Glutamine Synthetase: Localization Dictates Outcome



Abstract
:1. Introduction
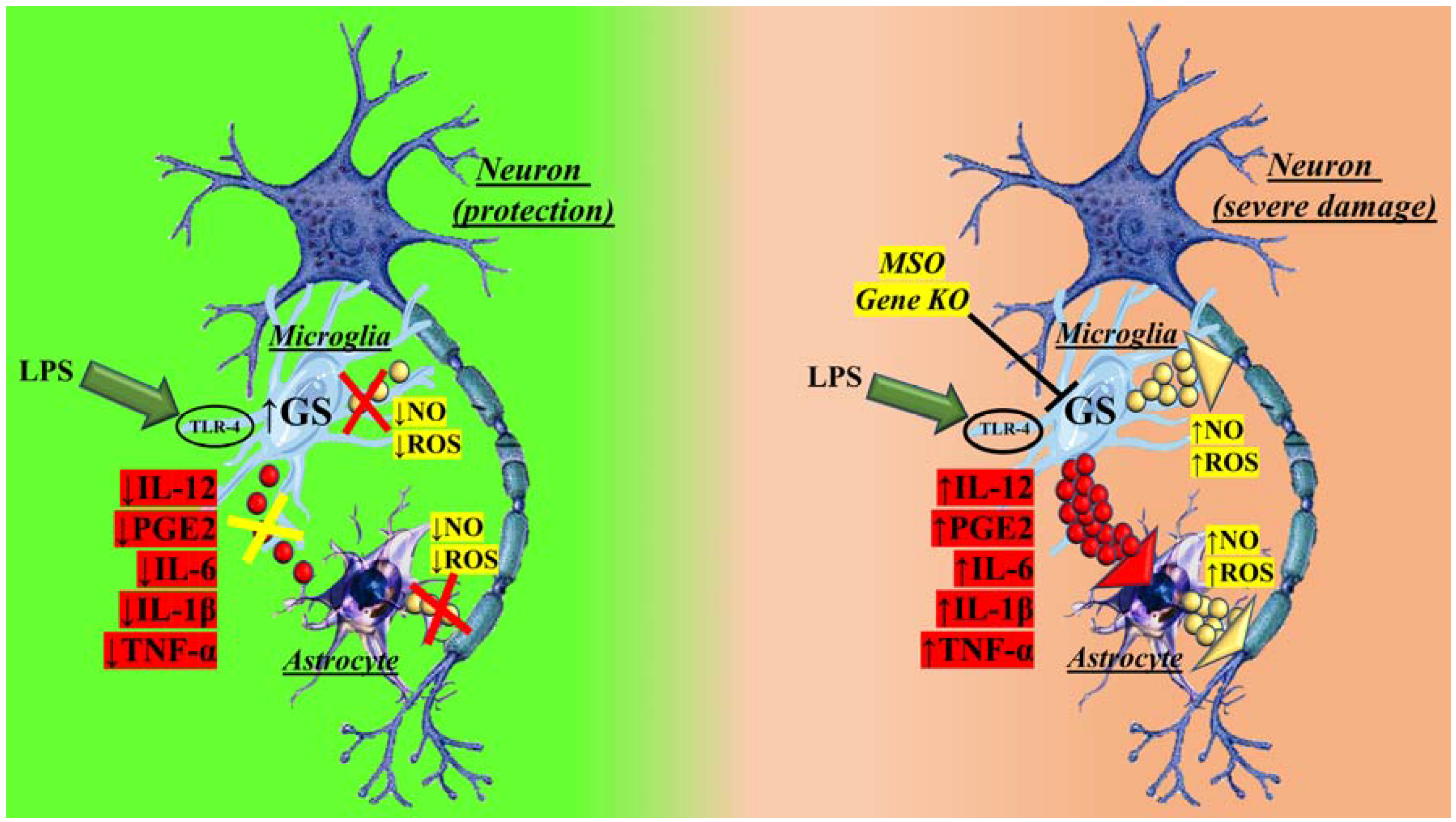
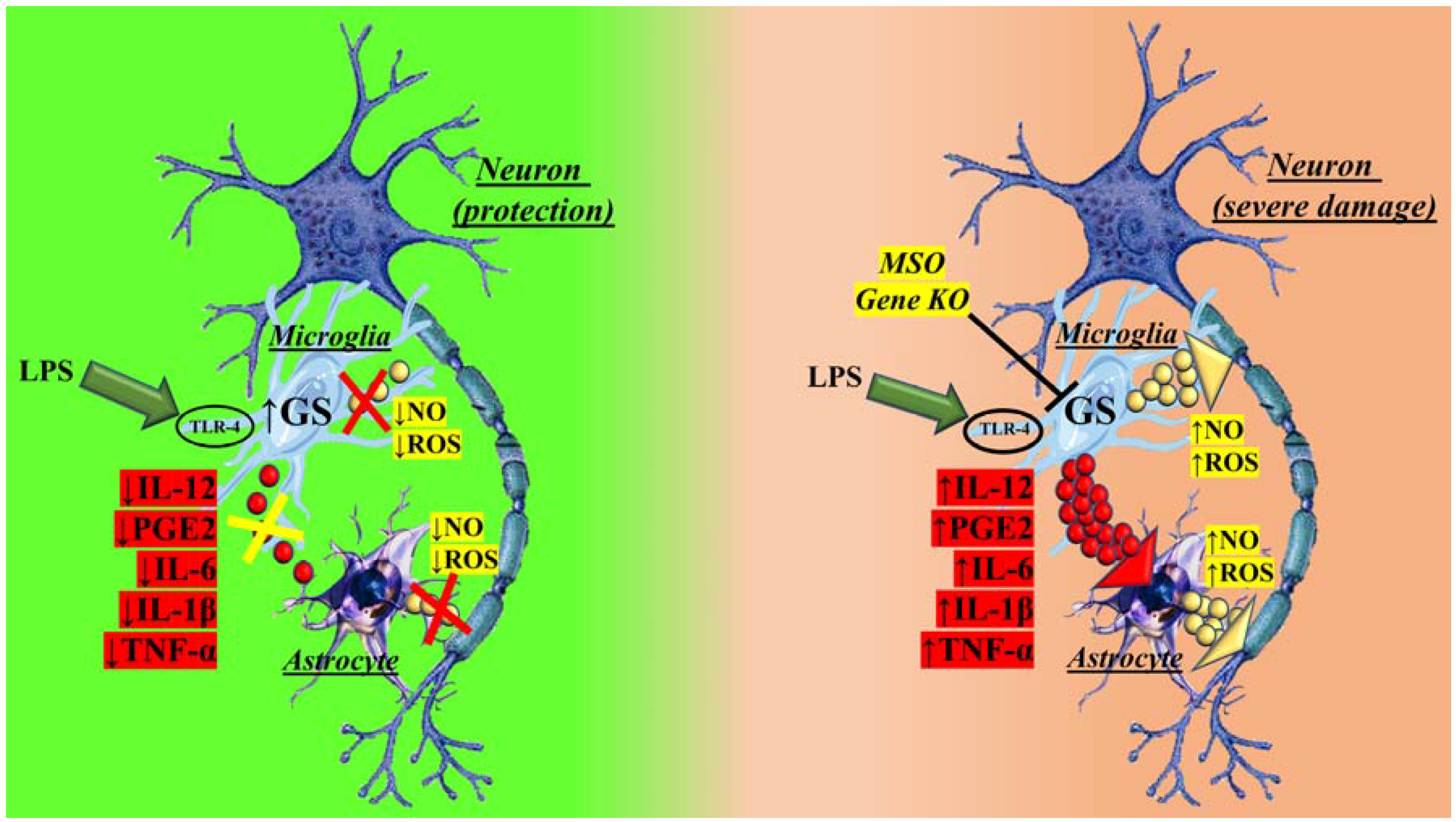
2. GS in Brain Physiological and Pathological Conditions
3. Glutamine Synthetase in Tumors
3.1. Glutamine Synthetase in Cancer Cells
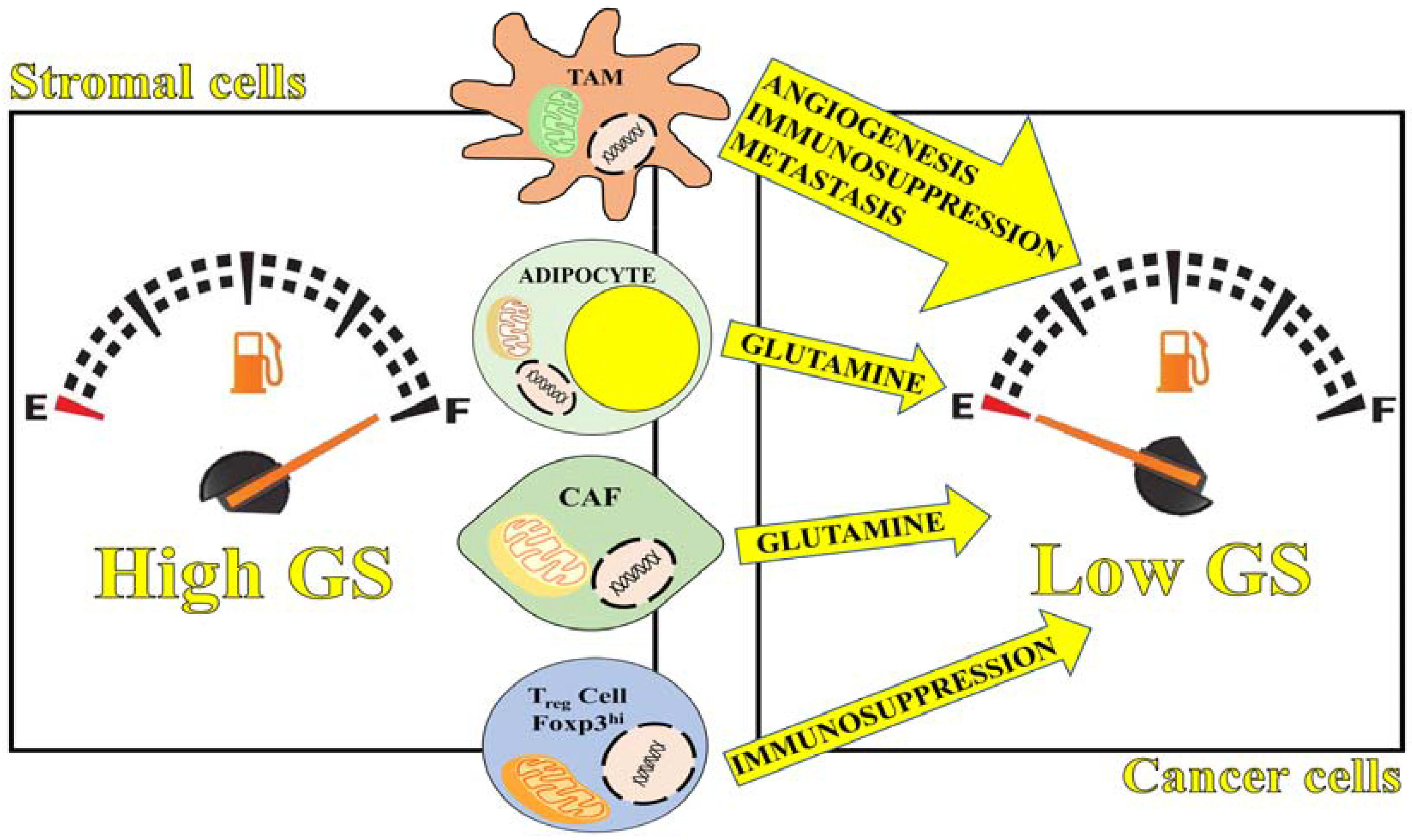
3.2. Glutamine Synthetase in the Tumor Microenvironment
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Stumvoll, M.; Perriello, G.; Meyer, C.; Gerich, J. Role of glutamine in human carbohydrate metabolism in kidney and other tissues. Kidney Int. 1999, 55, 778–792. [Google Scholar] [CrossRef] [PubMed]
- Schreier, H.J. Biosynthesis of Glutamine and Glutamate and the Assimilation of Ammonia. In Bacillus subtilis and Other Gram-Positive Bacteria; Sonenshein, A., Hoch, J., Losick, R., Eds.; ASM Press: Washington, DC, USA, 1993; pp. 281–298. [Google Scholar]
- Listrom, C.D.; Morizono, H.; Rajagopal, B.S.; McCann, M.T.; Tuchman, M.; Allewell, N.M. Expression, purification, and characterization of recombinant human glutamine synthetase. Biochem. J. 1997, 328, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L. The role of glutamine synthetase and glutamate dehydrogenase in cerebral ammonia homeostasis. Neurochem. Res. 2012, 37, 2439–2455. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.A.; Viña, J.R. How Glutamate Is Managed by the Blood-Brain Barrier. Biology 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.A. The blood-brain barrier and glutamate. Am. J. Clin. Nutr. 2009, 90, 867S–874S. [Google Scholar] [CrossRef] [PubMed]
- Walser, M.; Bodenlos, L.J. Urea metabolism in man. J. Clin. Investig. 1959, 38, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Gill, H.S.; Pfluegl, G.M.; Rotstein, S.H. Structure-function relationships of glutamine synthetases. Biochim. Biophys. Acta 2000, 1477, 122–145. [Google Scholar] [CrossRef]
- Cooper, A.; Freed, B. Metabolism of [13N]ammonia in rat lung. Neurochem. Int. 2005, 47, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Häussinger, D.; Schliess, F. Glutamine metabolism and signaling in the liver. Front. Biosci. 2007, 12, 371–391. [Google Scholar] [CrossRef] [PubMed]
- Qvartskhava, N.; Lang, P.A.; Görg, B.; Pozdeev, V.I.; Ortiz, M.P.; Lang, K.S.; Bidmon, H.J.; Lang, E.; Leibrock, C.B.; Herebian, D.; et al. Hyperammonemia in gene-targeted mice lacking functional hepatic glutamine synthetase. Proc. Natl. Acad. Sci. USA 2015, 112, 5521–5526. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hakvoort, T.B.M.; Köhler, S.E.; Vermeulen, J.L.M.; de Waart, D.R.; de Theije, C.; ten Have, G.A.M.; van Eijk, H.M.H.; Kunne, C.; Labruyere, W.T.; et al. Glutamine synthetase in muscle is required for glutamine production during fasting and extrahepatic ammonia detoxification. J. Biol. Chem. 2010, 285, 9516–9524. [Google Scholar] [CrossRef] [PubMed]
- Häberle, J.; Görg, B.; Rutsch, F.; Schmidt, E.; Toutain, A.; Benoist, J.-F.; Gelot, A.; Suc, A.-L.; Höhne, W.; Schliess, F.; et al. Congenital Glutamine Deficiency with Glutamine Synthetase Mutations. N. Engl. J. Med. 2005, 353, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Menga, A.; Lebrun, A.; Hooper, D.C.; Butterfield, D.A.; Mazzone, M.; Castegna, A. Blockade of Glutamine Synthetase Enhances Inflammatory Response in Microglial Cells. Antioxid. Redox Signal. 2016, 26, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Brusilow, S.W.; Koehler, R.C.; Traystman, R.J.; Cooper, A.J.L. Astrocyte glutamine synthetase: Importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics 2010, 7, 452–470. [Google Scholar] [CrossRef] [PubMed]
- Dadsetan, S.; Kukolj, E.; Bak, L.K.; Sørensen, M.; Ott, P.; Vilstrup, H.; Schousboe, A.; Keiding, S.; Waagepetersen, H.S. Brain alanine formation as an ammonia-scavenging pathway during hyperammonemia: Effects of glutamine synthetase inhibition in rats and astrocyte-neuron co-cultures. J. Cereb. Blood Flow Metab. 2013, 33, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Kanski, J. Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech. Ageing Dev. 2001, 122, 945–962. [Google Scholar] [CrossRef]
- Levine, R.L. Oxidative modification of glutamine synthetase. I. Inactivation is due to loss of one histidine residue. J. Biol. Chem. 1983, 258, 11823–11827. [Google Scholar] [PubMed]
- Rivett, A.J.; Levine, R.L. Metal-catalyzed oxidation of Escherichia coli glutamine synthetase: correlation of structural and functional changes. Arch. Biochem. Biophys. 1990, 278, 26–34. [Google Scholar] [CrossRef]
- Fisher, M.T.; Stadtman, E.R. Oxidative modification of Escherichia coli glutamine synthetase. Decreases in the thermodynamic stability of protein structure and specific changes in the active site conformation. J. Biol. Chem. 1992, 267, 1872–1880. [Google Scholar] [PubMed]
- Butterfield, D.A.; Hensley, K.; Cole, P.; Subramaniam, R.; Aksenov, M.; Aksenova, M.; Bummer, P.M.; Haley, B.E.; Carney, J.M. Oxidatively induced structural alteration of glutamine synthetase assessed by analysis of spin label incorporation kinetics: Relevance to Alzheimer’s disease. J. Neurochem. 1997, 68, 2451–2457. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Stadtman, E.R.; Floyd, R.A.; Markesbery, W.R. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 10540–10543. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 1995, 65, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Aksenov, M.; Aksenova, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: Creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic. Biol. Med. 2002, 33, 562–571. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D.; Castegna, A. Proteomics in Alzheimer’s disease: Insights into potential mechanisms of neurodegeneration. J. Neurochem. 2003, 86, 1313–1327. [Google Scholar] [CrossRef] [PubMed]
- Le Prince, G.; Delaere, P.; Fages, C.; Lefrançois, T.; Touret, M.; Salanon, M.; Tardy, M. Glutamine synthetase (GS) expression is reduced in senile dementia of the Alzheimer type. Neurochem. Res. 1995, 20, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Jiménez, F.J.; Molina, J.A.; Aguilar, M.V.; Meseguer, I.; Mateos-Vega, C.J.; González-Muñoz, M.J.; de Bustos, F.; Martínez-Salio, A.; Ortí-Pareja, M.; Zurdo, M.; et al. Cerebrospinal fluid levels of transition metals in patients with Parkinson’s disease. J. Neural Transm. 1998, 105, 497. [Google Scholar] [CrossRef] [PubMed]
- Csernansky, J.G.; Bardgett, M.E.; Sheline, Y.I.; Morris, J.C.; Olney, J.W. CSF excitatory amino acids and severity of illness in Alzheimer’s disease. Neurology 1996, 46, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Lauderback, C.M.; Hackett, J.M.; Huang, F.F.; Keller, J.N.; Szweda, L.I.; Markesbery, W.R.; Butterfield, D.A. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: The role of Abeta1-42. J. Neurochem. 2001, 78, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Hansen, L.; Alford, M.; Deteresa, R.; Mallory, M. Deficient glutamate tranport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Palmieri, L.; Spera, I.; Porcelli, V.; Palmieri, F.; Fabis-Pedrini, M.J.; Kean, R.B.; Barkhouse, D.A.; Curtis, M.T.; Hooper, D.C. Oxidative stress and reduced glutamine synthetase activity in the absence of inflammation in the cortex of mice with experimental allergic encephalomyelitis. Neuroscience 2011, 185, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Chrétien, F.; Vallat-Decouvelaere, A.-V.; Bossuet, C.; Rimaniol, A.-C.; Le Grand, R.; Le Pavec, G.; Créminon, C.; Dormont, D.; Gray, F.; Gras, G. Expression of excitatory amino acid transporter-2 (EAAT-2) and glutamine synthetase (GS) in brain macrophages and microglia of SIVmac251-infected macaques. Neuropathol. Appl. Neurobiol. 2002, 28, 410–417. [Google Scholar] [CrossRef] [PubMed]
- López-Redondo, F.; Nakajima, K.; Honda, S.; Kohsaka, S. Glutamate transporter GLT-1 is highly expressed in activated microglia following facial nerve axotomy. Brain Res. Mol. Brain Res. 2000, 76, 429–435. [Google Scholar] [CrossRef]
- Van Landeghem, F.K.; Stover, J.F.; Bechmann, I.; Brück, W.; Unterberg, A.; Bührer, C.; von Deimling, A. Early expression of glutamate transporter proteins in ramified microglia after controlled cortical impact injury in the rat. Glia 2001, 35, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Gras, G.; Chrétien, F.; Vallat-Decouvelaere, A.-V.; Le Pavec, G.; Porcheray, F.; Bossuet, C.; Léone, C.; Mialocq, P.; Dereuddre-Bosquet, N.; Clayette, P.; et al. Regulated expression of sodium-dependent glutamate transporters and synthetase: A neuroprotective role for activated microglia and macrophages in HIV infection? Brain Pathol. 2003, 13, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Vallat-Decouvelaere, A.V.; Chrétien, F.; Gras, G.; Le Pavec, G.; Dormont, D.; Gray, F. Expression of excitatory amino acid transporter-1 in brain macrophages and microglia of HIV-infected patients. A neuroprotective role for activated microglia? J. Neuropathol. Exp. Neurol. 2003, 62, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Chrétien, F.; Le Pavec, G.; Vallat-Decouvelaere, A.-V.; Delisle, M.-B.; Uro-Coste, E.; Ironside, J.W.; Gambetti, P.; Parchi, P.; Créminon, C.; Dormont, D.; et al. Expression of excitatory amino acid transporter-1 (EAAT-1) in brain macrophages and microglia of patients with prion diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Rimaniol, A.-C.; Mialocq, P.; Clayette, P.; Dormont, D.; Gras, G. Role of glutamate transporters in the regulation of glutathione levels in human macrophages. Am. J. Physiol. Physiol. 2001, 281, C1964–C1970. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Sandberg, M.; Hansson, E.; Rönnbäck, L. Microglial glutamate uptake is coupled to glutathione synthesis and glutamate release. Eur. J. Neurosci. 2006, 24, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Gras, G.; Porcheray, F.; Samah, B.; Leone, C. The glutamate-glutamine cycle as an inducible, protective face of macrophage activation. J. Leukoc. Biol. 2006, 80, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, E.M.; Cochrane, F.; Barclay, A.N.; van den Berg, T.K. Signal regulatory proteins in the immune system. J. Immunol. 2005, 175, 7781–7787. [Google Scholar] [CrossRef] [PubMed]
- Rosenstiel, P.; Lucius, R.; Deuschl, G.; Sievers, J.; Wilms, H. From theory to therapy: Implications from an in vitro model of ramified microglia. Microsc. Res. Tech. 2001, 54, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Schmidtmayer, J.; Jacobsen, C.; Miksch, G.; Sievers, J. Blood monocytes and spleen macrophages differentiate into microglia-like cells on monolayers of astrocytes: Membrane currents. Glia 1994, 12, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Sievers, J.; Parwaresch, R.; Wottge, H.-U. Blood monocytes and spleen macrophages differentiate into microglia-like cells on monolayers of astrocytes: Morphology. Glia 1994, 12, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Leone, C.; Le Pavec, G.; Même, W.; Porcheray, F.; Samah, B.; Dormont, D.; Gras, G. Characterization of human monocyte-derived microglia-like cells. Glia 2006, 54, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.A.; Bauer, J.; Flick, M.J.; Sikorski, S.L.; Nuriel, T.; Lassmann, H.; Degen, J.L.; Akassoglou, K. The fibrin-derived γ377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J. Exp. Med. 2007, 204, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Hage Hassan, R.; Bourron, O.; Hajduch, E. Defect of insulin signal in peripheral tissues: Important role of ceramide. World J. Diabetes 2014, 5, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Ruud, J.; Steculorum, S.M.; Brüning, J.C. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat. Commun. 2017, 8, 15259. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Feldman, E.L. Insulin resistance as a key link for the increased risk of cognitive impairment in the metabolic syndrome. Exp. Mol. Med. 2015, 47, e149. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 35–66. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers. Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Di Domenico, F.; Barone, E. Elevated risk of type 2 diabetes for development of Alzheimer disease: a key role for oxidative stress in brain. Biochim. Biophys. Acta 2014, 1842, 1693–1706. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.-C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease—Associated Aβ oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Spera, I.; Menga, A.; Infantino, V.; Iacobazzi, V.; Castegna, A. Glutamine synthetase desensitizes differentiated adipocytes to proinflammatory stimuli by raising intracellular glutamine levels. FEBS Lett. 2014, 588, 4807–4814. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.I.; Proenca, T.; Oliveira, C.R.; Santos, M.S.; Rego, A.C. Insulin Restores Metabolic Function in Cultured Cortical Neurons Subjected to Oxidative Stress. Diabetes 2006, 55, 2863–2870. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.I.; Santos, M.S.; Seiça, R.; de Oliveira, C.R. Insulin affects synaptosomal GABA and glutamate transport under oxidative stress conditions. Brain Res. 2003, 977, 23–30. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends in Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 2017, 358, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Sohn, B.H.; Park, I.Y.; Shin, J.-H.; Yim, S.Y.; Lee, J.-S. Glutamine synthetase mediates Sorafenib sensitivity in β-catenin-active hepatocellular carcinoma cells. Exp. Mol. Med. 2018, 50, e421. [Google Scholar] [CrossRef] [PubMed]
- Van der Vos, K.E.; Eliasson, P.; Proikas-Cezanne, T.; Vervoort, S.J.; van Boxtel, R.; Putker, M.; van Zutphen, I.J.; Mauthe, M.; Zellmer, S.; Pals, C.; et al. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat. Cell Biol. 2012, 14, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Kung, H.N.; Marks, J.R.; Chi, J.T. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.; Schraml, P.; Spagnoli, G.C.; Harder, F.; Heberer, M. Identification of genes differentially expressed in melanoma sublines derived from a single surgical specimen characterised by different sensitivity to cytotoxic T-lymphocyte activity. Schweiz. Med. Wochenschr. 2000, 130, 617–624. [Google Scholar] [PubMed]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- Bolzoni, M.; Chiu, M.; Accardi, F.; Vescovini, R.; Airoldi, I.; Storti, P.; Todoerti, K.; Agnelli, L.; Missale, G.; Andreoli, R.; et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: A new attractive target. Blood 2016, 128, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Stine, Z.E.; Xia, J.; Lu, Y.; O’Connor, R.S.; Altman, B.J.; Hsieh, A.L.; Gouw, A.M.; Thomas, A.G.; Gao, P.; et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Investig. 2015, 125, 2293–2306. [Google Scholar] [CrossRef] [PubMed]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.-L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of Tumor Metabolism Reveals Mitochondrial Glucose Oxidation in Genetically Diverse Human Glioblastomas in the Mouse Brain In Vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fan, S.; Lu, J.; Zhang, Z.; Wu, D.; Wu, Z.; Zheng, Y. GLUL Promotes Cell Proliferation in Breast Cancer. J. Cell. Biochem. 2017, 118, 2018–2025. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Albrechtsen, R.; Kronqvist, P.; Cox, J.; Mann, M.; Geiger, T. Proteomic maps of breast cancer subtypes. Nat. Commun. 2016, 7, 10259. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, M.O.; Fan, T.W.M.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Parlati, F.; Demo, S.D.; Gross, M.I.; Janes, J.R.; Lewis, E.R.; MacKinnon, A.L.; Rodriguez, M.L.M.; Shwonek, P.J.; Wang, T.; Yang, J.; et al. CB-839, a novel potent and selective glutaminase inhibitor, has broad antiproliferative activity in cell lines derived from both solid tumors and hematological malignancies. Cancer Res. 2014, 74, 839. [Google Scholar] [CrossRef]
- Kitajima, S.; Lee, K.L.; Hikasa, H.; Sun, W.; Huang, R.Y.-J.; Yang, H.; Matsunaga, S.; Yamaguchi, T.; Araki, M.; Kato, H.; et al. Hypoxia-inducible factor-1α promotes cell survival during ammonia stress response in ovarian cancer stem-like cells. Oncotarget 2017, 8, 114481–114494. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Lukey, M.J.; Greene, K.S.; Erickson, J.W.; Wilson, K.F.; Cerione, R.A. The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat. Commun. 2016, 7, 11321. [Google Scholar] [CrossRef] [PubMed]
- Nicolay, B.N.; Gameiro, P.A.; Tschöp, K.; Korenjak, M.; Heilmann, A.M.; Asara, J.M.; Stephanopoulos, G.; Iliopoulos, O.; Dyson, N.J. Loss of RBF1 changes glutamine catabolism. Genes Dev. 2013, 27, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014, 33, 556–566. [Google Scholar] [CrossRef] [PubMed]
- McGuirk, S.; Gravel, S.-P.; Deblois, G.; Papadopoli, D.J.; Faubert, B.; Wegner, A.; Hiller, K.; Avizonis, D.; Akavia, U.; Jones, R.G.; et al. PGC-1α supports glutamine metabolism in breast cancer. Cancer Metab. 2013, 1, 22. [Google Scholar] [CrossRef] [PubMed]
- Van Nguyen, T.; Lee, J.E.; Sweredoski, M.J.; Yang, S.-J.; Jeon, S.-J.; Harrison, J.S.; Yim, J.-H.; Lee, S.G.; Handa, H.; Kuhlman, B.; et al. Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon. Mol. Cell 2016, 61, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Oncogene-directed alterations in cancer cell metabolism. Trends Cancer 2016, 2, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Bott, A.J.; Peng, I.-C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D.; et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Hardie, D.G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Achreja, A.; Yeung, T.-L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.A.; Neeley, C.K.; Baker, N.A.; Washabaugh, A.R.; Flesher, C.G.; Nelson, B.S.; Frankel, T.L.; Lumeng, C.N.; Lyssiotis, C.A.; Wynn, M.L.; et al. Adipocytes promote pancreatic cancer cell proliferation via glutamine transfer. Biochem. Biophys. Rep. 2016, 7, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Ehsanipour, E.A.; Sheng, X.; Behan, J.W.; Wang, X.; Butturini, A.; Avramis, V.I.; Mittelman, S.D. Adipocytes Cause Leukemia Cell Resistance to L-Asparaginase via Release of Glutamine. Cancer Res. 2013, 73, 2998–3006. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine Uptake and Metabolism Are Coordinately Regulated by ERK/MAPK during T Lymphocyte Activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Klysz, D.; Tai, X.; Robert, P.A.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I.; et al. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 2015, 8, ra97. [Google Scholar] [CrossRef] [PubMed]
- Metzler, B.; Gfeller, P.; Guinet, E. Restricting Glutamine or Glutamine-Dependent Purine and Pyrimidine Syntheses Promotes Human T Cells with High FOXP3 Expression and Regulatory Properties. J. Immunol. 2016, 196, 3618–3630. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Tumor Type | GS Expression | Phenotype | References |
|---|---|---|---|
| Breast (luminal) Breast Breast (basal) | High | Low aggressiveness and therapy-resistance | [71] |
| High | (HER2+, ER+) High aggressiveness | [79,80] | |
| Low | High aggressiveness and therapy-resistance | [71] | |
| Liver | High | (MET-induced) Glutaminase inhibition-resistance | [81] |
| High | Sorafenib sensitivity | [69] | |
| Low | Sorafenib resistance | [69] | |
| Low | (MYC-induced) Glutaminase inhibition-sensitivity | [81] | |
| Glioblastoma multiforme | High | High aggressiveness, Glutaminase inhibition-resistance | [67] |
| Non-Small Cell | High | (MYC-induced) Glutaminase inhibition-resistance | [81] |
| Lung Carcinoma | Low | Glutaminase inhibition-sensitivity | [75] |
| Ovary | High | (CD90+ cancer stem-like cells) High tumorigenicity | [83] |
| High | Low invasiveness | [73] | |
| Low | High invasiveness | [73] | |
| Melanoma | High | Cytotoxic T lymphocyte killing sensitivity | [72] |
| Low | Cytotoxic T lymphocyte killing resistance | [72] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castegna, A.; Menga, A. Glutamine Synthetase: Localization Dictates Outcome. Genes 2018, 9, 108. https://doi.org/10.3390/genes9020108
Castegna A, Menga A. Glutamine Synthetase: Localization Dictates Outcome. Genes. 2018; 9(2):108. https://doi.org/10.3390/genes9020108
Chicago/Turabian StyleCastegna, Alessandra, and Alessio Menga. 2018. "Glutamine Synthetase: Localization Dictates Outcome" Genes 9, no. 2: 108. https://doi.org/10.3390/genes9020108
APA StyleCastegna, A., & Menga, A. (2018). Glutamine Synthetase: Localization Dictates Outcome. Genes, 9(2), 108. https://doi.org/10.3390/genes9020108