovic, A. Origins of mtDNA mutations in ageing. Essays Biochem. 2017, 61, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian mitochondria and aging: An update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, R.J.; Zsurka, G.; Kunz, W.S. Mitochondrial DNA damage and the aging process: Facts and imaginations. Free Radic. Res. 2006, 40, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Soong, N.W.; Hinton, D.R.; Cortopassi, G.; Arnheim, N. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat. Genet. 1992, 2, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Nekhaeva, E.; Bodyak, N.D.; Kraytsberg, Y.; McGrath, S.B.; Van Orsouw, N.J.; Pluzhnikov, A.; Wei, J.Y.; Vijg, J.; Khrapko, K. Clonally expanded mtDNA point mutations are abundant in individual cells of human tissues. Proc. Natl. Acad. Sci. USA 2002, 99, 5521–5526. [Google Scholar] [CrossRef] [PubMed]
- Coller, H.A.; Khrapko, K.; Bodyak, N.D.; Nekhaeva, E.; Herrero-Jimenez, P.; Thilly, W.G. High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat. Genet. 2001, 28, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Verkaart, S.; van Emst-de Vries, S.E.; Grefte, S.; Smeitink, J.A.; Nijtmans, L.G.; Willems, P.H. Mitigation of NADH: Ubiquinone oxidoreductase deficiency by chronic Trolox treatment. Biochim. Biophys. Acta 2008, 1777, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Menzies, K.J.; Robinson, B.H.; Hood, D.A. Effect of thyroid hormone on mitochondrial properties and oxidative stress in cells from patients with mtDNA defects. Am. J. Physiol. Cell Physiol. 2009, 296, C355–C362. [Google Scholar] [CrossRef] [PubMed]
- Itsara, L.S.; Kennedy, S.R.; Fox, E.J.; Yu, S.; Hewitt, J.J.; Sanchez-Contreras, M.; Cardozo-Pelaez, F.; Pallanck, L.J. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 2014, 10, e1003974. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Diseases of the mitochondrial DNA. Annu. Rev. Biochem. 1992, 61, 1175–1212. [Google Scholar] [CrossRef] [PubMed]
- Müller-Höcker, J. Cytochrome c oxidase deficient fibres in the limb muscle and diaphragm of man without muscular disease: An age-related alteration. J. Neurol. Sci. 1990, 100, 14–21. [Google Scholar] [CrossRef]
- Wanagat, J.; Cao, Z.; Pathare, P.; Aiken, J.M. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 2001, 15, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Müller-Höcker, J. Cytochrome-c-oxidase deficient cardiomyocytes in the human heart—An age-related phenomenon. A histochemical ultracytochemical study. Am. J. Pathol. 1989, 134, 1167–1173. [Google Scholar] [PubMed]
- Baris, O.R.; Ederer, S.; Neuhaus, J.F.; von Kleist-Retzow, J.C.; Wunderlich, C.M.; Pal, M.; Wunderlich, F.T.; Peeva, V.; Zsurka, G.; Kunz, W.S.; et al. Mosaic deficiency in mitochondrial oxidative metabolism promotes cardiac arrhythmia during aging. Cell Metab. 2015, 21, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K.; Turnbull, D. Mitochondrial DNA mutations in aging. Prog. Mol. Biol. Transl. Sci. 2014, 127, 29–62. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Malinska, D.; Kulawiak, B.; Kudin, A.P.; Kovacs, R.; Huchzermeyer, C.; Kann, O.; Szewczyk, A.; Kunz, W.S. Complex III-dependent superoxide production of brain mitochondria contributes to seizure-related ROS formation. Biochim. Biophys. Acta 2010, 1797, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Claude, J.; Linnartz-Gerlach, B.; Kudin, A.P.; Kunz, W.S.; Neumann, H. Microglial CD33-related Siglec-E inhibits neurotoxicity by preventing the phagocytosis-associated oxidative burst. J. Neurosci. 2013, 33, 18270–18276. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Henzler, T.; Steudle, E. Transport and metabolic degradation of hydrogen peroxide in Chara corallina: Model calculations and measurements with the pressure probe suggest transport of H2O2 across water channels. J. Exp. Bot. 2000, 51, 2053–2066. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Rovida, E. The role of iron in mitochondrial function. Biochim. Biophys. Acta 2009, 1790, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Chang, Y.Z. Mitochondrial ferritin in the regulation of brain iron homeostasis and neurodegenerative diseases. Front. Pharmacol. 2014, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Petrat, F.; Weisheit, D.; Lensen, M.; de Groot, H.; Sustmann, R.; Rauen, U. Selective determination of mitochondrial chelatable iron in viable cells with a new fluorescent sensor. Biochem. J. 2002, 362, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Rauen, U.; Springer, A.; Weisheit, D.; Petrat, F.; Korth, H.-G.; de Groot, H.; Sustmann, R. Assessment of chelatable mitochondrial Iron by using mitochondrion-selective fluorescent iron indicators with different iron-binding affinities. Chem. Bio. Chem. 2007, 8, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Petrat, F.; de Groot, H.; Rauen, U. Subcellular distribution of chelatable iron: A laser scanning microscopic study in isolated hepatocytes and liver endothelial cells. Biochem. J. 2001, 356, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Cantuti-Castelvetri, I.; Zheng, K.; Jackson, K.E.; Tan, Y.B.; Arzberger, T.; Lees, A.J.; Betensky, R.A.; Beal, M.F.; Simon, D.K. Somatic mitochondrial DNA mutations in early Parkinson and incidental Lewy body disease. Ann. Neurol. 2012, 71, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Genoud, S.; Roberts, B.R.; Gunn, A.P.; Halliday, G.M.; Lewis, S.J.G.; Ball, H.J.; Hare, D.J.; Double, K.L. Subcellular compartmentalisation of copper, iron, manganese, and zinc in the Parkinson’s disease brain. Metallomics 2017, 9, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Reichert, M.; Schaller, H.; Kunz, W.; Gerber, G. The dependence on the extramitochondrial ATP/ADP-ratio of the oxidative phosphorylation in mitochondria isolated by a new procedure from rat skeletal muscle. Acta Biol. Med. Ger. 1978, 37, 1167–1176. [Google Scholar] [PubMed]
- Guliaeva, N.A.; Kuznetsova, E.A.; Gaziev, A.I. Proteins associated with mitochondrial DNA protect it against the action of X-rays and hydrogen peroxide. Biofizika 2006, 51, 692–697. [Google Scholar] [PubMed]
- Halliwell, B.; Aruoma, O.I. DNA damage by oxygen-derived species. Its mechanism and measurement in mammalian systems. FEBS Lett. 1991, 281, 9–19. [Google Scholar] [CrossRef]
- Henle, E.S.; Luo, Y.; Gassmann, W.; Linn, S. Oxidative damage to DNA constituents by iron-mediated Fenton reactions. The deoxyguanosine family. J. Biol. Chem. 1996, 271, 21177–21186. [Google Scholar] [CrossRef] [PubMed]
- Bohr, V.A. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic. Biol. Med. 2002, 32, 804–812. [Google Scholar] [CrossRef]
- Wang, D.; Kreutzer, D.A.; Essigmann, J.M. Mutagenicity and repair of oxidative DNA damage: Insights from studies using defined lesions. Mutat. Res. 1998, 400, 99–115. [Google Scholar] [CrossRef]
- Pletjushkina, O.Y.; Fetisova, E.K.; Lyamzaev, K.G.; Ivanova, O.Y.; Domnina, L.V.; Vyssokikh, M.Y.; Pustovidko, A.V.; Alexeevski, A.V.; Alexeevski, D.A.; Vasiliev, J.M.; et al. Hydrogen peroxide produced inside mitochondria takes part in cell-to-cell transmission of apoptotic signal. Biochemistry 2006, 71, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Freudenthal, B.D.; Beard, W.A.; Perera, L.; Shock, D.D.; Kim, T.; Schlick, T.; Wilson, S.H. Uncovering the polymerase induced cytotoxicity of an oxidized nucleotide. Nature 2015, 517, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Minetti, C.A.; Remeta, D.P.; Iden, C.R.; Johnson, F.; Grollman, A.P.; Breslauer, K.J. Impact of thymine glycol damage on DNA duplex energetics: Correlations with lesion-induced biochemical and structural consequences. Biopolymers 2015, 103, 491–508. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013, 9, e1003794. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, J.G.; Hipp, M.J.; Montine, T.J.; Kennedy, S.R. Mitochondrial DNA mutations increase in early stage Alzheimer disease and are inconsistent with oxidative damage. Ann. Neurol. 2016, 80, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Graziewicz, M.A.; Bienstock, R.J.; Copeland, W.C. The DNA polymerase γ Y955C disease variant associated with PEO and parkinsonism mediates the incorporation and translesion synthesis opposite 7,8-dihydro-8-oxo-20-deoxyguanosine. Hum. Mol. Genet. 2007, 16, 2729–2739. [Google Scholar] [CrossRef] [PubMed]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Volmering, E.; Niehusmann, P.; Peeva, V.; Grote, A.; Zsurka, G.; Altmüller, J.; Nürnberg, P.; Becker, A.J.; Schoch, S.; Elger, C.E.; et al. Neuropathological signs of inflammation correlate with mitochondrial DNA deletions in mesial temporal lobe epilepsy. Acta Neuropathol. 2016, 132, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Johnson, K.A. Fidelity of the human mitochondrial DNA polymerase. J. Biol. Chem. 2006, 281, 36236–36240. [Google Scholar] [CrossRef] [PubMed]
- Dunn, D.C. Running on empty: Does mitochondrial DNA mutation limit replicative lifespan in yeast? Bioessays 2011, 33, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.M.; Stewart, J.B.; Hagström, E.; Brené, S.; Mourier, A.; Coppotelli, G.; Freyer, C.; Lagouge, M.; Hoffer, B.J.; Olson, L.; et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 2013, 501, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.M.; Coppotelli, G.; Hoffer, B.J.; Olson, L. Maternally transmitted mitochondrial DNA mutations can reduce lifespan. Sci. Rep. 2014, 4, 6569. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K.; Kraytsberg, Y.; de Grey, A.D.; Vijg, J.; Schon, E.A. Does premature aging of the mtDNA mutator mouse prove that mtDNA mutations are involved in natural aging? Aging Cell 2006, 5, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Huang, C.H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Foury, F.; Vanderstraeten, S. Yeast mitochondrial DNA mutators with deficient proofreading exonucleolytic activity. EMBO J. 1992, 11, 2717–2726. [Google Scholar] [PubMed]
- Vanderstraeten, S.; Van den Brule, S.; Hu, J.; Foury, F. The role of 3′-5′ exonucleolytic proofreading and mismatch repair in yeast mitochondrial DNA error avoidance. J. Biol. Chem. 1998, 273, 23690–23697. [Google Scholar] [CrossRef] [PubMed]
- Hiona, A.; Sanz, A.; Kujoth, G.C.; Pamplona, R.; Seo, A.Y.; Hofer, T.; Someya, S.; Miyakawa, T.; Nakayama, C.; Samhan-Arias, A.K.; et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE 2010, 5, e11468. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Wallace, D.C.; Martin, G.M. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc. Natl Acad. Sci. USA 2005, 102, 18769–18770. [Google Scholar] [CrossRef] [PubMed]
- Vermulst, M.; Wanagat, J.; Kujoth, G.C.; Bielas, J.H.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008, 40, 392–394. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chen, T.; Wanagat, J.; Laflamme, M.; Marcinek, D.J.; Emond, M.J.; Ngo, C.P.; Prolla, T.A.; Rabinovitch, P.S. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010, 9, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.J.; Hamalainen, R.H.; Yatsuga, S.; Uutela, M.; Terzioglu, M.; Gotz, A.; Forsstrom, S.; Salven, P.; Angers-Loustau, A.; Kopra, O.H.; et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012, 15, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Bourgeois, J.M.; Ogborn, D.I.; Little, J.P.; Hettinga, B.P.; Akhtar, M.; Thompson, J.E.; Melov, S.; Mocellin, N.J.; Kujoth, G.C.; et al. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc. Natl Acad. Sci. USA 2011, 108, 4135–4140. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, I.G.; Vyssokikh, M.Y.; Gibanova, N.; Csikasz, R.I.; Edgar, D.; Hallden-Waldemarson, A.; Rozhdestvenskaya, Z.; Bakeeva, L.E.; Vays, V.B.; Pustovidko, A.V.; et al. Improved health-span and lifespan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1. Aging 2017, 9, 315–339. [Google Scholar] [CrossRef] [PubMed]
- Logan, A.; Shabalina, I.G.; Prime, T.A.; Rogatti, S.; Kalinovich, A.V.; Hartley, R.C.; Budd, R.C.; Cannon, B.; Murphy, M.P. In vivo levels of mitochondrial hydrogen peroxide increase with age in mtDNA mutator mice. Aging Cell 2014, 13, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Weis, S.; Mehraein, P.; Müller-Höcker, J. Cytochrome c oxidase defects of the human substantia nigra in normal aging. Neurobiol. Aging 1996, 17, 843–848. [Google Scholar] [CrossRef]
- Taylor, R.W.; Barron, M.J.; Borthwick, G.M.; Gospel, A.; Chinnery, P.F.; Samuels, D.C.; Taylor, G.A.; Plusa, S.M.; Needham, S.J.; Greaves, L.C.; et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Invest. 2003, 112, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Fellous, T.G.; Islam, S.; Tadrous, P.J.; Elia, G.; Kocher, H.M.; Bhattacharya, S.; Mears, L.; Turnbull, D.M.; Taylor, R.W.; Greaves, L.C.; et al. Locating the stem cell niche and tracing hepatocyte lineages in human liver. Hepatology 2009, 49, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K.; Bodyak, N.; Thilly, W.G.; van Orsouw, N.J.; Zhang, X.; Coller, H.A.; Perls, T.T.; Upton, M.; Vijg, J.; Wei, J.Y. Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999, 27, 2434–2441. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fayet, G.; Jansson, M.; Sternberg, D.; Moslemi, A.R.; Blondy, P.; Lombès, A.; Fardeau, M.; Oldfors, A. Ageing muscle: Clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul. Disord. 2002, 12, 484–493. [Google Scholar] [CrossRef]
- Payne, B.A.; Wilson, I.J.; Hateley, C.A.; Horvath, R.; Santibanez-Koref, M.; Samuels, D.C.; Price, D.A.; Chinnery, P.F. Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtDNA mutations. Nat. Genet. 2011, 43, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Lott, M.T.; Voljavec, A.S.; Soueidan, S.A.; Costigan, D.A.; Wallace, D.C. Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: A slip-replication model and metabolic therapy. Proc. Natl. Acad. Sci. USA 1989, 86, 7952–7956. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Lightowlers, R.N.; Turnbull, D.M. What causes mitochondrial DNA deletions in human cells? Nat. Genet. 2008, 40, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Moraes, C.T. Double-strand breaks of mouse muscle mtDNA promote large deletions similar to multiple mtDNA deletions in humans. Hum. Mol. Genet. 2005, 14, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Popadin, K.Y.; Markuzon, N.; Orlov, Y.L.; Kraytsberg, Y.; Krishnan, K.J.; Zsurka, G.; Turnbull, D.M.; Kunz, W.S.; Khrapko, K. Repeats, longevity and the sources of mtDNA deletions: Evidence from ‘deletional spectra’. Trends Genet. 2010, 26, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Tadi, S.K.; Sebastian, R.; Dahal, S.; Babu, R.K.; Choudhary, B.; Raghavan, S.C. Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions. Mol. Biol. Cell 2016, 27, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Moraes, C.T. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum. Mol. Genet. 2009, 18, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Wanrooij, S.; Luoma, P.; van Goethem, G.; van Broeckhoven, C.; Suomalainen, A.; Spelbrink, J.N. Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 2004, 32, 3053–3064. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, T.J.; Zsurka, G.; Peeva, V.; Schöler, S.; Szczesny, R.J.; Cysewski, D.; Reyes, A.; Kornblum, C.; Sciacco, M.; Moggio, M.; et al. Linear mtDNA fragments and unusual mtDNA rearrangements associated with pathological deficiency of MGME1 exonuclease. Hum. Mol. Genet. 2014, 23, 6147–6162. [Google Scholar] [CrossRef] [PubMed]
- Moretton, A.; Morel, F.; Macao, B.; Lachaume, P.; Ishak, L.; Lefebvre, M.; Garreau-Balandier, I.; Vernet, P.; Falkenberg, M.; Farge, G. Selective mitochondrial DNA degradation following double-strand breaks. PLoS One 2017, 12, e0176795. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Kraytsberg, Y.; Krishnan, K.J.; Ohno, N.; Ziabreva, I.; Reeve, A.; Trapp, B.D.; Newcombe, J.; Reynolds, R.; Lassmann, H.; et al. Clonally expanded mitochondrial DNA deletions within the choroid plexus in multiple sclerosis. Acta Neuropathol. 2012, 124, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Bailey, L.J.; Cluett, T.J.; Reyes, A.; Prolla, T.A.; Poulton, J.; Leeuwenburgh, C.; Holt, I.J. Mice expressing an error-prone DNA polymerase in mitochondria display elevated replication pausing and chromosomal breakage at fragile sites of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Vila, M.; Perier, C. The Parkinson disease mitochondrial hypothesis: Where are we at? Neuroscientist 2016, 22, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef] [PubMed]
- Nido, G.S.; Dölle, C.; Flønes, I.; Tuppen, H.A.; Alves, G.; Tysnes, O.B.; Haugarvoll, K.; Tzoulis, C. Ultradeep mapping of neuronal mitochondrial deletions in Parkinson’s disease. Neurobiol. Aging. 2017, 63, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Vögtle, F.N.; Taskin, A.A.; Teixeira, P.F.; Ring, J.; Burkhart, J.M.; Burger, N.; Pinho, C.M.; Tadic, J.; Loreth, D.; et al. Amyloid-β peptide induces mitochondrial dysfunction by inhibition of preprotein maturation. Cell Metab. 2014, 20, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Simon, D.K.; Ahn, C.H.; Kim, L.M.; Beal, M.F. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum. Mol. Genet 2002, 11, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Hanes, J.W.; Thal, D.M.; Johnson, K.A. Incorporation and replication of 8-oxo-deoxyguanosine by the human mitochondrial DNA polymerase. J. Biol. Chem. 2006, 281, 36241–36248. [Google Scholar] [CrossRef] [PubMed]
- Kunz, W.S.; Kudin, A.P.; Vielhaber, S.; Blümcke, I.; Zuschratter, W.; Schramm, J.; Beck, H.; Elger, C.E. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann. Neurol. 2000, 48, 766–773. [Google Scholar] [CrossRef]
- Baron, M.; Kudin, A.P.; Kunz, W.S. Mitochondrial dysfunction in neurodegenerative disorders. Biochem. Soc. Trans. 2007, 35, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Mahad, D.J. Mitochondrial changes associated with demyelination: Consequences for axonal integrity. Mitochondrion 2012, 12, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zsurka, G.; Peeva, V.; Kotlyar, A.; Kunz, W.S. Is There Still Any Role for Oxidative Stress in Mitochondrial DNA-Dependent Aging? Genes 2018, 9, 175. https://doi.org/10.3390/genes9040175
Zsurka G, Peeva V, Kotlyar A, Kunz WS. Is There Still Any Role for Oxidative Stress in Mitochondrial DNA-Dependent Aging? Genes. 2018; 9(4):175. https://doi.org/10.3390/genes9040175
Chicago/Turabian StyleZsurka, Gábor, Viktoriya Peeva, Alexander Kotlyar, and Wolfram S. Kunz. 2018. "Is There Still Any Role for Oxidative Stress in Mitochondrial DNA-Dependent Aging?" Genes 9, no. 4: 175. https://doi.org/10.3390/genes9040175
APA StyleZsurka, G., Peeva, V., Kotlyar, A., & Kunz, W. S. (2018). Is There Still Any Role for Oxidative Stress in Mitochondrial DNA-Dependent Aging? Genes, 9(4), 175. https://doi.org/10.3390/genes9040175