Genomic Characterization of Listeria monocytogenes Isolates Associated with Clinical Listeriosis and the Food Production Environment in Ireland


 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Isolates
2.2. Pulsed Field Gel Electrophoresis
2.3. Genome Sequencing and Annotation
2.4. Multilocus Sequence Typing and Clonal Complex
2.5. Single Nucleotide Polymorphisms Analysis
2.6. Public Data Sources
2.7. Pan- and Core-Genomic Profiling of Protein-Coding Genes
3. Results and Discussion
3.1. Serotypes and Clonal Complexes of Clinical Isolates
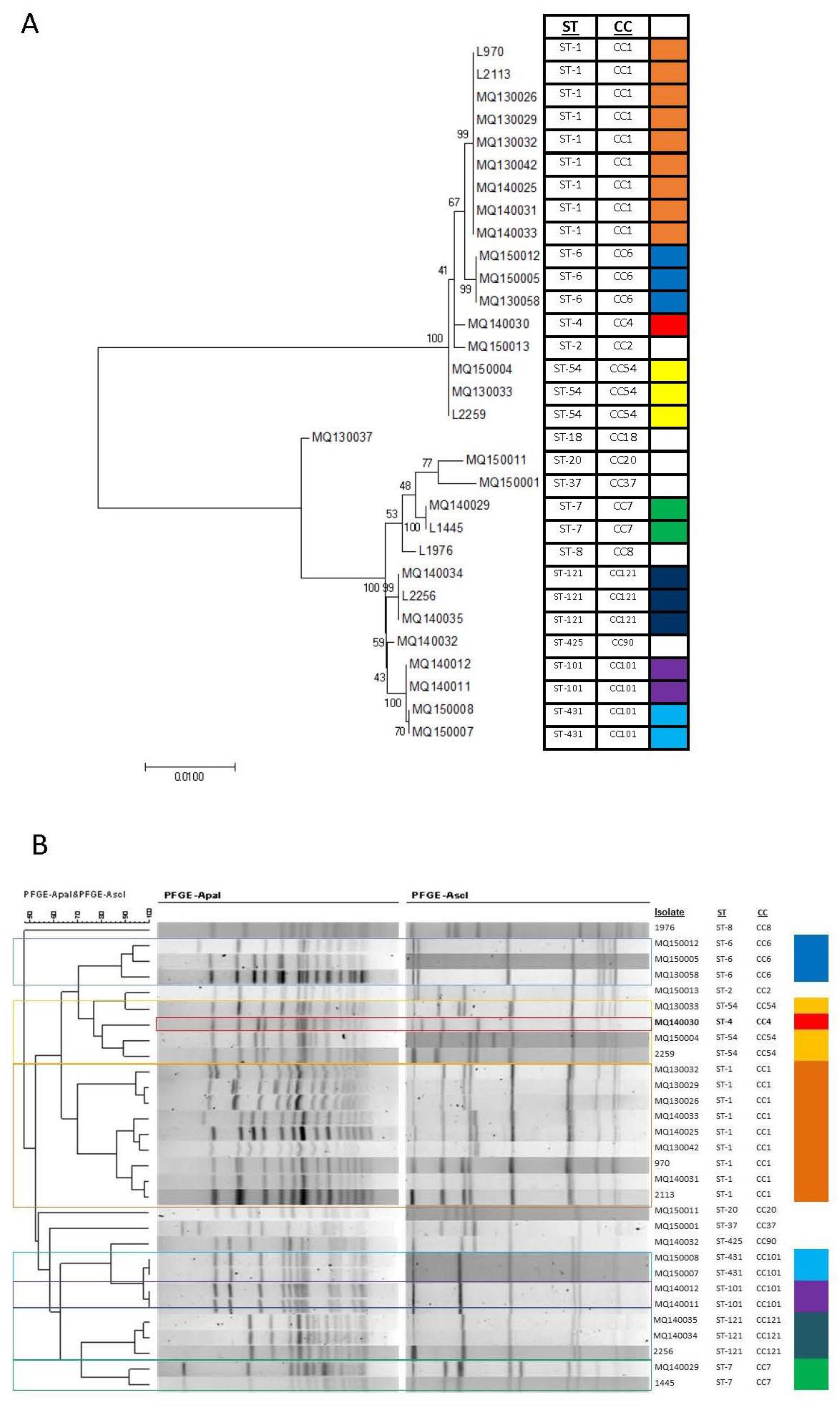
3.2. Relationship between Sequence Type and PFGE Profiles
3.3. Single-Nucleotide Polymorphism Analysis of Isolates
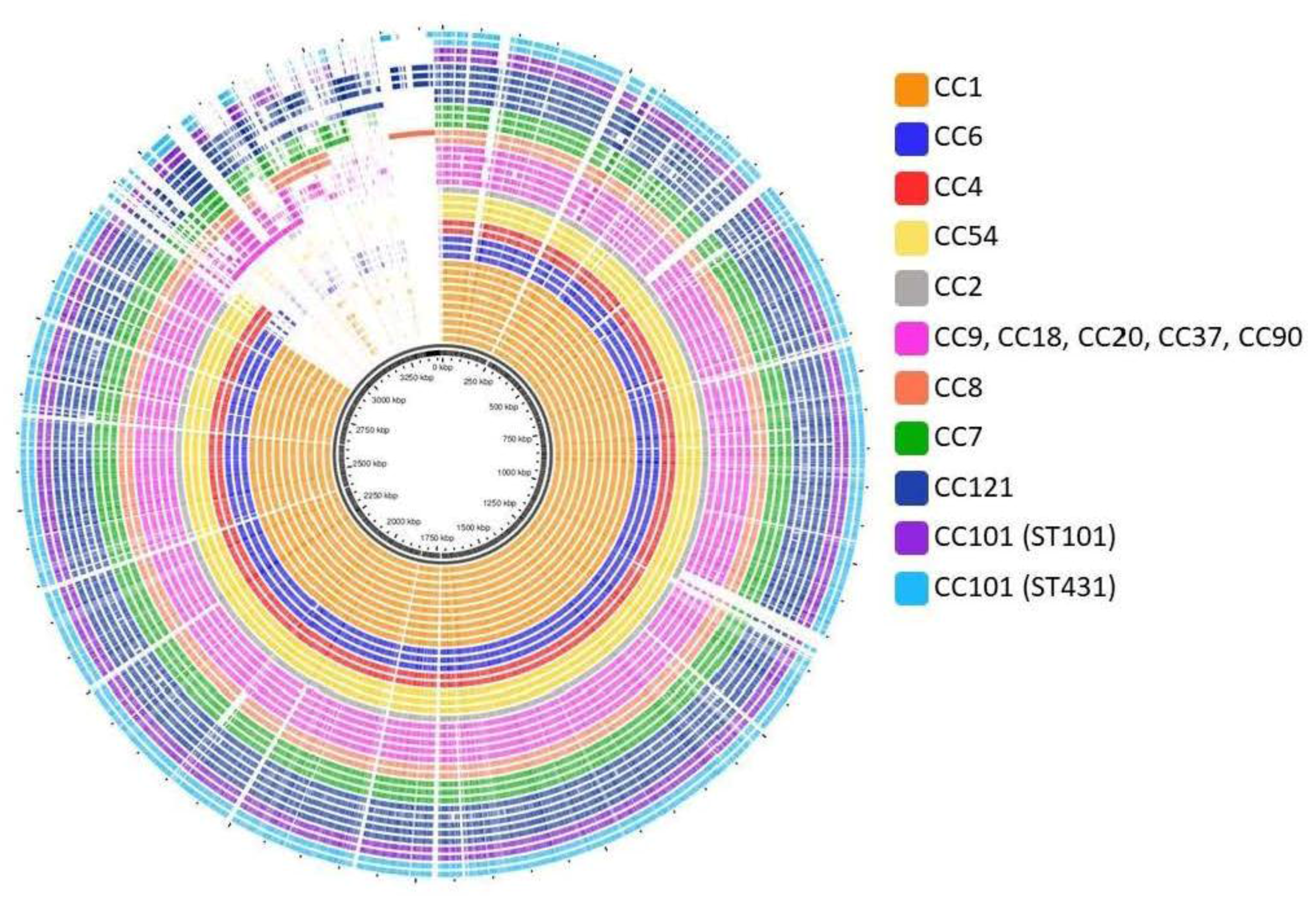
3.4. Pan- and Core-Genome Analysis
3.5. Listeria Pathogenicity Islands
3.6. Internalins
3.7. Stress Survival Islet (SSI-1)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Noordhout, C.M.; Devleesschauwer, B.; Angulo, F.J.; Verbeke, G.; Haagsma, J.; Kirk, M.; Havelaar, A.; Speybroeck, N. The global burden of listeriosis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 1073–1082. [Google Scholar] [CrossRef]
- Vazquez-Boland, J.A.; Kuhn, M.; Berche, P.; Chakraborty, T.; Dominguez-Bernal, G.; Goebel, W.; Gonzalez-Zorn, B.; Wehland, J.; Kreft, J. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 2001, 14, 584–640. [Google Scholar] [CrossRef] [PubMed]
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States—Major pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2014. EFSA J. 2015, 13, 4329. [Google Scholar]
- Health Protection Surveillance Centre (HPSC). HPSC Annual Epidemiological Report 2015; Health Protection Surveillance Centre: Dublin, Ireland, 2016; Volume 75. [Google Scholar]
- Lomonaco, S.; Nucera, D. Molecular subtyping methods for Listeria monocytogenes: Tools for tracking and control. In DNA Methods in Food Safety: Molecular Typing of Foodborne and Waterborne Bacterial Pathogens; Wiley: Hoboken, NJ, USA, 2014; pp. 303–336. [Google Scholar]
- Maury, M.M.; Tsai, Y.H.; Charlier, C.; Touchon, M.; Chenal-Francisque, V.; Leclercq, A.; Criscuolo, A.; Gaultier, C.; Roussel, S.; Brisabois, A.; et al. Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nat. Genet. 2016, 48, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Moura, A.; Criscuolo, A.; Pouseele, H.; Maury, M.M.; Leclercq, A.; Tarr, C.; Bjorkman, J.T.; Dallman, T.; Reimer, A.; Enouf, V.; et al. Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbiol. 2016, 2, 16185. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Perez-Osorio, A.; Wang, Y.; Eckmann, K.; Glover, W.A.; Allard, M.W.; Brown, E.W.; Chen, Y. Whole genome sequencing analyses of Listeria monocytogenes that persisted in a milkshake machine for a year and caused illnesses in Washington state. BMC Microbiol. 2017, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Kvistholm Jensen, A.; Nielsen, E.M.; Bjorkman, J.T.; Jensen, T.; Muller, L.; Persson, S.; Bjerager, G.; Perge, A.; Krause, T.G.; Kiil, K.; et al. Whole-genome sequencing used to investigate a nationwide outbreak of listeriosis caused by ready-to-eat delicatessen meat, Denmark, 2014. Clin. Infect. Dis. 2016, 63, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, J.L.; Berche, P.; Frehel, C.; Gouin, E.; Cossart, P. Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell 1991, 65, 1127–1141. [Google Scholar] [CrossRef]
- Disson, O.; Grayo, S.; Huillet, E.; Nikitas, G.; Langa-Vives, F.; Dussurget, O.; Ragon, M.; Le Monnier, A.; Babinet, C.; Cossart, P.; et al. Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 2008, 455, 1114–1118. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, M.; Vandormael-Pournin, S.; Lefort, J.; Huerre, M.; Gounon, P.; Dupuy, C.; Babinet, C.; Cossart, P. A transgenic model for listeriosis: Role of internalin in crossing the intestinal barrier. Science 2001, 292, 1722–1725. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, C.; Doumith, M.; Gordon, J.I.; Martin, P.M.; Cossart, P.; Lecuit, M. A molecular marker for evaluating the pathogenic potential of foodborne Listeria monocytogenes. J. Infect. Dis 2004, 189, 2094–2100. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, K.K.; Ivy, R.A.; Ho, A.J.; Fortes, E.D.; Njaa, B.L.; Peters, R.M.; Wiedmann, M. InlA premature stop codons are common among Listeria monocytogenes isolates from foods and yield virulence-attenuated strains that confer protection against fully virulent strains. Appl. Environ. Microbiol. 2008, 74, 6570–6583. [Google Scholar] [CrossRef] [PubMed]
- Gouin, E.; Mengaud, J.; Cossart, P. The virulence gene cluster of Listeria monocytogenes is also present in Listeria ivanovii, an animal pathogen, and Listeria seeligeri, a nonpathogenic species. Infect. Immun. 1994, 62, 3550–3553. [Google Scholar] [PubMed]
- Cotter, P.D.; Draper, L.A.; Lawton, E.M.; Daly, K.M.; Groeger, D.S.; Casey, P.G.; Ross, R.P.; Hill, C. Listeriolysin S, a novel peptide haemolysin associated with a subset of lineage I Listeria monocytogenes. PLoS Pathog. 2008, 4, e1000144. [Google Scholar] [CrossRef] [PubMed]
- Quereda, J.J.; Dussurget, O.; Nahori, M.A.; Ghozlane, A.; Volant, S.; Dillies, M.A.; Regnault, B.; Kennedy, S.; Mondot, S.; Villoing, B.; et al. Bacteriocin from epidemic Listeria strains alters the host intestinal microbiota to favor infection. Proc. Natl. Acad. Sci. USA 2016, 113, 5706–5711. [Google Scholar] [CrossRef] [PubMed]
- Gahan, C.G.; Hill, C. Listeria monocytogenes: Survival and adaptation in the gastrointestinal tract. Front. Cell. Infect. Microbiol. 2014, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.; Begley, M.; Hill, C.; Gahan, C.G. A five-gene stress survival islet (SSI-1) that contributes to the growth of Listeria monocytogenes in suboptimal conditions. J. Appl. Microbiol. 2010, 109, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Leong, D.; NicAogain, K.; Luque-Sastre, L.; McManamon, O.; Hunt, K.; Alvarez-Ordonez, A.; Scollard, J.; Schmalenberger, A.; Fanning, S.; O’Byrne, C.; et al. A 3-year multi-food study of the presence and persistence of Listeria monocytogenes in 54 small food businesses in Ireland. Int. J. Food Microbiol. 2017, 249, 18–26. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, A.; Hilliard, A.; Morgan, C.A.; Culligan, E.P.; Leong, D.; DeLappe, N.; Hill, C.; Jordan, K.; Cormican, M.; Gahan, C.G.M. Draft genome sequences of 25 Listeria monocytogenes isolates associated with human clinical listeriosis in Ireland. Genome Announc. 2017, 5, e00184-17. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-mem. arXiv, 2013; arXiv:1303.3997. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Rissman, A.I.; Mau, B.; Biehl, B.S.; Darling, A.E.; Glasner, J.D.; Perna, N.T. Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 2009, 25, 2071–2073. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Ragon, M.; Wirth, T.; Hollandt, F.; Lavenir, R.; Lecuit, M.; Le Monnier, A.; Brisse, S. A new perspective on Listeria monocytogenes evolution. PLoS Pathog. 2008, 4, e1000146. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Kaas, R.S.; Leekitcharoenphon, P.; Aarestrup, F.M.; Lund, O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS ONE 2014, 9, e104984. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, J.; Yu, P.; Ge, P.; Jiang, Y.; Xu, R.; Chen, R.; Liu, X. Identification of antibiotic resistance genes in the multidrug-resistant Acinetobacter baumannii strain, MDR-SHH02, using whole-genome sequencing. Int. J. Mol. Med. 2017, 39, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Petkau, A.; Stuart-Edwards, M.; Stothard, P.; Van Domselaar, G. Interactive microbial genome visualization with GView. Bioinformatics 2010, 26, 3125–3126. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Bhardwaj, A.; Bag, S.K.; Sokurenko, E.V.; Chattopadhyay, S. Pancoregen—Profiling, detecting, annotating protein-coding genes in microbial genomes. Genomics 2015, 106, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Haase, J.K.; Didelot, X.; Lecuit, M.; Korkeala, H.; Achtman, M. The ubiquitous nature of Listeria monocytogenes clones: A large-scale multilocus sequence typing study. Environ. Microbiol. 2014, 16, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Linnan, M.J.; Mascola, L.; Lou, X.D.; Goulet, V.; May, S.; Salminen, C.; Hird, D.W.; Yonekura, M.L.; Hayes, P.; Weaver, R.; et al. Epidemic listeriosis associated with mexican-style cheese. N. Engl. J. Med. 1988, 319, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Leong, D.; Alvarez-Ordonez, A.; Zaouali, S.; Jordan, K. Examination of Listeria monocytogenes in seafood processing facilities and smoked salmon in the Republic of Ireland. J. Food Prot. 2015, 78, 2184–2190. [Google Scholar] [CrossRef] [PubMed]
- Henri, C.; Leekitcharoenphon, P.; Carleton, H.A.; Radomski, N.; Kaas, R.S.; Mariet, J.F.; Felten, A.; Aarestrup, F.M.; Gerner Smidt, P.; Roussel, S.; et al. An assessment of different genomic approaches for inferring phylogeny of Listeria monocytogenes. Front. Microbiol. 2017, 8, 2351. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.C.; Mercoulia, K.; Tomita, T.; Easton, M.; Li, H.Y.; Bulach, D.M.; Stinear, T.P.; Seemann, T.; Howden, B.P. Prospective whole-genome sequencing enhances national surveillance of Listeria monocytogenes. J. Clin. Microbiol. 2016, 54, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Leekitcharoenphon, P.; Nielsen, E.M.; Kaas, R.S.; Lund, O.; Aarestrup, F.M. Evaluation of whole genome sequencing for outbreak detection of Salmonella enterica. PLoS ONE 2014, 9, e87991. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.J.; Lappi, V.; Wolfgang, W.J.; Lapierre, P.; Palumbo, M.J.; Medus, C.; Boxrud, D. Characterization of foodborne outbreaks of Salmonella enterica serovar enteritidis with whole-genome sequencing single nucleotide polymorphism-based analysis for surveillance and outbreak detection. J. Clin. Microbiol. 2015, 53, 3334–3340. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, V.; Denayer, S.; Roosens, N.H.; Mattheus, W.; Bertrand, S.; Marchal, K.; Dierick, K.; De Keersmaecker, S.C. Whole genome sequence analysis of Salmonella enteritidis PT4 outbreaks from a national reference laboratory's viewpoint. PLoS Curr. 2015, 7, 159. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Phillippy, A.M.; Li, Z.; Salzberg, S.L.; Zhang, W. Probing the pan-genome of Listeria monocytogenes: New insights into intraspecific niche expansion and genomic diversification. BMC Genomics 2010, 11, 500. [Google Scholar] [CrossRef] [PubMed]
- Den Bakker, H.C.; Cummings, C.A.; Ferreira, V.; Vatta, P.; Orsi, R.H.; Degoricija, L.; Barker, M.; Petrauskene, O.; Furtado, M.R.; Wiedmann, M. Comparative genomics of the bacterial genus Listeria: Genome evolution is characterized by limited gene acquisition and limited gene loss. BMC Genomics 2010, 11, 688. [Google Scholar] [CrossRef] [PubMed]
- Dunn, K.A.; Bielawski, J.P.; Ward, T.J.; Urquhart, C.; Gu, H. Reconciling ecological and genomic divergence among lineages of Listeria under an “extended mosaic genome concept”. Mol. Biol. Evol. 2009, 26, 2605–2615. [Google Scholar] [CrossRef] [PubMed]
- Kuenne, C.; Billion, A.; Mraheil, M.A.; Strittmatter, A.; Daniel, R.; Goesmann, A.; Barbuddhe, S.; Hain, T.; Chakraborty, T. Reassessment of the Listeria monocytogenes pan-genome reveals dynamic integration hotspots and mobile genetic elements as major components of the accessory genome. BMC Genomics 2013, 14, 47. [Google Scholar] [CrossRef] [PubMed]
- Moura, A.; Tourdjman, M.; Leclercq, A.; Hamelin, E.; Laurent, E.; Fredriksen, N.; van Cauteren, D.; Bracq-Dieye, H.; Thouvenot, P.; Vales, G.; et al. . Real-time whole-genome sequencing for surveillance of Listeria monocytogenes, France. Emerg. Infect. Dis. 2017, 23, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Cantinelli, T.; Chenal-Francisque, V.; Diancourt, L.; Frezal, L.; Leclercq, A.; Wirth, T.; Lecuit, M.; Brisse, S. “Epidemic clones” of Listeria monocytogenes are widespread and ancient clonal groups. J. Clin. Microbiol. 2013, 51, 3770–3779. [Google Scholar] [CrossRef]
- Knudsen, G.M.; Nielsen, J.B.; Marvig, R.L.; Ng, Y.; Worning, P.; Westh, H.; Gram, L. Genome-wide-analyses of Listeria monocytogenes from food-processing plants reveal clonal diversity and date the emergence of persisting sequence types. Environ. Microbiol. Rep. 2017, 9, 428–440. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Genbank Accession Number | ST 1 | CC 2 | Lineage | Serotype | Year of Isolation | Sample Type | Pulsotype |
|---|---|---|---|---|---|---|---|---|
| MQ130026 | MUZG00000000 | ST-1 | CC1 | I | 4b | 2013 | Blood | P2 * |
| L970 | PJJD00000000 | ST-1 | CC1 | I | 4b | 2013 | Food production ** | P2 * |
| MQ130029 | MVED00000000 | ST-1 | CC1 | I | 4b | 2013 | CSF 3 | P2 * |
| MQ130032 | MVEE00000000 | ST-1 | CC1 | I | 4b | 2013 | Blood | P2 * |
| MQ130042 | MVEG00000000 | ST-1 | CC1 | I | 4b | 2013 | Pleural Swab | P1 * |
| MQ140025 | MVEK00000000 | ST-1 | CC1 | I | 4b | 2014 | Ear Swab | P68 |
| MQ140031 | MVEN00000000 | ST-1 | CC1 | I | 4b | 2014 | Blood | P2 * |
| MQ140033 | MVEP00000000 | ST-1 | CC1 | I | 4b | 2014 | Blood | P1 * |
| L2113 | PJJE00000000 | ST-1 | CC1 | I | 4b | 2015 | Food production | P2 * |
| MQ150012 | MVEY00000000 | ST-6 | CC6 | I | 4b | 2015 | Blood | P13 * |
| MQ150005 | MVEU00000000 | ST-6 | CC6 | I | 4b | 2015 | Blood | P13 * |
| MQ130058 | MVEH00000000 | ST-6 | CC6 | I | 4b | 2013 | Blood | P13 * |
| MQ140030 | MVEM00000000 | ST-4 | CC4 | I | 4b | 2014 | CSF 3 | NC 4 |
| MQ150004 | MVET00000000 | ST-54 | CC54 | I | 4b | 2015 | Placental Swab | P6 * |
| MQ130033 | MVEF00000000 | ST-54 | CC54 | I | 4b | 2013 | Blood | P12 |
| L2259 | PJJF00000000 | ST-54 | CC54 | I | 4b | 2015 | Food production | P6 * |
| MQ150013 | MVEZ00000000 | ST-2 | CC2 | I | 4b | 2015 | Blood | P16 * |
| MQ130037 | MVFA00000000 | ST-18 | CC18 | II | 1/2a | 2013 | Blood | P32 * |
| MQ150011 | MVEX00000000 | ST-20 | CC20 | II | 1/2a | 2015 | Nasal Swab | NC |
| MQ150001 | MVES00000000 | ST-37 | CC37 | II | 1/2a | 2015 | Blood | P32 * |
| MQ140029 | MVEL00000000 | ST-7 | CC7 | II | 1/2a | 2014 | Blood | P31 * |
| L1445 | PJJG00000000 | ST-7 | CC7 | II | 1/2a | 2014 | Food production | P31 * |
| L1976 | PJJI00000000 | ST-8 | CC8 | II | 1/2c | 2015 | Food production | P48 * |
| MQ140034 | MVEQ00000000 | ST-121 | CC121 | II | 1/2a | 2014 | Blood—Mother/Infant | P59 * |
| L2256 | PJJH00000000 | ST-121 | CC121 | II | 1/2c | 2015 | Food production | P59 * |
| MQ140035 | MVER00000000 | ST-121 | CC121 | II | 1/2a | 2014 | Ear Swab—Mother/Infant | P59 * |
| MQ140032 | MVEO00000000 | ST-425 | CC90 | II | 1/2a | 2014 | Blood | NC |
| MQ140012 | MVEJ00000000 | ST-101 | CC101 | II | 1/2a | 2014 | Blood—Mother/Infant | P30 |
| MQ140011 | MVEI00000000 | ST-101 | CC101 | II | 1/2a | 2014 | Placental Surface Swab—Mother/Infant | P30 |
| MQ150008 | MVEW00000000 | ST-431 | CC101 | II | 1/2a | 2015 | Blood | P30 |
| MQ150007 | MVEV00000000 | ST-431 | CC101 | II | 1/2a | 2015 | CSF 3 | P30 |
| Sequence Type | Isolates | Reference Genome | Minimum SNPs | Maximum SNPs |
|---|---|---|---|---|
| ST1 | L970, L2113, MQ130026, MQ130029, MQ130032 *, MQ130042, MQ140025, MQ140031, MQ140033 | F2365 | 43 | 261 |
| F2365, L2113, MQ130026, MQ130029, MQ130032 *, MQ130042, MQ140025, MQ140031, MQ140033 | L970 | 42 | 256 | |
| F2365, L970, MQ130026, MQ130029, MQ130032 *, MQ130042, MQ140025, MQ140031, MQ140033 | L2113 | 42 | 254 | |
| F2365, L970, MQ130029, MQ130032 *, MQ130042 | MQ130026 | 44 | 190 | |
| F2365, L2113, MQ140031, MQ140033 * | MQ140025 | 66 | 259 | |
| ST6 | H7858, MQ130058, MQ150012 * | MQ150005 | 199 | 373 |
| ST54 | LM07-01337, MQ130033, MQ150004 * | L2259 | 65 | 115 |
| ST7 | J2692, L1846, L2676, MQ140029 * | L1445 | 1 | 409 |
| ST121 | 4423, 6179, L2256, La111, Lm1880, N53-1, MQ140035 * | MQ140034 | 3 | 461 |
| ST101 | 2012-L5240, 2012-L5323, Lm1840, MQ140012, MQ150007, MQ150008 * | MQ140011 | 1 | 145 |
| 2012-L5240, 2012-L5323, Lm1840, MQ150008 * | MQ150007 | 2 | 146 |
| Isolate | ST 1 | CC 2 | LIPI1 | LIPI3 | LIPI4 | InlA | SSI-1 |
|---|---|---|---|---|---|---|---|
| MQ130026 | ST-1 | CC1 | + 3 | + | − 4 | + 5 | − |
| L970 | ST-1 | CC1 | + | + | − | + | − |
| MQ130029 | ST-1 | CC1 | + | + | − | + | − |
| MQ130032 | ST-1 | CC1 | + | + | − | + | − |
| MQ130042 | ST-1 | CC1 | + | + | − | + | − |
| MQ140025 | ST-1 | CC1 | + | + | − | + | − |
| MQ140031 | ST-1 | CC1 | + | + | − | + | − |
| MQ140033 | ST-1 | CC1 | + | + | − | + | − |
| L2113 | ST-1 | CC1 | + | + | − | + | − |
| MQ150012 | ST-6 | CC6 | + | + | − | + | − |
| MQ150005 | ST-6 | CC6 | + | + | − | + | − |
| MQ130058 | ST-6 | CC6 | + | + | − | + | − |
| MQ140030 | ST-4 | CC4 | + | + | + | + | − |
| MQ150004 | ST-54 | CC54 | + | + | − | + | − |
| MQ130033 | ST-54 | CC54 | + | + | − | + | − |
| L2259 | ST-54 | CC54 | + | + | − | + | − |
| MQ150013 | ST-2 | CC2 | + | − | − | + | − |
| MQ130037 | ST-18 | CC18 | + | − | − | + | + |
| MQ150011 | ST-20 | CC20 | + | − | − | + | − |
| MQ150001 | ST-37 | CC37 | + | − | − | + | − |
| MQ140029 | ST-7 | CC7 | + | − | − | + | + |
| L1445 | ST-7 | CC7 | + | − | − | + | + |
| L1976 | ST-8 | CC8 | + | − | − | + | + |
| MQ140034 | ST-121 | CC121 | + | − | − | + | − |
| L2256 | ST-121 | CC121 | + | − | − | − | − |
| MQ140035 | ST-121 | CC121 | + | − | − | + | − |
| MQ140032 | ST-425 | CC90 | + | − | − | + | − |
| MQ140012 | ST-101 | CC101 | + | − | − | + | − |
| MQ140011 | ST-101 | CC101 | + | − | − | + | − |
| MQ150008 | ST-431 | CC101 | + | − | − | + | − |
| MQ150007 | ST-431 | CC101 | + | − | − | + | − |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hilliard, A.; Leong, D.; O’Callaghan, A.; Culligan, E.P.; Morgan, C.A.; DeLappe, N.; Hill, C.; Jordan, K.; Cormican, M.; Gahan, C.G.M. Genomic Characterization of Listeria monocytogenes Isolates Associated with Clinical Listeriosis and the Food Production Environment in Ireland. Genes 2018, 9, 171. https://doi.org/10.3390/genes9030171
Hilliard A, Leong D, O’Callaghan A, Culligan EP, Morgan CA, DeLappe N, Hill C, Jordan K, Cormican M, Gahan CGM. Genomic Characterization of Listeria monocytogenes Isolates Associated with Clinical Listeriosis and the Food Production Environment in Ireland. Genes. 2018; 9(3):171. https://doi.org/10.3390/genes9030171
Chicago/Turabian StyleHilliard, Amber, Dara Leong, Amy O’Callaghan, Eamonn P. Culligan, Ciara A. Morgan, Niall DeLappe, Colin Hill, Kieran Jordan, Martin Cormican, and Cormac G.M. Gahan. 2018. "Genomic Characterization of Listeria monocytogenes Isolates Associated with Clinical Listeriosis and the Food Production Environment in Ireland" Genes 9, no. 3: 171. https://doi.org/10.3390/genes9030171
APA StyleHilliard, A., Leong, D., O’Callaghan, A., Culligan, E. P., Morgan, C. A., DeLappe, N., Hill, C., Jordan, K., Cormican, M., & Gahan, C. G. M. (2018). Genomic Characterization of Listeria monocytogenes Isolates Associated with Clinical Listeriosis and the Food Production Environment in Ireland. Genes, 9(3), 171. https://doi.org/10.3390/genes9030171