Covalent Strategies for Targeting Messenger and Non-Coding RNAs: An Updated Review on siRNA, miRNA and antimiR Conjugates



Abstract
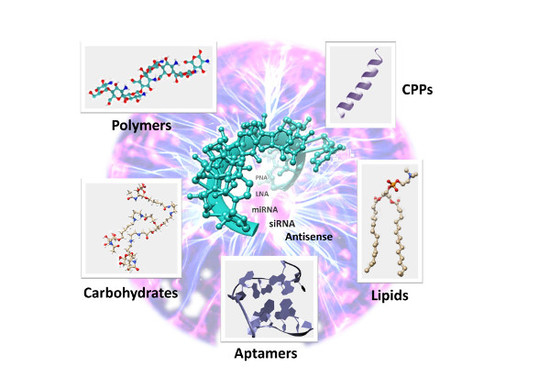
1. Introduction
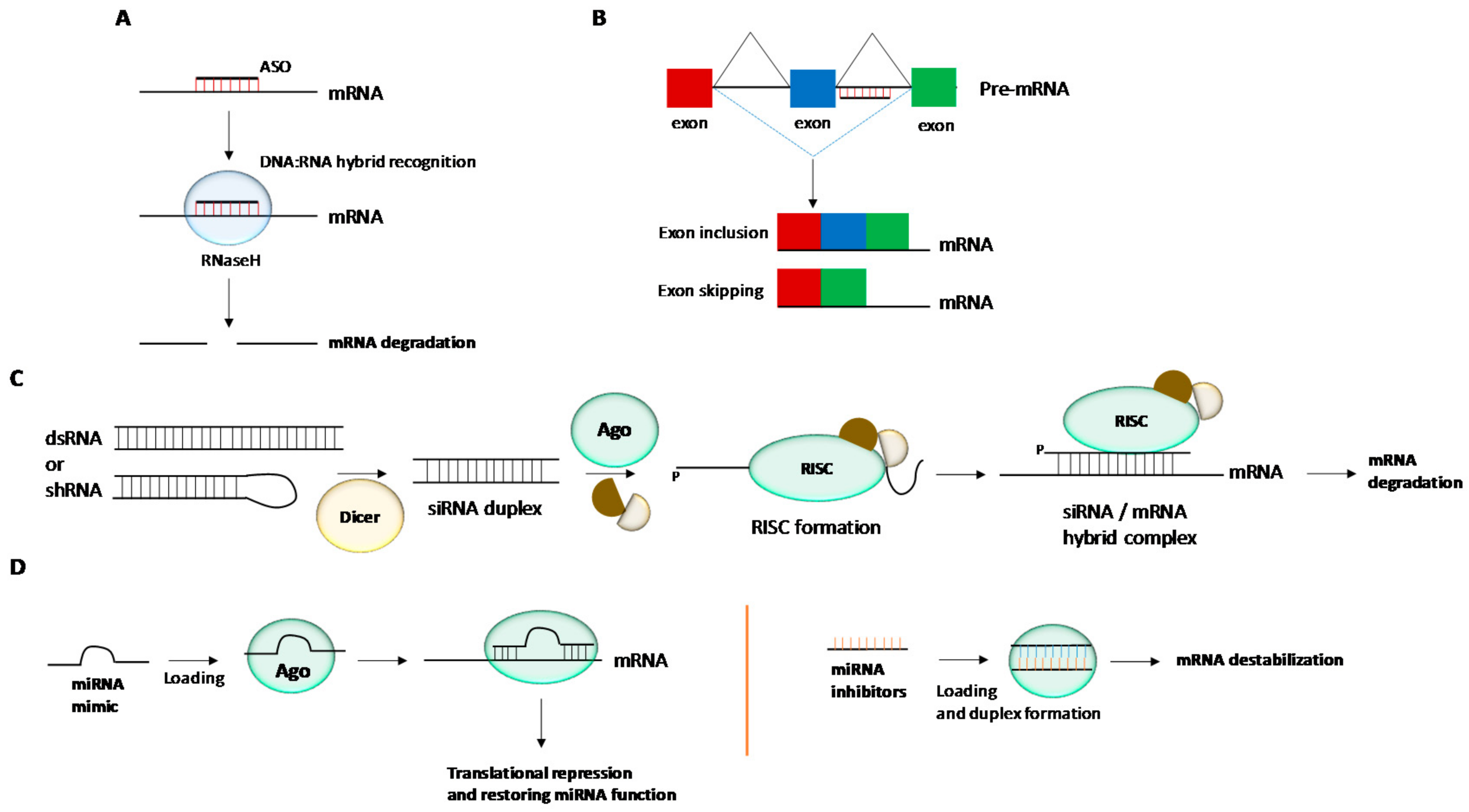
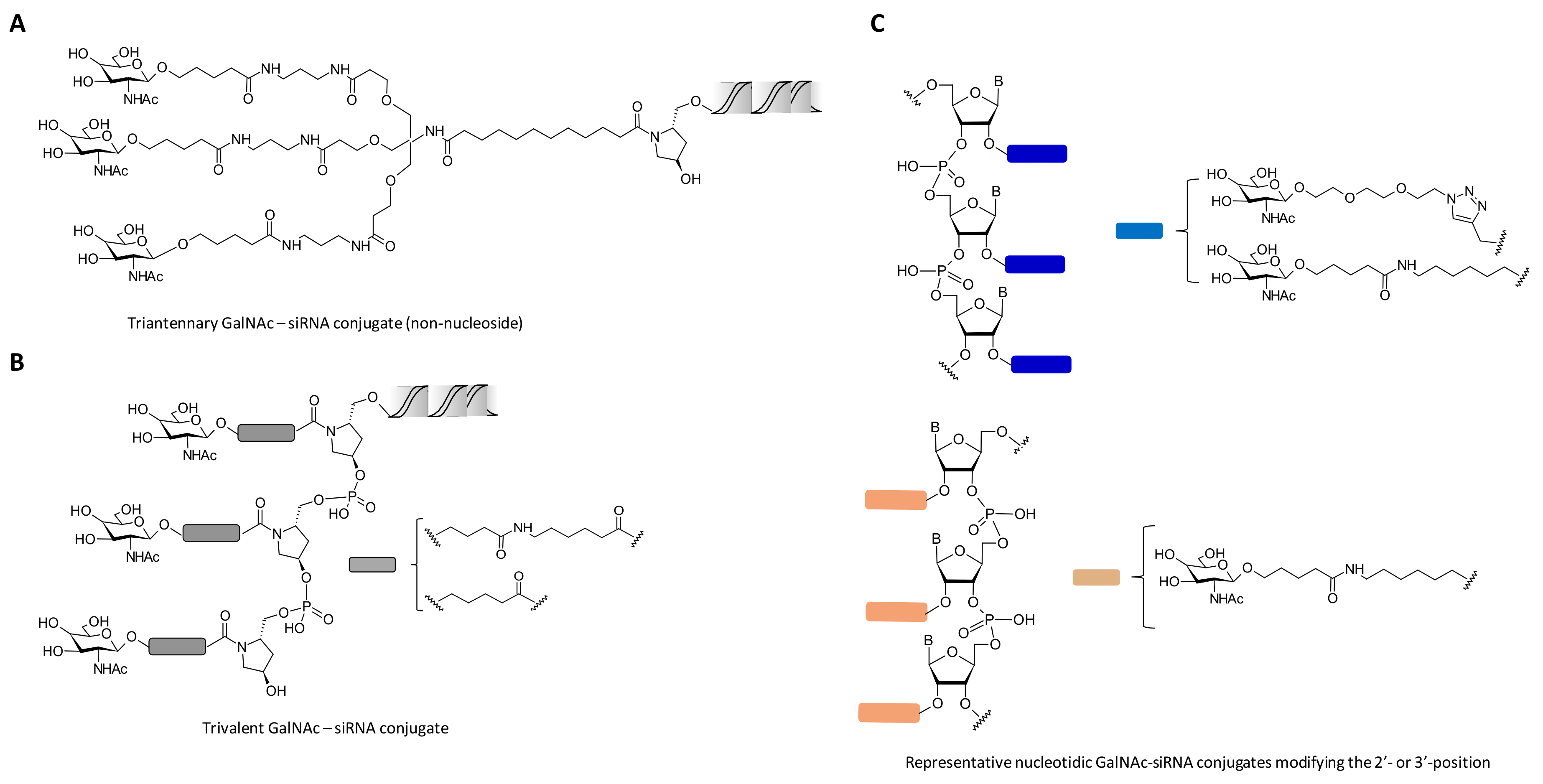
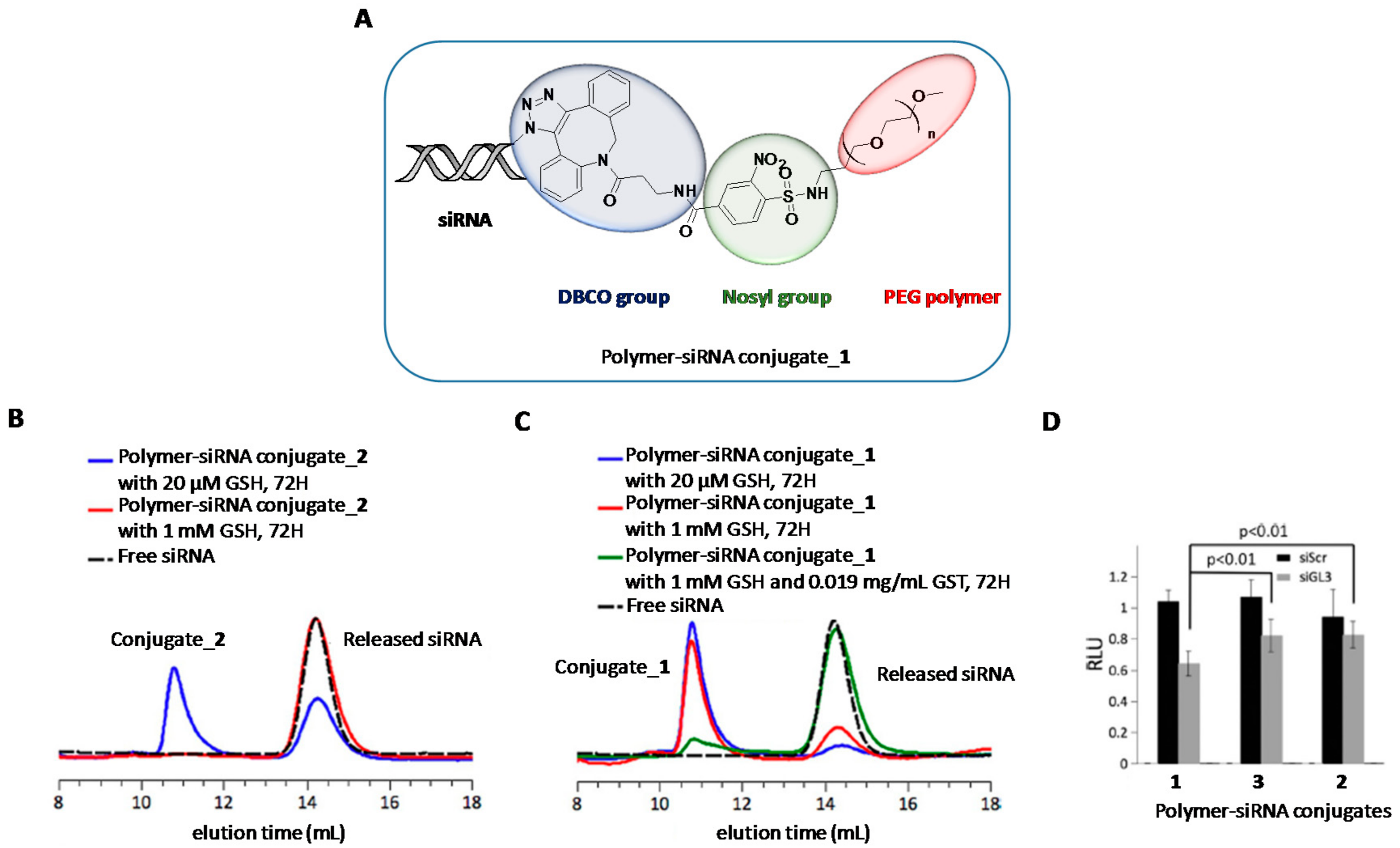
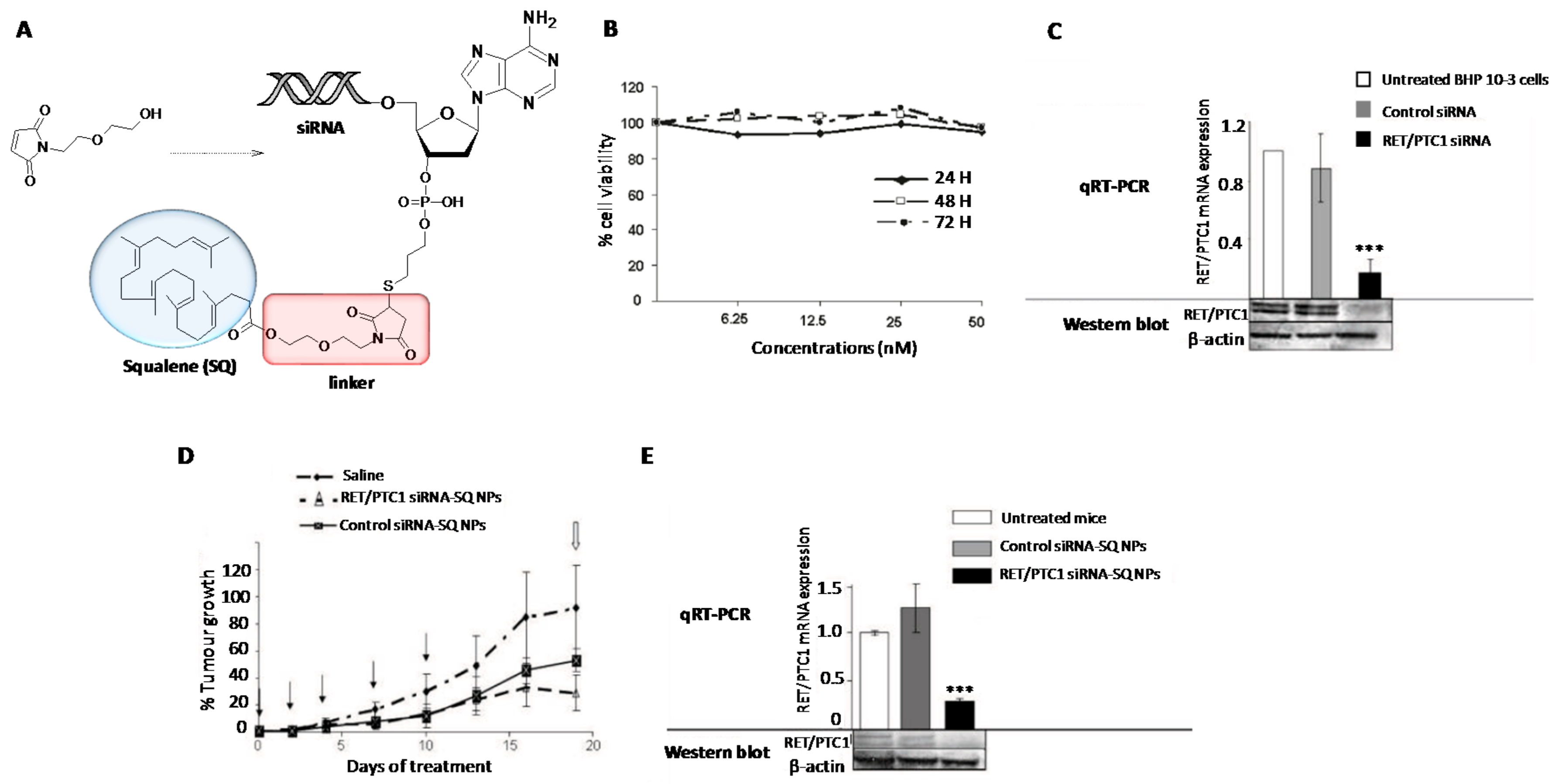
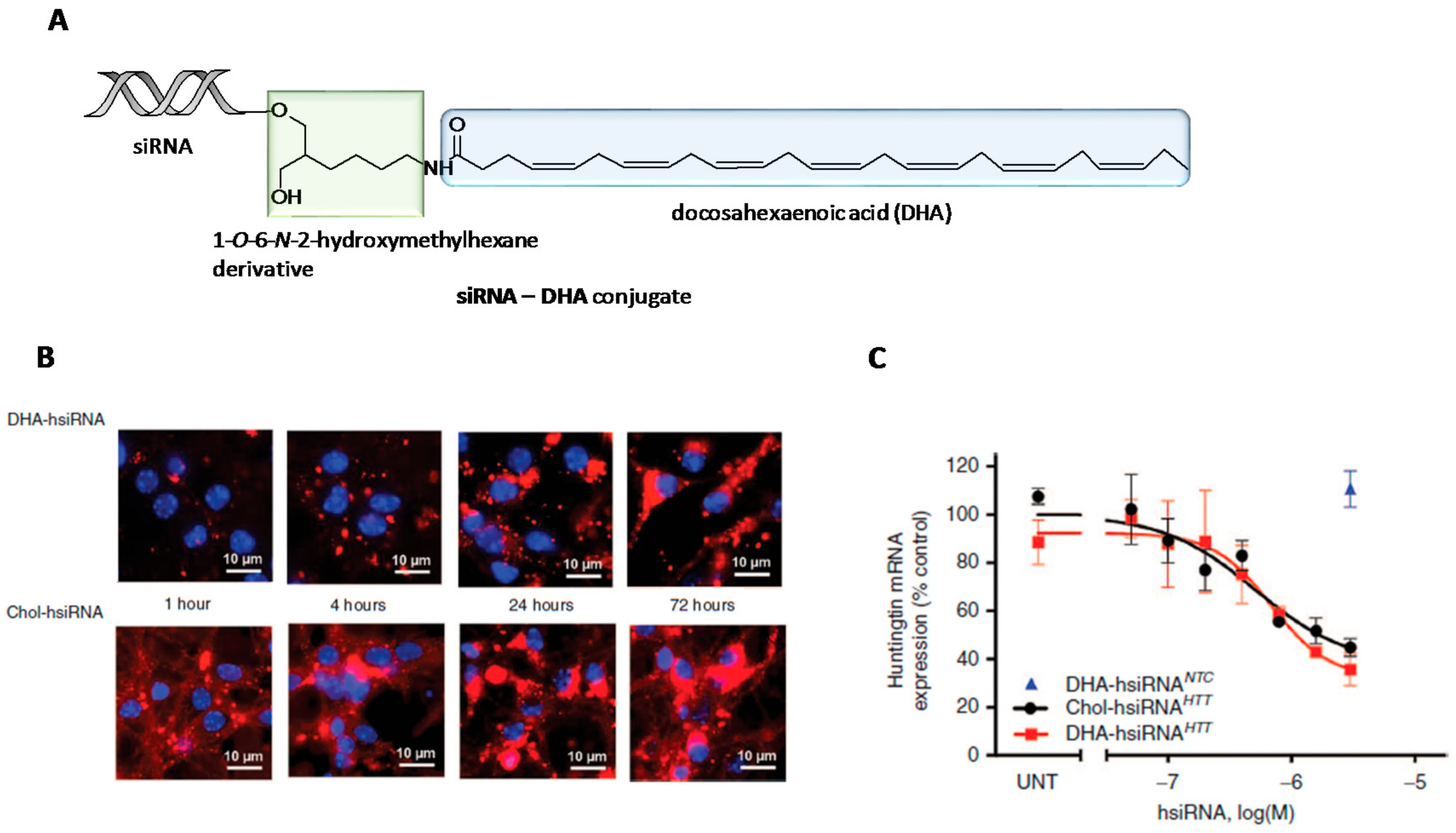
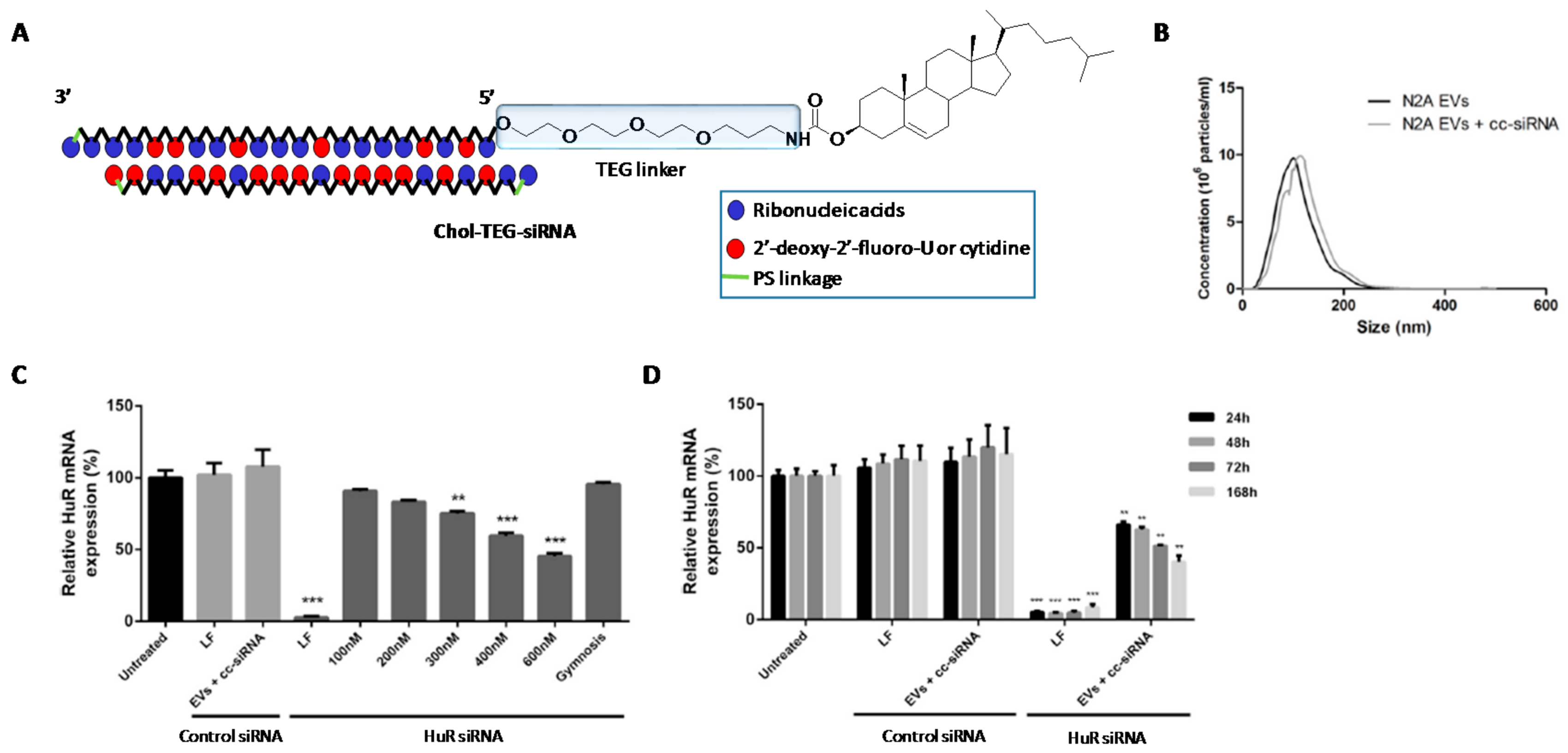
2. Chemical Modifications and siRNA Delivery
3. Chemical Modifications and miRNA Delivery
4. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Swayze, E.E. RNA Targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2016, 16, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Hüttenhofer, A.; Schattner, P.; Polacek, N. Non-coding RNAs: Hope or hype? Trends Genet. 2005, 21, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Rungta, P.; Prasad, A.K. Nucleic acid therapeutics: Basic concepts and recent developments. RSC Adv. 2014, 4, 16618. [Google Scholar] [CrossRef]
- Deleavey, G.F.; Damha, M.J. Designing chemically modified oligonucleotides for targeted gene silencing. Chem. Biol. 2012, 19, 937–954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, Y.; Zhi, D.; Zhang, S. Non-viral vectors for the mediation of RNAi. Bioorg. Chem. 2012, 40, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.H.; Jo, S.D.; Lee, Y.K.; Kim, K.; Kim, S.H. Chemical and structural modifications of RNAi therapeutics. Adv. Drug Deliv. Rev. 2016, 104, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Gurav, B.; Srinivasan, G. Antisense oligonucleotides as therapeutics and their delivery. Curr. Sci. 2017, 112, 490–498. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Agrawal, Y.K. Targeting nanocarriers containing antisense oligonucleotides to cancer cell. J. Drug Deliv. Sci. Technol. 2017, 37, 97–114. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.-S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther.-Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kurreck, J. Antisense technologies: Improvement through novel chemical modifications. Eur. J. Biochem. 2003, 270, 1628–1644. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.; Stein, C. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Alagia, A.; Eritja, R. siRNA and RNAi optimization. Wiley Interdiscip. Rev. RNA 2016, 7, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Jaskiewicz, L.; Kolb, F.A.; Pillai, R.S. Post-transcriptional gene silencing by siRNAs and miRNAs. Curr. Opin. Struct. Biol. 2005, 15, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Wood, M.J.A. Therapeutic targeting of non-coding RNAs. Essays Biochem. 2013, 54, 127–145. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Kauppinen, S. Development of microRNA therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Lennox, K.A.; Behlke, M.A. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011, 18, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Rodrigues, P.M.; Borralho, P.M.; Rodrigues, C.M.P. Delivering the promise of miRNA cancer therapeutics. Drug Discov. Today 2013, 18, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Hesse, M.; Arenz, C.A. Rapid and versatile assay for Ago2-mediated cleavage by using branched rolling circle amplification. ChemBioChem 2016, 17, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, D.A.; Song, J.M.; Garner, A.L. High-throughput platform assay technology for the discovery of pre-microRNA-selective small molecule probes. Bioconjug. Chem. 2015, 26, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, A.; Tran, T.P.A.; Duca, M. Small-molecule approaches toward the targeting of oncogenic MiRNAs: Roadmap for the discovery of RNA modulators. Future Med. Chem. 2016, 8, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Gooding, M.; Malhotra, M.; Evans, J.C.; Darcy, R.; O’Driscoll, C.M. Oligonucleotide conjugates—Candidates for gene silencing therapeutics. Eur. J. Pharm. Biopharm. 2016, 107, 321–340. [Google Scholar] [CrossRef] [PubMed]
- Gambari, R.; Brognara, E.; Spandidos, D.A.; Fabbri, E. Targeting oncomiRNAs and mimicking tumour suppressor miRNAs: New trends in the development of miRNA therapeutic strategies in oncology (Review). Int. J. Oncol. 2016, 49, 5–32. [Google Scholar] [CrossRef] [PubMed]
- Asghari, F.; Haghnavaz, N.; Baradaran, B.; Hemmatzadeh, M.; Kazemi, T. Tumour suppressor microRNAs: Targeted molecules and signaling pathways in breast cancer. Biomed. Pharmacother. 2016, 81, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.M.; Horton, K.L.; Kelley, S.O. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org. Biomol. Chem. 2008, 6, 2242. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.B.; Pereira, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 2009, 61, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Cooley, C.B.; Geihe, E.I. Beyond cell penetrating peptides: Designed molecular transporters. Drug Discov. Today Technol. 2012, 9, e49–e55. [Google Scholar] [CrossRef] [PubMed]
- Wyman, T.B.; Nicol, F.; Zelphati, O.; Scaria, P.V.; Plank, C.; Szoka, F.C. Design, synthesis and characterization of a cationic peptide that binds to nucleic acids and permeabilizes bilayers. Biochemistry 1997, 36, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Lehto, T.; Ezzat, K.; Wood, M.J.A.; EL Andaloussi, S. Peptides for nucleic acid delivery. Adv. Drug Deliv. Rev. 2016, 106, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Arukuusk, P.; Pärnaste, L.; Hällbrink, M.; Langel, Ü. PepFects and NickFects for the intracellular delivery of nucleic acids. In Cell-Penetrating Peptides: Methods and Protocols; Springer: New York, NY, USA, 2015; pp. 303–315. ISBN 978-1-4939-2806-4. [Google Scholar]
- Pärnaste, L.; Arukuusk, P.; Langel, K.; Tenson, T.; Langel, Ü. The formation of nanoparticles between small interfering RNA and amphipathic cell-penetrating peptides. Mol. Ther.-Nucleic Acids 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.A.; Jones, S.W.; Perry, M.M.; Williams, A.E.; Erjefalt, J.S.; Turner, J.J.; Barnes, P.J.; Sproat, B.S.; Gait, M.J.; Lindsay, M.A. Lung delivery studies using siRNA conjugated to TAT(48-60) and penetratin reveal peptide induced reduction in gene expression and induction of innate immunity. Bioconjug. Chem. 2007, 18, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Goto, M.; Kodera, K.; Kanazawa, H.; Murakami, Y.; Mizushima, Y.; Higaki, M. Intracellular delivery of siRNA by cell-penetrating peptides modified with cationic oligopeptides. Drug Deliv. 2009, 16, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Muratovska, A.; Eccles, M.R. Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett. 2004, 558, 63–68. [Google Scholar] [CrossRef]
- Meyer, M.; Dohmen, C.; Philipp, A.; Kiener, D.; Maiwald, G.; Scheu, C.; Ogris, M.; Wagner, E. Synthesis and biological evaluation of a bioresponsive and endosomolytic siRNA−polymer conjugate. Mol. Pharm. 2009, 6, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Castagner, B.; Leroux, J.C. Is there a future for cell-penetrating peptides in oligonucleotide delivery? Eur. J. Pharm. Biopharm. 2013, 85, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Liu, E.; Gong, J.; Wang, J.; Huang, Y.; He, H.; Yang, V.C. High-yield synthesis of monomeric LMWP(CPP)-siRNA covalent conjugate for effective cytosolic delivery of siRNA. Theranostics 2017, 7, 2495–2508. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Feng, F.; Meng, F.; Deng, C.; Feijen, J.; Zhong, Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J. Control. Release 2011, 152, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Telomerase therapeutics for cancer: Challenges and new directions. Nat. Rev. Drug Discov. 2006, 5, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Seo, E.H.; Lee, S.H.; Kim, B.J. The telomerase-derived anticancer peptide vaccine GV1001 as an extracellular heat shock protein-mediated cell-penetrating peptide. Int. J. Mol. Sci. 2016, 17, 2054. [Google Scholar] [CrossRef] [PubMed]
- Shaw, V.E.; Naisbitt, D.J.; Costello, E.; Greenhalf, W.; Park, B.K.; Neoptelomos, J.P.; Middleton, G.M. Current status of GV1001 and other telomerase vaccination strategies in the treatment of cancer. Expert Rev. Vaccines 2010, 9, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Park, H.H.; Yu, H.J.; Kim, S.; Kim, G.; Choi, N.Y.; Lee, E.H.; Lee, Y.J.; Yoon, M.Y.; Lee, K.Y.; Koh, S.H. Neural stem cells injured by oxidative stress can be rejuvenated by GV1001, a novel peptide, through scavenging free radicals and enhancing survival signals. Neurotoxicology 2016, 55, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.Y.; Yan, J.-J.; Yang, J. Protective effect of peptide GV1001 against renal ischemia-reperfusion injury in mice. Transplant. Proc. 2014, 46, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.-J.; Kwon, K.-Y.; Kum, K.-Y.; Lee, W.-C.; Baek, S.-H.; Kang, M.K.; Shon, W.-J. The anti-inflammatory effect of human telomerase-derived peptide on P. gingivalis lipopolysaccharide-induced inflammatory cytokine production and its mechanism in human dental pulp cells. Mediators Inflamm. 2015, 2015, 385127. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, M.-S.; Inn, K.-S.; Kim, B.-J. Inhibition of HIV-1 reactivation by a telomerase-derived peptide in a HSP90-dependent manner. Sci. Rep. 2016, 6, 28896. [Google Scholar] [CrossRef] [PubMed]
- Park, H.H.; Lee, K.Y.; Park, D.W.; Choi, N.Y.; Lee, Y.J.; Son, J.W.; Kim, S.; Moon, C.; Kim, H.W.; Rhyu, I.J.; et al. Tracking and protection of transplanted stem cells using a ferrocenecarboxylic acid-conjugated peptide that mimics hTERT. Biomaterials 2018, 155, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Kim, B.R.; Kim, B.K.; Kim, D.W.; Shon, W.J.; Lee, N.R.; Inn, K.S.; Kim, B.J. Heat shock protein-mediated cell penetration and cytosolic delivery of macromolecules by a telomerase-derived peptide vaccine. Biomaterials 2013, 34, 7495–7505. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Zhang, Y.; Liu, X.; He, H.; Ma, Y.; Sun, J.; Huang, Y.; Wang, X.; Wu, Y.; Zhang, L.; et al. Biological properties of a 3′,3′′-bis-peptide-siRNA conjugate in vitro and in vivo. Bioconjug. Chem. 2016, 27, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xie, X.; Xu, X.; Xia, X.; Wang, H.; Li, L.; Dong, W.; Ma, P.; Yang, Y.; Liu, Y.; et al. Thermal and magnetic dual-responsive liposomes with a cell-penetrating peptide-siRNA conjugate for enhanced and targeted cancer therapy. Colloids Surf. B Biointerfaces 2016, 146, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Xie, X.; Xu, X.; Xia, X.; Wang, H.; Li, L.; Dong, W.; Ma, P.; Liu, Y. Dual stimulus of hyperthermia and intracellular redox environment triggered release of siRNA for tumour-specific therapy. Int. J. Pharm. 2016, 506, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xia, X.; Dong, W.; Wang, H.; Li, L.; Ma, P.; Sheng, W.; Xu, X.; Liu, Y. Acid sensitive polymeric micelles combining folate and bioreducible conjugate for specific intracellular siRNA Delivery. Macromol. Biosci. 2016, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef] [PubMed]
- Gabius, H.J.; Siebert, H.C.; André, S.; Jiménez-Barbero, J.; Rüdiger, H. Chemical biology of the sugar code. ChemBioChem 2004, 5, 740–764. [Google Scholar] [CrossRef] [PubMed]
- Zatsepin, T.S.; Oretskaya, T.S. Synthesis and applications of oligonucleotide-carbohydrate conjugates. Chem. Biodivers. 2004, 1, 1401–1417. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, J.; De Champdoré, M.; De Napoli, L.; Montesarchio, D.; Di Fabio, G. Glycomimetics as decorating motifs for oligonucleotides: Solid-phase synthesis, stability and hybridization properties of carbopeptoid- oligonucleotide conjugates. Bioconjug. Chem. 2005, 16, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Kubota, D.; Nagasaki, Y. Simple solid-phase synthesis and biological properties of carbohydrate−oligonucleotide conjugates modified at the 3′-terminus. Bioconjug. Chem. 2010, 21, 1685–1690. [Google Scholar] [CrossRef] [PubMed]
- Vengut-Climent, E.; Terrazas, M.; Lucas, R.; Arévalo-Ruiz, M.; Eritja, R.; Morales, J.C. Synthesis, RNAi activity and nuclease-resistant properties of apolar carbohydrates siRNA conjugates. Bioorg. Med. Chem. Lett. 2013, 23, 4048–4051. [Google Scholar] [CrossRef] [PubMed]
- Aviñó, A.; Ocampo, S.M.; Lucas, R.; Reina, J.J.; Morales, J.C.; Perales, J.C.; Eritja, R. Synthesis and in vitro inhibition properties of siRNA conjugates carrying glucose and galactose with different presentations. Mol. Divers. 2011, 15, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Grewal, P.K. The Ashwell-Morell receptor. Methods Enzymol. 2010, 479, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Zelensky, A.N.; Gready, J.E. The C-type lectin-like domain superfamily. FEBS J. 2005, 272, 6179–6217. [Google Scholar] [CrossRef] [PubMed]
- Mamidyala, S.K.; Dutta, S.; Chrunyk, B.A.; Préville, C.; Wang, H.; Withka, J.M.; McColl, A.; Subashi, T.A.; Hawrylik, S.J.; Griffor, M.C.; et al. Glycomimetic ligands for the human asialoglycoprotein receptor. J. Am. Chem. Soc. 2012, 134, 1978–1981. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.S.; Karsten, V.; Chan, A.; Chiesa, J.; Boyce, M.; Bettencourt, B.R.; Hutabarat, R.; Nochur, S.; Vaishnaw, A.; Gollob, J. Clinical proof of concept for a novel hepatocyte-targeting GalNAc-siRNA conjugate. Mol. Ther. 2017, 25, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y. Preclinical and clinical advances of GalNAc-decorated nucleic acid therapeutics. Mol. Ther.-Nucleic Acids 2017, 6, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Willoughby, J.L.S.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kelin, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, K.; Monge, J.C.; Lee, N.; Law, S.W.; Brewer, H.B. ApoB-100 is encoded by a single copy gene in the human genome. Biochem. Biophys. Res. Commun. 1987, 144, 1332–1339. [Google Scholar] [CrossRef]
- Yan, C.; Costa, R.H.; Darnell, J.E.; Chen, J.D.; Van Dyke, T. Distinct positive and negative elements control the limited hepatocyte and choroid plexus expression of transthyretin in transgenic mice. EMBO J. 1990, 9, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Rajeev, K.G.; Nair, J.K.; Jayaraman, M.; Charisse, K.; Taneja, N.; O’Shea, J.; Willoughby, J.L.S.; Yucius, K.; Nguyen, T.; Shulga-Morskaya, S.; et al. Hepatocyte-specific delivery of siRNAs conjugated to novel non-nucleosidic trivalent N-acetylgalactosamine elicits robust gene silencing in vivo. ChemBioChem 2015, 16, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Keiser, K.; Nair, J.K.; Charisse, K.; Manoharan, R.M.; Kretschmer, P.; Peng, C.G.; Kel’in, A.V.; Kandasamy, P.; Willoughby, J.L.S.; et al. siRNA conjugates carrying sequentially assembled trivalent N-acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem. Biol. 2015, 10, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Weitzer, S.; Martinez, J. The human RNA kinase hClp1 is active on 3′ transfer RNA exons and short interfering RNAs. Nature 2007, 447, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Heydrick, S.J.; Lardeux, B.R.; Mortimore, G.E. Uptake and degradation of cytoplasmic RNA by hepatic lysosomes: Quantitative relationship to RNA turnover. J. Biol. Chem. 1991, 266, 8790–8796. [Google Scholar] [PubMed]
- Parmar, R.; Willoughby, J.L.S.; Liu, J.; Foster, D.J.; Brigham, B.; Theile, C.S.; Charisse, K.; Akinc, A.; Guidry, E.; Pei, Y.; et al. 5′-(E)-Vinylphosphonate: A stable phosphate mimic can improve the RNAi activity of siRNA-GalNAc conjugates. ChemBioChem 2016, 17, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Kinberger, G.A.; Murray, H.M.; Chappell, A.; Riney, S.; Graham, M.J.; Lima, W.F.; Swayze, E.E.; Seth, P.P. Synergistic effect of phosphorothioate, 5′-vinylphosphonate and GalNAc modifications for enhancing activity of synthetic siRNA. Bioorg. Med. Chem. Lett. 2016, 26, 2817–2820. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E. Functional Polymer Conjugates for Medicinal Nucleic Acid Delivery. In Polymers in Nanomedicine, 1st ed.; Kunugi, S., Yamaoka, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 247, pp. 1–29. ISBN 978-3-642-27855-6. [Google Scholar]
- Morille, M.; Passirani, C.; Vonarbourg, A.; Clavreul, A.; Benoit, J.P. Progress in developing cationic vectors for non-viral systemic gene therapy against cancer. Biomaterials 2008, 29, 3477–3496. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.F.; Sarraguça, J.M.G.; Dias, R.S.; Pais, A.A.C.C. Polyelectrolyte compaction by pH-responsive agents. Phys. Chem. Chem. Phys. 2009, 11, 10890. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P.; Jorge, A.F.; Martins, R.; Pais, A.A.C.C.; Sousa, F.; Figueiras, A. Characterization of polyplexes involving small RNA. J. Colloid Interface Sci. 2012, 387, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Sun, J.; Sun, B.; He, Z. Prodrug-based nanoparticulate drug delivery strategies for cancer therapy. Trends Pharmacol. Sci. 2014, 35, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, T.; Edinger, D.; Russ, V.; Wagner, E. Stabilization of polyplexes via polymer crosslinking for efficient siRNA delivery. Eur. J. Pharm. Sci. 2012, 47, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Philipp, A.; Zhao, X.; Tarcha, P.; Wagner, E.; Zintchenko, A. Hydrophobically modified oligoethylenimines as highly efficient transfection agents for siRNA delivery. Bioconjug. Chem. 2009, 20, 2055–2061. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Takemoto, H.; Nomoto, T.; Tomoda, K.; Matsui, M.; Nishiyama, N. Utility of the 2-nitrobenzenesulfonamide group as a chemical linker for enhanced extracellular stability and cytosolic cleavage in siRNA-conjugated polymer systems. ChemMedChem 2017, 12, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Jewett, J.C.; Bertozzi, C.R. Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 2010, 39, 1272. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, H.; Miyata, K.; Hattori, S.; Ishii, T.; Suma, T.; Uchida, S.; Nishiyama, N.; Kataoka, K. Acidic pH-responsive siRNA conjugate for reversible carrier stability and accelerated endosomal escape with reduced IFNα-associated immune response. Angew. Chem. Int. Ed. 2013, 52, 6218–6221. [Google Scholar] [CrossRef] [PubMed]
- Miyata, K.; Oba, M.; Nakanishi, M.; Fukushima, S.; Yamasaki, Y.; Koyama, H.; Nishiyama, N.; Kataoka, K. Polyplexes from poly(aspartamide) bearing 1,2-diaminoethane side chains induce pH-selective, endosomal membrane destabilization with amplified transfection and negligible cytotoxicity. J. Am. Chem. Soc. 2008, 130, 16287–16294. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kim, Y.; Song, Y.; Choi, J.U.; Park, E.; Choi, W.; Park, J.; Lee, Y. Comparison of pH-sensitive degradability of maleic acid amide derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 2364–2367. [Google Scholar] [CrossRef] [PubMed]
- Parmar, R.G.; Poslusney, M.; Busuek, M.; Williams, J.M.; Garbaccio, R.; Leander, K.; Walsh, E.; Howell, B.; Sepp-Lorenzino, L.; Riley, S.; et al. Novel endosomolytic poly(amido amine) polymer conjugates for systemic delivery of siRNA to hepatocytes in rodents and nonhuman primates. Bioconjug. Chem. 2014, 25, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Parmar, R.G.; Busuek, M.; Walsh, E.S.; Leander, K.R.; Howell, B.J.; Sepp-Lorenzino, L.; Kemp, E.; Crocker, L.S.; Leone, A.; Kochansky, C.J.; et al. Endosomolytic bioreducible poly(amido amine disulfide) polymer conjugates for the in vivo systemic delivery of siRNA therapeutics. Bioconjug. Chem. 2013, 24, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P.S. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005, 4, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S.E.; Abrams, M.T.; Burke, R.; Carr, B.A.; Crocker, L.S.; Garbaccio, R.M.; Howell, B.J.; Kemp, E.A.; Kowtoniuk, R.A.; Latham, A.H.; et al. An in vivo evaluation of amphiphilic, biodegradable peptide copolymers as siRNA delivery agents. Int. J. Pharm. 2014, 466, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Guidry, E.N.; Farand, J.; Soheili, A.; Parish, C.A.; Kevin, N.J.; Pipik, B.; Calati, K.B.; Ikemoto, N.; Waldman, J.H.; Latham, A.H.; et al. Improving the in vivo therapeutic index of siRNA polymer conjugates through increasing pH responsiveness. Bioconjug. Chem. 2014, 25, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Mok, H.; Lee, Y.; Park, T.G. Self-assembled siRNA–PLGA conjugate micelles for gene silencing. J. Control. Release 2011, 152, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Park, T.G. Novel polymer−DNA hybrid polymeric micelles composed of hydrophobic poly (d,l-lactic-co-glycolic acid) and hydrophilic Oligonucleotides. Bioconjug. Chem. 2001, 12, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, J.Y.; Kim, J.S.; Park, T.G.; Mok, H. Amphiphilic siRNA conjugates for co-delivery of nucleic acids and hydrophobic drugs. Bioconjug. Chem. 2017, 28, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Gallas, A.; Alexander, C.; Davies, M.C.; Puri, S.; Allen, S. Chemistry and formulations for siRNA therapeutics. Chem. Soc. Rev. 2013, 42, 7983. [Google Scholar] [CrossRef] [PubMed]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Hadwiger, P.; John, M.; Vornlocher, H.P.; Unverzagt, C. Steroid and lipid conjugates of siRNAs to enhance cellular uptake and gene silencing in liver cells. Bioorg. Med. Chem. Lett. 2004, 14, 4975–4977. [Google Scholar] [CrossRef] [PubMed]
- Alterman, J.F.; Hall, L.M.; Coles, A.H.; Hassler, M.R.; Didiot, M.-C.; Chase, K.; Abraham, J.; Sottosanti, E.; Johnson, E.; Sapp, E.; et al. Hydrophobically modified siRNAs silence Huntingtin mRNA in primary neurons and mouse brain. Mol. Ther.-Nucleic Acids 2015, 4, e266. [Google Scholar] [CrossRef] [PubMed]
- Haraszti, R.A.; Roux, L.; Coles, A.H.; Turanov, A.A.; Alterman, J.F.; Echeverria, D.; Godinho, B.M.D.C.; Aronin, N.; Khvorova, A. 5′-Vinylphosphonate improves tissue accumulation and efficacy of conjugated siRNAs in vivo. Nucleic Acids Res. 2017, 45, 7581–7592. [Google Scholar] [CrossRef] [PubMed]
- Baigude, H.; McCarroll, J.; Yang, C.S.; Swain, P.M.; Rana, T.M. Design and creation of new nanomaterials for therapeutic RNAi. ACS Chem. Biol. 2007, 2, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.S.; Lee, A.C.H.; Akinc, A.; Bramlage, B.; Bumcrot, D.; Fedoruk, M.N.; Harborth, J.; Heyes, J.A.; Jeffs, L.B.; John, M.; et al. RNAi-mediated gene silencing in non-human primates. Nature 2006, 441, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Rozema, D.B.; Lewis, D.L.; Wakefield, D.H.; Wong, S.C.; Klein, J.J.; Roesch, P.L.; Bertin, S.L.; Reppen, T.W.; Chu, Q.; Blokhin, A.V.; et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 12982–12987. [Google Scholar] [CrossRef] [PubMed]
- Trubetskoy, V.S.; Griffin, J.B.; Nicholas, A.L.; Nord, E.M.; Xu, Z.; Peterson, R.M.; Wooddell, C.I.; Rozema, D.B.; Wakefield, D.H.; Lewis, D.L.; et al. Phosphorylation-specific status of RNAi triggers in pharmacokinetic and biodistribution analyses. Nucleic Acids Res. 2017, 45, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Gish, R.G.; Yuen, M.F.; Chan, H.L.Y.; Given, B.D.; Lai, C.L.; Locarnini, S.A.; Lau, J.Y.N.; Wooddell, C.I.; Schluep, T.; Lewis, D.L. Synthetic RNAi triggers and their use in chronic hepatitis B therapies with curative intent. Antiviral Res. 2015, 121, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.I.; Rozema, D.B.; Hossbach, M.; John, M.; Hamilton, H.L.; Chu, Q.; Hegge, J.O.; Klein, J.J.; Wakefield, D.H.; Oropeza, C.E.; et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol. Ther. 2013, 21, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Zannis, V.I.; Chroni, A.; Krieger, M. Role of apoA-I, ABCA1, LCAT and SR-BI in the biogenesis of HDL. J. Mol. Med. 2006, 84, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wang, W.; Feng, M.; Wang, Y.; Zhou, J.; Ding, X.; Zhou, X.; Liu, C.; Wang, R.; Zhang, Q. A biomimetic nanovector-mediated targeted cholesterol-conjugated siRNA delivery for tumor gene therapy. Biomaterials 2012, 33, 8893–8905. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Zhou, J.P.; Liu, Y.P.; Liu, J.J.; Yang, X.N.; Jazag, A.; Zhang, Z.P.; Guleng, B.; Ren, J.L. The silencing of Pokemon attenuates the proliferation of hepatocellular carcinoma cells in vitro and in vivo by inhibiting the PI3K/Akt Pathway. PLoS ONE 2012, 7, e51916. [Google Scholar] [CrossRef] [PubMed]
- Couvreur, P.; Stella, B.; Reddy, L.H.; Hillaireau, H.; Dubernet, C.; Desmaěie, D.; Lepêtre-Mouelhi, S.; Rocco, F.; Dereuddre-Bosquet, N.; Clayette, P.; et al. Squalenoyl nanomedicines as potential therapeutics. Nano Lett. 2006, 6, 2544–2548. [Google Scholar] [CrossRef] [PubMed]
- Raouane, M.; Desmaele, D.; Gilbert-Sirieix, M.; Gueutin, C.; Zouhiri, F.; Bourgaux, C.; Lepeltier, E.; Gref, R.; Ben Salah, R.; Clayman, G.; et al. Synthesis, characterization and in vivo delivery of siRNA-squalene nanoparticles targeting fusion oncogene in papillary thyroid carcinoma. J. Med. Chem. 2011, 54, 4067–4076. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.S. Squalene and its potential clinical uses. Altern. Med. Rev. 1999, 4, 29–36. [Google Scholar] [PubMed]
- Nishina, K.; Unno, T.; Uno, Y.; Kubodera, T.; Kanouchi, T.; Mizusawa, H.; Yokota, T. Efficient in vivo delivery of siRNA to the liver by conjugation of α-Tocopherol. Mol. Ther. 2008, 16, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nishina, K.; Watanabe, C.; Yoshida-Tanaka, K.; Piao, W.; Kuwahara, H.; Horikiri, Y.; Miyata, K.; Nishiyama, N.; Kataoka, K.; et al. Enteral siRNA delivery technique for therapeutic gene silencing in the liver via the lymphatic route. Sci. Rep. 2015, 5, 17035. [Google Scholar] [CrossRef] [PubMed]
- Uno, Y.; Piao, W.; Miyata, K.; Nishina, K.; Mizusawa, H.; Yokota, T. High-density lipoprotein facilitates in vivo delivery of α-tocopherol–conjugated short-interfering RNA to the brain. Hum. Gene Ther. 2011, 22, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Grijalvo, S.; Ocampo, S.M.; Perales, J.C.; Eritja, R. Synthesis of lipid-oligonucleotide conjugates for RNA interference studies. Chem. Biodivers. 2011, 8, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Grijalvo, S.; Ocampo, S.M.; Perales, J.C.; Eritja, R. Synthesis of oligonucleotides carrying amino lipid groups at the 3′-end for RNA interference studies. J. Org. Chem. 2010, 75, 6806–6813. [Google Scholar] [CrossRef] [PubMed]
- Ugarte-Uribe, B.; Grijalvo, S.; Busto, J.V.; Martín, C.; Eritja, R.; Goñi, F.M.; Alkorta, I. Double-tailed lipid modification as a promising candidate for oligonucleotide delivery in mammalian cells. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 4872–4884. [Google Scholar] [CrossRef] [PubMed]
- Nikan, M.; Osborn, M.F.; Coles, A.H.; Godinho, B.M.; Hall, L.M.; Haraszti, R.A.; Hassler, M.R.; Echeverria, D.; Aronin, N.; Khvorova, A. Docosahexaenoic acid conjugation enhances distribution and safety of siRNA upon local administration in mouse brain. Mol. Ther.-Nucleic Acids 2016, 5, e344. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Hossain, S.; Shimada, T.; Sugioka, K.; Yamasaki, H.; Fujii, Y.; Ishibashi, Y.; Oka, J.I.; Shido, O. Docosahexaenoic acid provides protection from impairment of learning ability in Alzheimer’s disease model rats. J. Neurochem. 2002, 81, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, M.; Descamps, L.; Allain, F.; Denys, A.; Durieux, S.; Fenart, L.; Kieda, C.; Cecchelli, R.; Spik, G. Receptor-mediated transcytosis of cyclophilin B through the blood-brain barrier. J. Neurochem. 1999, 73, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Nikan, M.; Osborn, M.F.; Coles, A.H.; Biscans, A.; Godinho, B.M.D.C.; Haraszti, R.A.; Sapp, E.; Echeverria, D.; Difiglia, M.; Aronin, N.; et al. Synthesis and evaluation of parenchymal retention and efficacy of a metabolically stable O-phosphocholine-N-docosahexaenoyl-l-serine siRNA conjugate in mouse brain. Bioconjug. Chem. 2017, 28, 1758–1766. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, M.; Bernoud, N.; Brossard, N.; Lemaitre-Delaunay, D.; Thiès, F.; Croset, M.; Lecerf, J. Lysophosphatidylcholine as a preferred carrier form of docosahexaenoic acid to the brain. J. Mol. Neurosci. 2001, 16, 201–4. [Google Scholar] [CrossRef]
- Kanada, M.; Bachmann, M.H.; Hardy, J.W.; Frimannson, D.O.; Bronsart, L.; Wang, A.; Sylvester, M.D.; Schmidt, T.L.; Kaspar, R.L.; Butte, M.J.; et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc. Natl. Acad. Sci. USA 2015, 201418401. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, A.J.; Mäger, I.; de Jong, O.G.; Varela, M.A.; Schiffelers, R.M.; El Andaloussi, S.; Wood, M.J.A.; Vader, P. Functional Delivery of Lipid-Conjugated siRNA by Extracellular Vesicles. Mol. Ther. 2017, 25, 1580–1587. [Google Scholar] [CrossRef] [PubMed]
- Famulok, M. Oligonucleotide aptamers that recognize small molecules. Curr. Opin. Struct. Biol. 1999, 9, 324–329. [Google Scholar] [CrossRef]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX-A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.L.; Wang, W.; Mao, Z.; Kang, T.S.; Han, Q.B.; Chan, P.W.H.; Leung, C.H. Utilization of G-quadruplex-forming aptamers for the construction of luminescence sensing platforms. Chempluschem 2017, 82, 8–17. [Google Scholar] [CrossRef]
- De Almeida, C.E.B.; Alves, L.N.; Rocha, H.F.; Cabral-Neto, J.B.; Missailidis, S. Aptamer delivery of siRNA, radiopharmaceutics and chemotherapy agents in cancer. Int. J. Pharm. 2017, 525, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wei, T.; Zhao, J.; Huang, Y.; Deng, H.; Kumar, A.; Wang, C.; Liang, Z.; Ma, X.; Liang, X.J. Multifunctional aptamer-based nanoparticles for targeted drug delivery to circumvent cancer resistance. Biomaterials 2016, 91, 44–56. [Google Scholar] [CrossRef] [PubMed]
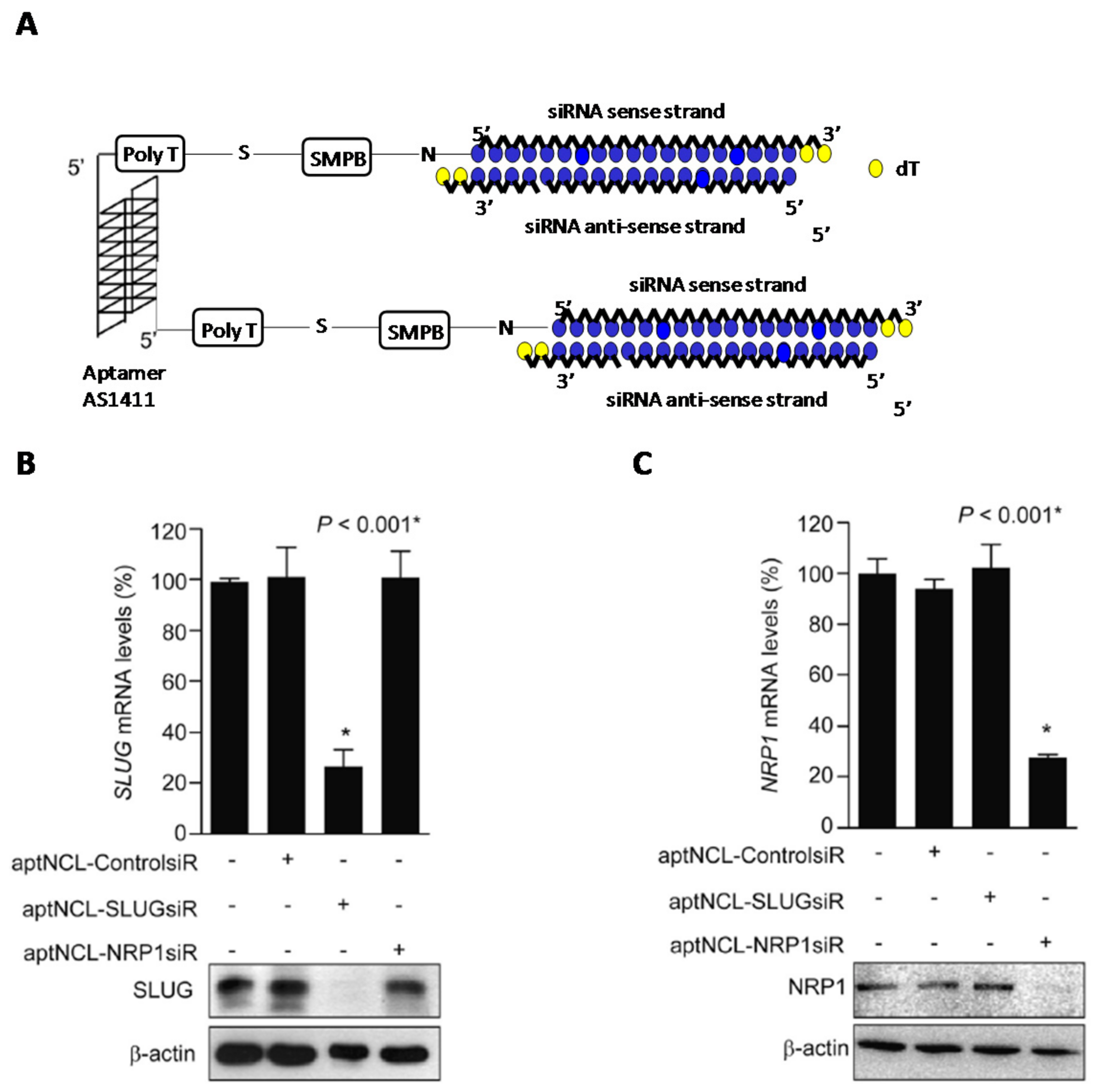
- Chu, T.C.; Twu, K.Y.; Ellington, A.D.; Levy, M. Aptamer mediated siRNA delivery. Nucleic Acids Res. 2006, 34, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.B.; Dellamaggiore, K.R.; Ouellette, C.P.; Sedano, C.D.; Lizadjohry, M.; Chernis, G.A.; Gonzales, M.; Baltasar, F.E.; Fan, A.L.; Myerowitz, R.; et al. Aptamer-based endocytosis of a lysosomal enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 15908–15913. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J.J. Cell-type-specific, Aptamer-functionalized Agents for Targeted Disease Therapy. Mol. Ther.-Nucleic Acids 2014, 3, e169. [Google Scholar] [CrossRef] [PubMed]
- Lupold, S.E.; Hicke, B.J.; Lin, Y.; Coffey, D.S. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen 1. Cancer Res. 2002, 62, 4029–4033. [Google Scholar] [PubMed]
- Ni, X.; Zhang, Y.; Zennami, K.; Castanares, M.; Mukherjee, A.; Raval, R.R.; Zhou, H.; DeWeese, T.L.; Lupold, S.E. Systemic administration and targeted radiosensitization via chemically synthetic aptamer-siRNA chimeras in human tumor xenografts. Mol. Cancer Ther. 2015, 14, 2797–2804. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, A.; Brenneman, R.; Bajgelman, M.; Seales, D.; Gilboa, E. Thermal stability of siRNA modulates aptamer-conjugated siRNA inhibition. Mol. Ther.-Nucleic Acids 2012, 1, e51. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Yu, X.; Liu, H.; Wu, D.; She, J.-X. Co-targeting EGFR and survivin with a bivalent aptamer-dual siRNA chimera effectively suppresses prostate cancer. Sci. Rep. 2016, 6, 30346. [Google Scholar] [CrossRef] [PubMed]
- Woodburn, J.R. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol. Ther. 1999, 82, 241–50. [Google Scholar] [CrossRef]
- Mita, A.C.; Mita, M.M.; Nawrocki, S.T.; Giles, F.J. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 2008, 14, 5000–5005. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xue, S.; Mao, Z.-W. Nanoparticle delivery systems for siRNA-based therapeutics. J. Mater. Chem. B 2016, 4, 6620–6639. [Google Scholar] [CrossRef]
- Janib, S.M.; Moses, A.S.; MacKay, J.A. Imaging and drug delivery using theranostic nanoparticles. Adv. Drug Deliv. Rev. 2010, 62, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Duan, H.; Mohs, A.M.; Nie, S. Bioconjugated quantum dots for in vivo molecular and cellular imaging. Adv. Drug Deliv. Rev. 2008, 60, 1226–1240. [Google Scholar] [CrossRef] [PubMed]
- Bagalkot, V.; Gao, X. SiRNA-aptamer chimeras on nanoparticles: Preserving targeting functionality for effective gene silencing. ACS Nano 2011, 5, 8131–8139. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A.; von Zastrow, M. Endocytosis and signalling: Intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2010, 10, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Jung, H.; Kim, S.A.; Mok, H. Multivalent comb-type aptamer-siRNA conjugates for efficient and selective intracellular delivery. Chem. Commun. 2014, 50, 6765–6767. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Castanares, M.; Mukherjee, A.; Lupold, S.E. Nucleic acid aptamers: Clinical applications and promising new horizons. Curr. Med. Chem. 2011, 18, 4206–14. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Lee, S.H.; Hwang, Y.; Yoo, H.; Jung, H.; Kim, S.H.; Mok, H. Multivalent aptamer–RNA conjugates for simple and efficient delivery of doxorubicin/siRNA into multidrug-resistant cells. Macromol. Biosci. 2017, 17, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.Y.; Wang, W.Y.; Chang, Y.C.; Chang, C.J.; Yang, P.C.; Peck, K. Synergistic inhibition of lung cancer cell invasion, tumor growth and angiogenesis using aptamer-siRNA chimeras. Biomaterials 2014, 35, 2905–2914. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Sun, N.; Liu, M.; Wang, J.; Pei, R. Building a chimera of aptamer–antisense oligonucleotide for silencing galectin-1 gene. RSC Adv. 2016, 6, 112445–112450. [Google Scholar] [CrossRef]
- Bates, P.J.; Reyes-Reyes, E.M.; Malik, M.T.; Murphy, E.M.; O’Toole, M.G.; Trent, J.O. G-quadruplex oligonucleotide AS1411 as a cancer-targeting agent: Uses and mechanisms. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 1414–1428. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Meng, F.; Deng, C.; Zhong, Z. Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules 2014, 15, 1955–1969. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of Tissue-Specific MicroRNAs from Mouse. Curr. Biol. 2017, 12, 735–739. [Google Scholar] [CrossRef]
- Kato, M.; Slack, F.J. microRNAs: Small molecules with big roles—C. elegans to human cancer. Biol. Cell 2008, 100, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Barh, D.; Malhotra, R.; Ravi, B.; Sindhurani, P. MicroRNA let-7: An emerging next-generation cancer therapeutic. Curr. Oncol. 2010, 17, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Sun, B.; Su, C. Targeting microRNAs in cancer gene therapy. Genes 2017, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.; Wu, W. MicroRNA-based therapeutics for cancer. BioDrugs 2009, 23, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.W.; Nemunaitis, J. Modulation of miRNA activity in human cancer: A new paradigm for cancer gene therapy? Cancer Gene Ther. 2008, 15, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Xu, Y.; Hao, J.; Wang, S.; Li, C.; Meng, S. MiR-122 in hepatic function and liver diseases. Protein Cell 2012, 3, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Kuwajima, S.; Braich, R.; Rajeev, K.G.; Pena, J.; Tuschl, T.; Manoharan, M.; Stoffel, M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007, 35, 2885–2892. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Stelma, F.; van der Ree, M.H.; Sinnige, M.J.; Brown, A.; Swadling, L.; de Vree, J.M.L.; Willemse, S.B.; van der Valk, M.; Grint, P.; Neben, S.; et al. Immune phenotype and function of natural killer and T cells in chronic hepatitis C patients who received a single dose of anti-MicroRNA-122, RG-101. Hepatology 2017, 66, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.D.; Esquela-Kerscher, A.; Stefani, G.; Byrom, M.; Kelnar, K.; Ovcharenko, D.; Wilson, M.; Wang, X.; Shelton, J.; Shingara, J.; et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007, 67, 7713–7722. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Huang, X.; Shao, Q.; Huang, M.; Deng, L. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. RNA 2008, 14, 2348–2360. [Google Scholar] [CrossRef] [PubMed]
- Gramantieri, L.; Ferracin, M.; Fornari, F.; Veronese, A.; Sabbioni, S.; Liu, C.G.; Calin, G.A.; Giovannini, C.; Ferrazzi, E.; Grazi, G.L.; et al. Cyclin G1 is a target of miR-122a, a MicroRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007, 67, 6092–6099. [Google Scholar] [CrossRef] [PubMed]
- Faraoni, I.; Antonetti, F.R.; Cardone, J.; Bonmassar, E. miR-155 gene: A typical multifunctional microRNA. Biochim. Biophys. Acta-Mol. Basis Dis. 2009, 1792, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Puisségur, M.-P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011, 18, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Quintavalle, C.; Romano, G.; Croce, C.M.; Condorelli, G. miR221/222 in cancer: Their role in tumor progression and response to therapy. Curr. Mol. Med. 2012, 12, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Gandellini, P.; Folini, M.; Longoni, N.; Pennati, M.; Binda, M.; Colecchia, M.; Samoni, R.; Supino, R.; Moretti, R.; Limonta, P.; et al. MiR-205 exerts tumor-suppressive functions in human prostate through down-regulation of protein kinase Cε. Cancer Res. 2009, 69, 2287–2295. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Yao, D.; Chen, J.; Ding, N.; Ren, F. MiR-20a promotes cervical cancer proliferation and metastasis in vitro and in vivo. PLoS ONE 2015, 10, e0120905. [Google Scholar] [CrossRef] [PubMed]
- Mott, J.L.; Kobayashi, S.; Bronk, S.F.; Gores, G.J. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 2007, 26, 6133–6140. [Google Scholar] [CrossRef] [PubMed]
- Loyer, X.; Mallat, Z.; Boulanger, C.M.; Tedgui, A. MicroRNAs as therapeutic targets in atherosclerosis. Expert Opin. Ther. Targets 2015, 19, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Son, D.J.; Kumar, S.; Takabe, W.; Kim, C.W.; Ni, C.-W.; Alberts-Grill, N.; Jang, I.-H.; Kim, S.; Kim, W.; Kang, S.W.; et al. The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nat. Commun. 2013, 4, 3000. [Google Scholar] [CrossRef] [PubMed]
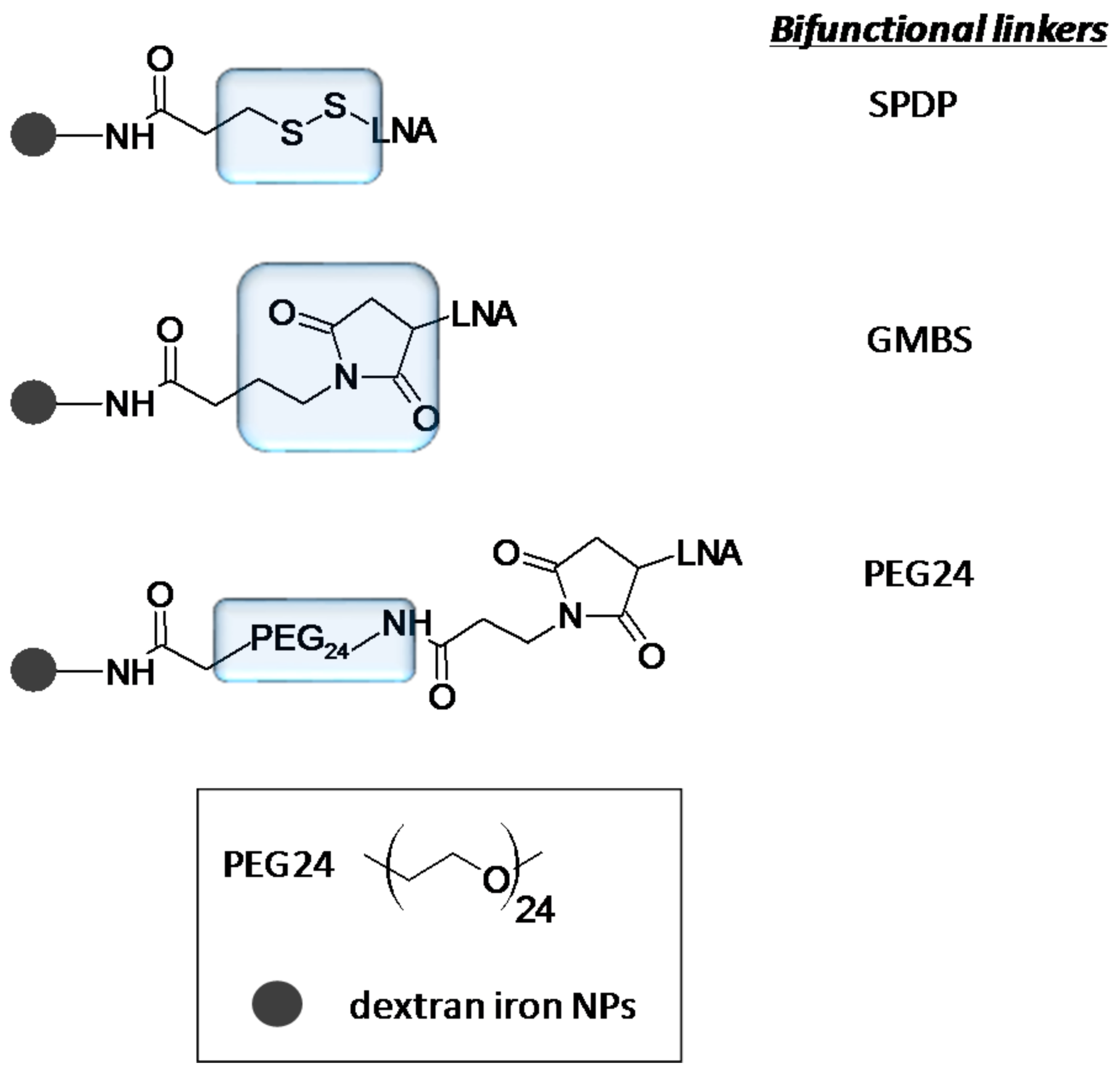
- Petersen, M.; Wengel, J. LNA: A versatile tool for therapeutics and genomics. Trends Biotechnol. 2003, 21, 74–81. [Google Scholar] [CrossRef]
- Petersen, M.; Bondensgaard, K.; Wengel, J.; Jacobsen, J.P. Locked nucleic acid (LNA) recognition of RNA: NMR solution structures of LNA:RNA hybrids. J. Am. Chem. Soc. 2002, 124, 5974–5982. [Google Scholar] [CrossRef] [PubMed]
- Stenvang, J.; Lindow, M.; Kauppinen, S. Targeting of microRNAs for therapeutics. Biochem. Soc. Trans. 2008, 36, 1197–200. [Google Scholar] [CrossRef] [PubMed]
- Nedaeinia, R.; Avan, A.; Ahmadian, M.; Nia, S.N.; Ranjbar, M.; Sharifi, M.; Goli, M.; Piroozmand, A.; Nourmohammadi, E.; Manian, M.; et al. Current Status and Perspectives Regarding LNA-Anti-miR Oligonucleotides and microRNA miR-21 Inhibitors as a Potential Therapeutic Option in Treatment of Colorectal Cancer. J. Cell. Biochem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Elmén, J.; Lindow, M.; Schütz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjärn, M.; Hansen, H.F.; Berger, U.; et al. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-P.; Gottwein, J.M.; Scheel, T.K.; Jensen, T.B.; Bukh, J. MicroRNA-122 antagonism against hepatitis C virus genotypes 1-6 and reduced efficacy by host RNA insertion or mutations in the HCV 5′ UTR. Proc. Natl. Acad. Sci. USA 2011, 108, 4991–4996. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Ørum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Ottosen, S.; Parsley, T.B.; Yang, L.; Zeh, K.; Van Doorn, L.J.; Van Der Veer, E.; Raney, A.K.; Hodges, M.R.; Patick, A.K. In Vitro antiviral activity and preclinical and clinical resistance profile of miravirsen, a novel anti-hepatitis C virus therapeutic targeting the human factor miR-122. Antimicrob. Agents Chemother. 2015, 59, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Van Der Ree, M.H.; Van Der Meer, A.J.; De Bruijne, J.; Maan, R.; Van Vliet, A.; Welzel, T.M.; Zeuzem, S.; Lawitz, E.J.; Rodriguez-Torres, M.; Kupcova, V.; et al. Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antiviral Res. 2014, 111, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Selcuklu, S.D.; Donoghue, M.T.A.; Spillane, C. miR-21 as a key regulator of oncogenic processes. Biochem. Soc. Trans. 2009, 37, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Nedaeinia, R.; Sharifi, M.; Avan, A.; Kazemi, M.; Rafiee, L.; Ghayour-Mobarhan, M.; Salehi, R. Locked nucleic acid anti-miR-21 inhibits cell growth and invasive behaviors of a colorectal adenocarcinoma cell line: LNA-anti-miR as a novel approach. Cancer Gene Ther. 2016, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.; Ghosh, S.K.; Kumar, M.; Moore, A.; Yigit, M.V.; Medarova, Z. Design of nanodrugs for miRNA targeting in tumor cells. J. Biomed. Nanotechnol. 2014, 10, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Yigit, M.V.; Ghosh, S.K.; Kumar, M.; Petkova, V.; Kavishwar, A.; Moore, A.; Medarova, Z. Context-dependent differences in miR-10b breast oncogenesis can be targeted for the prevention and arrest of lymph node metastasis. Oncogene 2013, 32, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E. Gene targeting and expression modulation by peptide nucleic acids (PNA). Curr. Pharm. Des. 2010, 16, 3118–3123. [Google Scholar] [CrossRef] [PubMed]
- Chiarantini, L.; Cerasi, A.; Fraternale, A.; Millo, E.; Benatti, U.; Sparnacci, K.; Laus, M.; Ballestri, M.; Tondelli, L. Comparison of novel delivery systems for antisense peptide nucleic acids. J. Control. Release 2005, 109, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.; Egholm, M.; Berg, R.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, E.; Brognara, E.; Borgatti, M.; Lampronti, I.; Finotti, A.; Bianchi, N.; Sforza, S.; Tedeschi, T.; Manicardi, A.; Marchelli, R.; et al. miRNA therapeutics: Delivery and biological activity of peptide nucleic acids targeting miRNAs. Epigenomics 2011, 3, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Demidov, V.V.; Potaman, V.N.; Frank-Kamenetskil, M.D.; Egholm, M.; Buchard, O.; Sönnichsen, S.H.; Nlelsen, P.E. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem. Pharmacol. 1994, 48, 1310–1313. [Google Scholar] [CrossRef]
- Koppelhus, U.; Nielsen, P.E. Cellular delivery of peptide nucleic acid (PNA). Adv. Drug Deliv. Rev. 2003, 55, 267–280. [Google Scholar] [CrossRef]
- Oh, S.Y.; Ju, Y.; Kim, S.; Park, H. PNA-based antisense oligonucleotides for micrornas inhibition in the absence of a transfection reagent. Oligonucleotides 2010, 20, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Fabani, M.M.; Gait, M.J. miR-122 targeting with LNA/2′-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA) and PNA-peptide conjugates. RNA 2007, 14, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.G.; Fabani, M.M.; Vigorito, E.; Williams, D.; Al-Obaidi, N.; Wojciechowski, F.; Hudson, R.H.E.; Seitz, O.; Gait, M.J. Chemical structure requirements and cellular targeting of microRNA-122 by peptide nucleic acids anti-miRs. Nucleic Acids Res. 2012, 40, 2152–2167. [Google Scholar] [CrossRef] [PubMed]
- Fabani, M.M.; Abreu-Goodger, C.; Williams, D.; Lyons, P.A.; Torres, A.G.; Smith, K.G.C.; Enright, A.J.; Gait, M.J.; Vigorito, E. Efficient inhibition of miR-155 function in vivo by peptide nucleic acids. Nucleic Acids Res. 2010, 38, 4466–4475. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Croce, C.M.; Michaille, J.-J. miR-155: On the crosstalk between inflammation and cancer. Int. Rev. Immunol. 2009, 28, 264–284. [Google Scholar] [CrossRef] [PubMed]
- Corsten, M.F.; Papageorgiou, A.; Verhesen, W.; Carai, P.; Lindow, M.; Obad, S.; Summer, G.; Coort, S.L.M.; Hazebroek, M.; Van Leeuwen, R.; et al. MicroRNA profiling identifies microRNA-155 as an adverse mediator of cardiac injury and dysfunction during acute viral myoCarditis. Circ. Res. 2012, 111, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, J.; Han, L.; Zhang, A.; Zhang, C.; Zheng, Y.; Jiang, T.; Pu, P.; Jiang, C.; Kang, C. Downregulation of miR-221/222 sensitizes glioma cells to temozolomide by regulating apoptosis independently of p53 status. Oncol. Rep. 2012, 27, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, A.M.; Phillips-Mason, P.J.; Burden-Gulley, S.M.; Robinson, S.; Sloan, A.E.; Miller, R.H.; Brady-Kalnay, S.M. Proteolytic cleavage of protein tyrosine phosphatase μ regulates glioblastoma cell migration. Cancer Res. 2009, 69, 6960–6968. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.T.; Rossi, M.; Caracciolo, D.; Gullà, A.; Tagliaferri, P.; Tassone, P. Mir-221/222 are promising targets for innovative anticancer therapy. Expert Opin. Ther. Targets 2016, 20, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Brognara, E.; Fabbri, E.; Montagner, G.; Gasparello, J.; Manicardi, A.; Corradini, R.; Bianchi, N.; Finotti, A.; Breveglieri, G.; Borgatti, M.; et al. High levels of apoptosis are induced in human glioma cell lines by co-administration of peptide nucleic acids targeting miR-221 and miR-222. Int. J. Oncol. 2016, 48, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Brognara, E.; Fabbri, E.; Bazzoli, E.; Montagner, G.; Ghimenton, C.; Eccher, A.; Cantù, C.; Manicardi, A.; Bianchi, N.; Finotti, A.; et al. Uptake by human glioma cell lines and biological effects of a peptide-nucleic acids targeting miR-221. J. Neurooncol. 2014, 118, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Huang, M.; Li, Z.; Jia, F.; Ghosh, Z.; Lijkwan, M.A.; Fasanaro, P.; Sun, N.; Wang, X.; Martelli, F.; et al. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation 2010, 122. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Le, Q.T.; Giaccia, A.J. MiR-210—Micromanager of the hypoxia pathway. Trends Mol. Med. 2010, 16, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, E.; Manicardi, A.; Tedeschi, T.; Sforza, S.; Bianchi, N.; Brognara, E.; Finotti, A.; Breveglieri, G.; Borgatti, M.; Corradini, R.; et al. Modulation of the biological activity of microRNA-210 with peptide nucleic acids (PNAs). ChemMedChem 2011, 6, 2192–2202. [Google Scholar] [CrossRef] [PubMed]
- Manicardi, A.; Fabbri, E.; Tedeschi, T.; Sforza, S.; Bianchi, N.; Brognara, E.; Gambari, R.; Marchelli, R.; Corradini, R. Cellular uptakes, biostabilities and anti-miR-210 activities of chiral arginine-PNAs in leukaemic K562 Cells. ChemBioChem 2012, 13, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Avitabile, C.; Accardo, A.; Ringhieri, P.; Morelli, G.; Saviano, M.; Montagner, G.; Fabbri, E.; Gallerani, E.; Gambari, R.; Romanelli, A. Incorporation of naked peptide nucleic acids into liposomes leads to fast and efficient delivery. Bioconjug. Chem. 2015, 26, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Giljohann, D.A.; Seferos, D.S.; Daniel, W.L.; Massich, M.D.; Patel, P.C.; Mirkin, C.A. Gold nanoparticles for biology and medicine. Angew. Chem. Int. Ed. 2010, 49, 3280–3294. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Han, G.; De, M.; Kim, C.K.; Rotello, V.M. Gold nanoparticles in delivery applications. Adv. Drug Deliv. Rev. 2008, 60, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Ki, J.; Jang, E.; Han, S.; Shin, M.K.; Kang, B.; Huh, Y.M.; Haam, S. Instantaneous pH-boosted functionalization of stellate gold nanoparticles for intracellular imaging of miRNA. ACS Appl. Mater. Interfaces 2017, 9, 17702–17709. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, S.; Pedrero, M.; Pingarrón, J.M. Electrochemical genosensors for the detection of cancer-related miRNAs. Anal. Bioanal. Chem. 2014, 406, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Muthiah, M.; Park, I.-K.; Cho, C.-S. Nanoparticle-mediated delivery of therapeutic genes: Focus on miRNA therapeutics. Expert Opin. Drug Deliv. 2013, 10, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Hazarika, P.; Giorgi, T.; Reibner, M.; Ceyhan, B.; Niemeyer, C.M. Synthesis and characterization of deoxyribonucleic acid-conjugated gold nanoparticles. Bioconjugation Protoc. 2004, 283, 295–304. [Google Scholar]
- Hao, L.; Patel, P.C.; Alhasan, A.H.; Giljohann, D.A.; Mirkin, C.A. Nucleic acid-gold nanoparticle conjugates as mimics of microRNA. Small 2011, 7, 3158–3162. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Jang, H.H.; Ryou, S.-M.; Kim, S.; Bae, J.; Lee, K.; Han, M.S. A functionalized gold nanoparticles-assisted universal carrier for antisense DNA. Chem. Commun. 2010, 46, 4151–4153. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Yeom, J.H.; Ko, J.J.; Han, M.S.; Lee, K.; Na, S.Y.; Bae, J. Effective delivery of anti-miRNA DNA oligonucleotides by functionalized gold nanoparticles. J. Biotechnol. 2011, 155, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Simmons, R.; Huo, D.; Pang, B.; Zhao, X.; Kim, C.W.; Jo, H.; Xia, Y. Targeted delivery of anti-miR-712 by VCAM1-binding Au nanospheres for atherosclerosis therapy. ChemNanoMat 2016, 2, 400–406. [Google Scholar] [CrossRef]
- Fotis, L.; Agrogiannis, G.; Vlachos, I.S.; Pantopoulou, A.; Margoni, A.; Kostaki, M.; Verikokos, C.; Tzivras, D.; Mikhailidis, D.P.; Perrea, D. Intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 at the early stages of atherosclerosis in a rat model. In Vivo (Brooklyn) 2012, 26, 243–250. [Google Scholar]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Company | Nucleic Acid | Target | Clinical Trial |
|---|---|---|---|---|
| Oblimersen | Genta | ASO | Bcl-2 | Phase III |
| Apatorsen | Ionis Pharm | ASO | Hsp27 | Phase II |
| Resten-MP | AVI Biopharm | ASO | c-myc | Phase II |
| ALN-TTR02 | Alnylam | siRNA | TTR | Phase III |
| Bevasiranib | OPKO Health | siRNA | VEGF | Phase III |
| SYL040012 | Sylentis | siRNA | ADRB2 | Phase II |
| AZD9150 | AstraZeneca | siRNA | STAT3 | Phase II |
| PF-655 | Quark Pharm | siRNA | RTP801 | Phase II |
| DCR-MYC | Dicerna Pharm | siRNA | c-myc | Phase II |
| Fitusiran | Alnylam | siRNA | AT3 | Phase II |
| Atu027 | Silence Ther | siRNA | PKN3 | Phase I |
| RXI-109 | RXi Pharm | siRNA | CTGF | Phase I |
| ALN-VSP | Alnylam | siRNA | VEGF | Phase I |
| IMO-2125 | Idera | siRNA | TLR9 | Phase I |
| RG-101 | Regulus | miRNA | HCV | Phase II |
| Miravirsen | Roche | miRNA | HCV | Phase II |
| RG-125 | Regulus | miRNA | miR-103/107 | Phase I |
| MRG-106 | miRagen | miRNA | miR-155 | Phase I |
| MRX-34 | Mirna | miRNA | miR-34 | Phase I |
| miRNA | Target | Biologic Function | Reference |
|---|---|---|---|
| Let-7 | c-myc, HMGA2, RAS | Tumour suppressor | [163,171] |
| miR-10b | HOXD10 | Oncogene | [172] |
| miR-21 | PTEN, TPM1, PDCD4 | Oncogene | [173] |
| miR-122a | Cyclin G1 | Tumour suppressor | [174] |
| miR-155 | AT1R, TP53INP1 | Oncogene | [175] |
| miR-210 | HIF-1α | Oncogene | [176] |
| miR-221/222 | CD117 | Oncogene | [177] |
| miR-205 | PRKCε | Tumour suppressor | [178] |
| miR-20a | TIMP2, ATG7 | Oncogene | [179] |
| miR-29b | Mcl-1 | Oncogene | [180] |
| miR-712 | TIMP3 | Oncogene | [181,182] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grijalvo, S.; Alagia, A.; Jorge, A.F.; Eritja, R. Covalent Strategies for Targeting Messenger and Non-Coding RNAs: An Updated Review on siRNA, miRNA and antimiR Conjugates. Genes 2018, 9, 74. https://doi.org/10.3390/genes9020074
Grijalvo S, Alagia A, Jorge AF, Eritja R. Covalent Strategies for Targeting Messenger and Non-Coding RNAs: An Updated Review on siRNA, miRNA and antimiR Conjugates. Genes. 2018; 9(2):74. https://doi.org/10.3390/genes9020074
Chicago/Turabian StyleGrijalvo, Santiago, Adele Alagia, Andreia F. Jorge, and Ramon Eritja. 2018. "Covalent Strategies for Targeting Messenger and Non-Coding RNAs: An Updated Review on siRNA, miRNA and antimiR Conjugates" Genes 9, no. 2: 74. https://doi.org/10.3390/genes9020074
APA StyleGrijalvo, S., Alagia, A., Jorge, A. F., & Eritja, R. (2018). Covalent Strategies for Targeting Messenger and Non-Coding RNAs: An Updated Review on siRNA, miRNA and antimiR Conjugates. Genes, 9(2), 74. https://doi.org/10.3390/genes9020074