Multiple Sequence Alignments Enhance Boundary Definition of RNA Structures


Abstract
1. Introduction
2. Materials and Methods
2.1. Fitness Function for Boundary Detection
2.2. Benchmark Data Set
2.3. Benchmarking
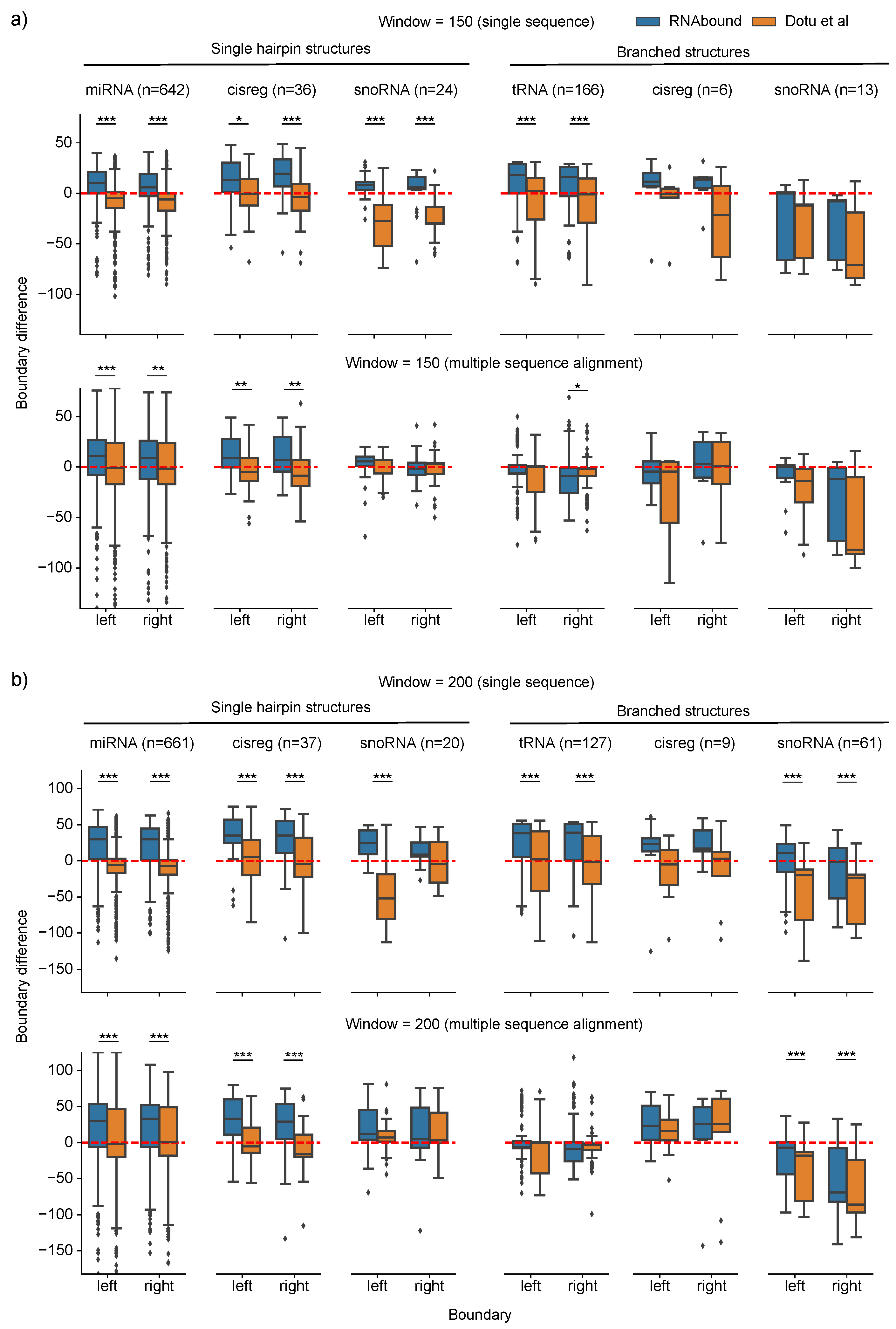
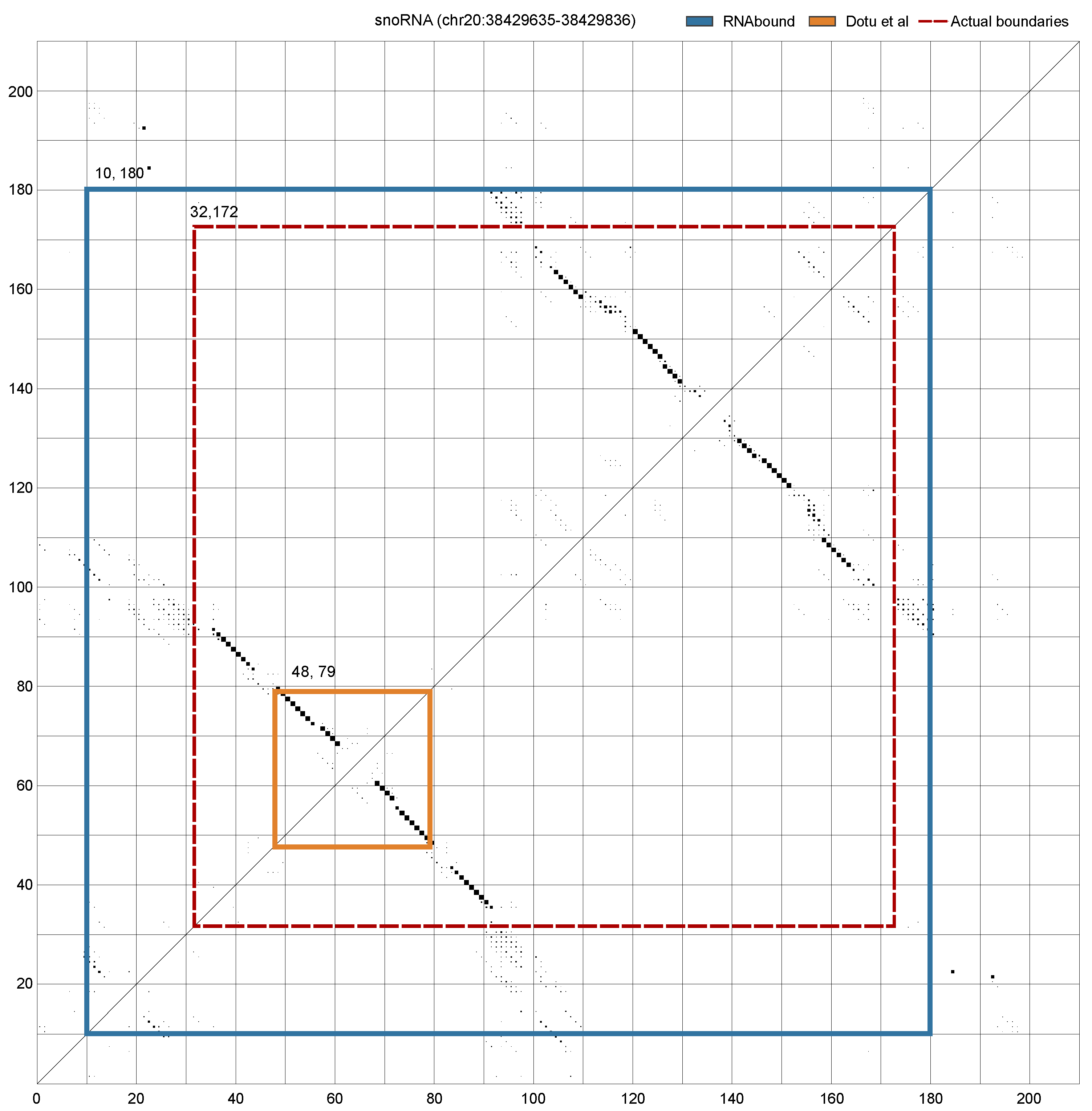
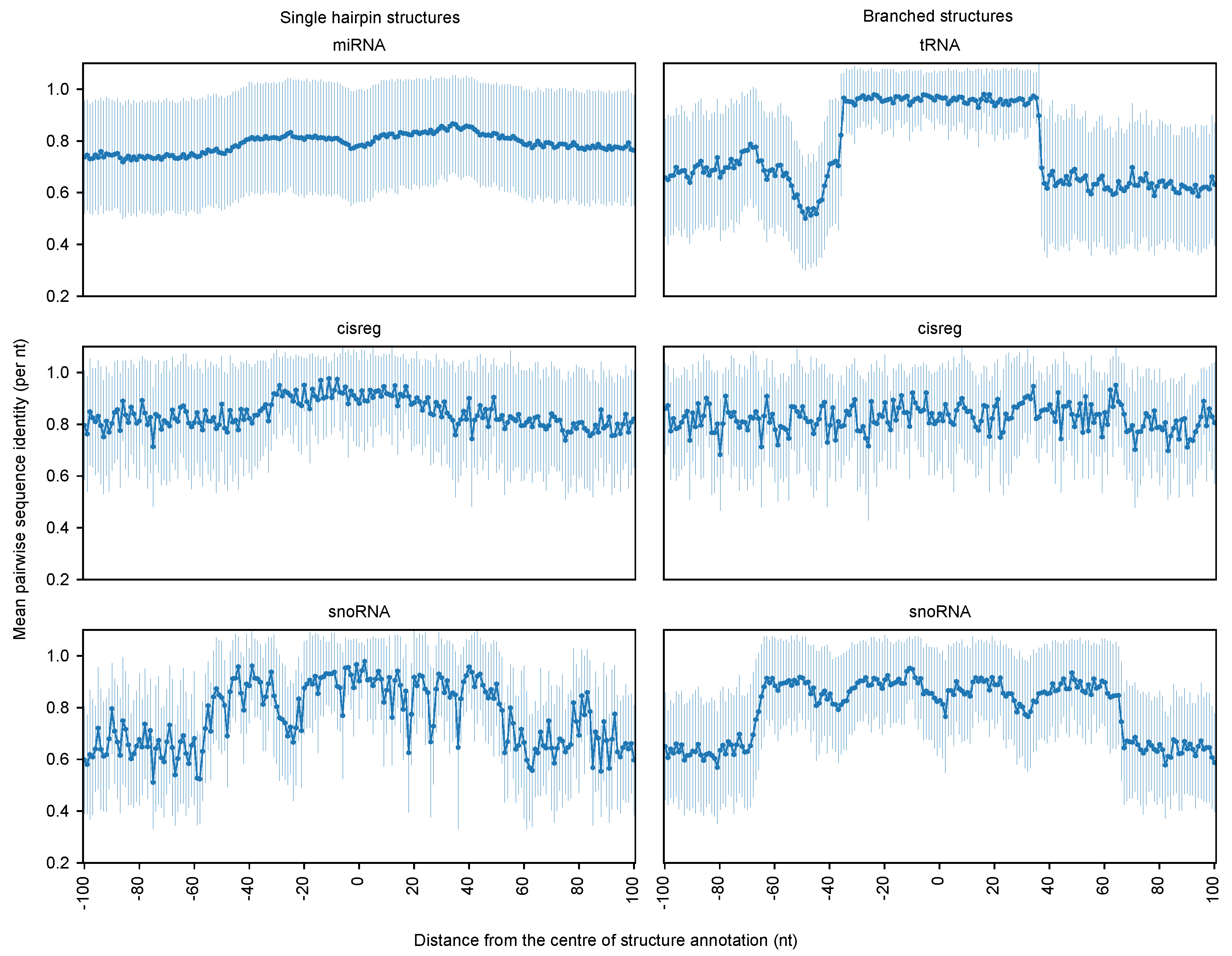
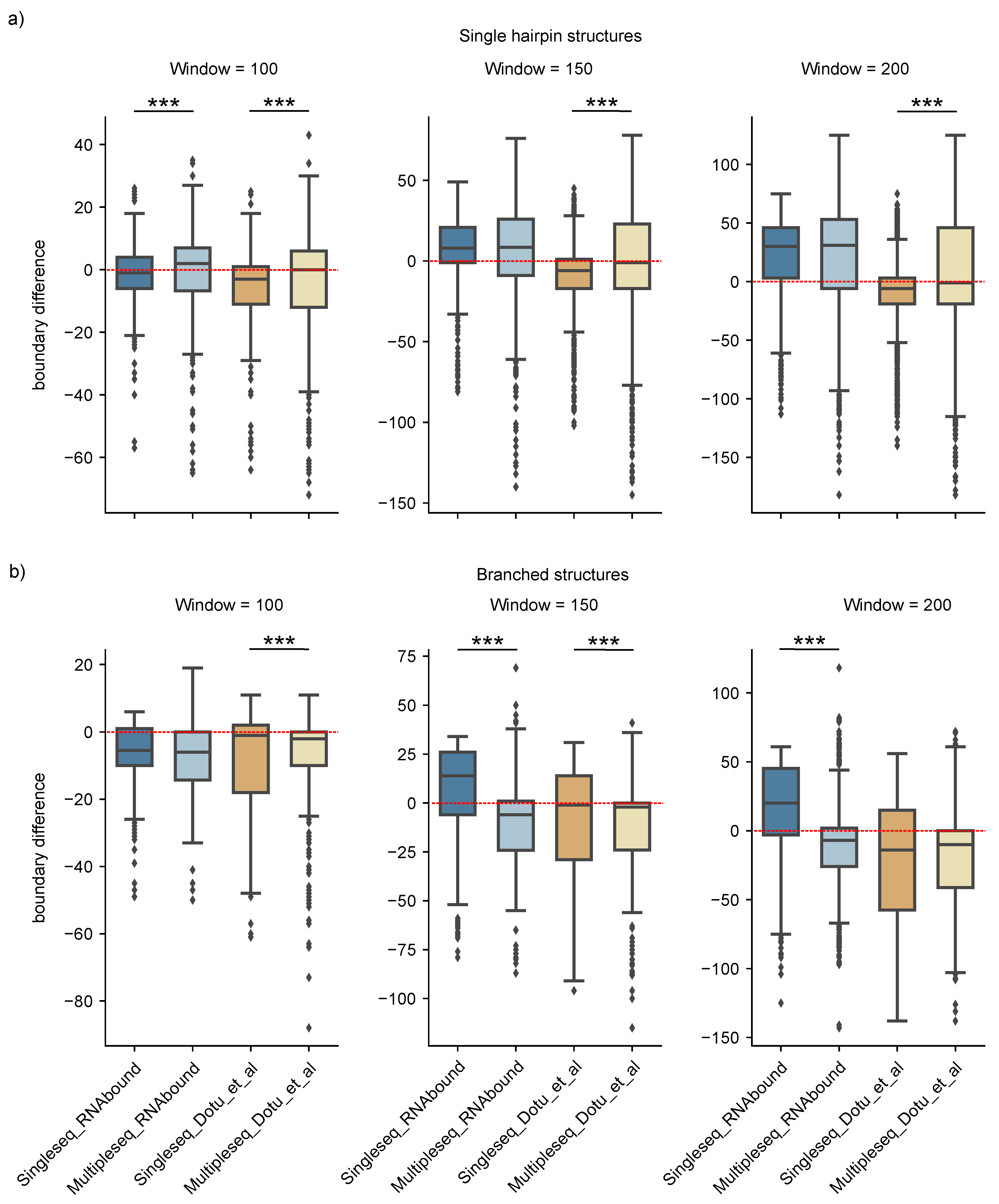
3. Results
3.1. Boundary Detection of Branched RNA Structures Is Improved with Multiple Sequence Alignment
3.2. RNAbound Predictions Are Sensitive to both Single Hairpin and Branched Structures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Window | Family (n) | RNAbound | Dotu et al. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Left | Right | Left | Right | ||||||
| Median | std | Median | std | Median | std | Median | std | ||
| 100 | Single hairpin structures | ||||||||
| miRNA (244) | 4.0 | 12.50 | 1.0 | 14.34 | 1.0 | 16.85 | 0.0 | 14.30 | |
| cisreg (28) | 5.0 | 10.23 | −2.0 | 14.83 | −8.5 | 14.58 | −15.5 | 18.21 | |
| snoRNA (6) | 1.5 | 10.57 | −6.0 | 15.80 | −18.0 | 15.20 | −17.5 | 18.86 | |
| all (279) | 4.0 | 12.47 | 0.0 | 14.39 | 1.0 | 16.67 | −1.0 | 14.93 | |
| Branched structures | |||||||||
| tRNA (185) | −5.0 | 11.07 | −9.0 | 12.38 | 0.0 | 21.19 | −2.0 | 11.17 | |
| cisreg (2) | 7.5 | 3.54 | 7.5 | 3.54 | 5.0 | 0.0 | 7.5 | 2.12 | |
| ribozyme (2) | −8.0 | 15.56 | −10.5 | 2.12 | −25.0 | 35.36 | −8.0 | 7.07 | |
| snRNA (1) | −33.0 | 0.00 | −12.0 | 0.00 | −33.0 | 0.00 | −13.0 | 0.00 | |
| all (190) | −5.0 | 11.07 | −9.0 | 12.41 | 0.0 | 21.23 | −2.0 | 11.16 | |
| 150 | Single hairpin structures | ||||||||
| miRNA (642) | 11.0 | 27.29 | 9.0 | 26.62 | −1.0 | 33.91 | −1.5 | 29.71 | |
| cisreg (36) | 9.0 | 18.35 | 7.0 | 21.42 | −5.0 | 21.14 | −8.5 | 24.76 | |
| snoRNA (24) | 5.5 | 19.48 | −1.5 | 16.01 | 7.0 | 15.46 | 3.0 | 21.32 | |
| all (709) | 9.0 | 26.61 | 8.0 | 26.04 | −1.0 | 32.86 | −2.0 | 29.17 | |
| Branched structures | |||||||||
| tRNA (166) | −6.0 | 18.34 | −9.0 | 20.09 | 0.0 | 22.96 | −2.0 | 14.53 | |
| cisreg (6) | −4.5 | 24.70 | 3.0 | 39.39 | −4.5 | 50.49 | 1.0 | 40.18 | |
| snoRNA (13) | 0.0 | 21.45 | −12.0 | 36.90 | −14.0 | 32.78 | −82.0 | 44.22 | |
| ribozyme (3) | −51.0 | 32.97 | −14.0 | 15.39 | −50.0 | 33.26 | −13.0 | 15.28 | |
| snRNA (3) | −17.0 | 15.01 | −50.0 | 33.60 | −80.0 | 32.75 | −13.0 | 9.02 | |
| all (190) | −6.0 | 19.23 | −9.5 | 23.24 | 0.0 | 26.23 | −3.0 | 21.68 | |
| 200 | Single hairpin structures | ||||||||
| miRNA (661) | 30.0 | 43.96 | 33.0 | 39.94 | −2.0 | 49.87 | 1.0 | 45.70 | |
| cisreg (37) | 33.0 | 31.60 | 29.0 | 41.10 | −5.0 | 29.04 | −16.0 | 34.79 | |
| snoRNA (20) | 12.0 | 34.34 | 5.0 | 44.38 | 7.0 | 28.95 | 3.0 | 33.36 | |
| all (727) | 29.0 | 43.47 | 32.0 | 40.06 | −1.0 | 45.58 | 0.0 | 45.27 | |
| Branched structures | |||||||||
| tRNA (127) | −6.0 | 30.38 | −9.0 | 34.29 | 0.0 | 29.02 | −3.0 | 19.67 | |
| cisreg (9) | 23.0 | 32.20 | 26.0 | 61.57 | 16.0 | 35.36 | 26 | 75.80 | |
| snoRNA (61) | −7.0 | 36.62 | −69.0 | 44.87 | −18.0 | 36.99 | −86.0 | 39.29 | |
| ribozyme (2) | −35.0 | 49.50 | −14.5 | 31.82 | −31.5 | 44.55 | −18.0 | 21.21 | |
| snRNA (6) | −22.0 | 38.90 | −48.5 | 29.95 | −86.0 | 44.46 | −12.0 | 32.94 | |
| all (206) | −6.0 | 34.85 | −11.5 | 43.77 | −14.0 | 35.76 | −9.0 | 40.63 | |

| Window | Family (n) | RNAbound | Dotu et al. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Left | Right | Left | Right | ||||||
| Median | std | Median | std | Median | std | Median | std | ||
| 100 | Single hairpin structures | ||||||||
| miRNA (244) | 0.0 | 7.35 | −2.0 | 9.12 | −2.0 | 10.70 | −3.0 | 10.08 | |
| cisreg (28) | 6.5 | 12.01 | −1.0 | 12.57 | −5.0 | 12.12 | −5.0 | 17.51 | |
| snoRNA (6) | 2.0 | 17.71 | −2.5 | 21.92 | −19.0 | 12.04 | −16.0 | 15.95 | |
| all (279) | 0.0 | 8.63 | −2.0 | 10.00 | −2.0 | 11.11 | −4.0 | 11.19 | |
| Branched structures | |||||||||
| tRNA (185) | −5.0 | 11.53 | −7.0 | 11.37 | 1.0 | 15.90 | −2.0 | 14.47 | |
| cisreg (2) | −5.0 | 15.56 | −10.5 | 21.92 | −5.5 | 7.78 | −9.5 | 17.68 | |
| ribozyme (2) | −1.5 | 6.36 | −5.5 | 0.71 | −23.0 | 35.36 | −6.0 | 9.90 | |
| snRNA (1) | −33.0 | 0.00 | −2.0 | 0.00 | −33.0 | 0.00 | −4.0 | 0.00 | |
| all (190) | −5.0 | 11.48 | −7.0 | 11.34 | 1.0 | 16.06 | −2.0 | 14.41 | |
| 150 | Single hairpin structures | ||||||||
| miRNA (642) | 10.0 | 17.39 | 6.0 | 18.57 | −5.0 | 19.90 | −6.0 | 18.42 | |
| cisreg (36) | 13.0 | 24.99 | 19.5 | 21.81 | −0.5 | 21.95 | −3.5 | 23.33 | |
| snoRNA (24) | 8.0 | 12.44 | 6.0 | 19.65 | −27.5 | 26.06 | −29.5 | 24.07 | |
| all (709) | 10.0 | 17.64 | 7.0 | 18.91 | −6.0 | 20.62 | −7.0 | 19.21 | |
| Branched structures | |||||||||
| tRNA (166) | 18.0 | 21.07 | 16.0 | 22.31 | 2.0 | 30.55 | −1.0 | 28.52 | |
| cisreg (6) | 11.5 | 35.74 | 13.5 | 22.69 | −0.5 | 32.62 | −21.5 | 46.07 | |
| snoRNA (13) | 0.0 | 35.47 | −8.0 | 30.95 | −12.0 | 34.08 | −71.0 | 40.03 | |
| ribozyme (3) | 2.0 | 40.81 | 1.0 | 25.17 | −48.0 | 30.75 | −13.0 | 15.01 | |
| snRNA (3) | 7.0 | 8.08 | −22.0 | 40.93 | −81.0 | 32.91 | −4.0 | 6.82 | |
| all (190) | 14.5 | 24.63 | 14.0 | 25.12 | 1.0 | 32.05 | −2.0 | 31.34 | |
| 200 | Single hairpin structures | ||||||||
| miRNA (661) | 30.0 | 29.63 | 30.0 | 29.15 | −6.0 | 29.80 | −7.0 | 29.62 | |
| cisreg (37) | 35.0 | 32.03 | 35.0 | 35.66 | 5.0 | 38.48 | −4.0 | 41.38 | |
| snoRNA (20) | 24.5 | 19.53 | 8.5 | 18.64 | −52.0 | 48.98 | −4.5 | 31.54 | |
| all (727) | 30.0 | 29.63 | 29.0 | 29.45 | −6.0 | 32.30 | −7.0 | 30.54 | |
| Branched structures | |||||||||
| tRNA (127) | 38.0 | 31.42 | 39.0 | 33.16 | 2.0 | 47.27 | −2.0 | 43.57 | |
| cisreg (9) | 23.0 | 54.76 | 17.0 | 23.21 | −5.0 | 45.72 | −2.0 | 43.57 | |
| snoRNA (61) | 11.0 | 36.62 | −2.0 | 39.95 | −20.0 | 44.11 | −24.0 | 40.40 | |
| ribozyme (2) | 15.5 | 19.09 | 13.0 | 15.56 | −17.0 | 52.33 | −51.0 | 31.11 | |
| snRNA (6) | −3.0 | 39.36 | −6.5 | 35.15 | −94.5 | 14.96 | −8.5 | 23.94 | |
| all (206) | 23.0 | 37.03 | 18.5 | 39.14 | −14.5 | 50.82 | −12.5 | 46.18 | |
References
- Kino, T.; Hurt, D.; Ichijo, T.; Nader, N.; Chrousos, G. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal. 2010, 3, ra8. [Google Scholar] [CrossRef] [PubMed]
- Dotu, I.; Lorenz, W.; Van Hentenryck, P.; Clote, P. RNA structural segmentation. Pac. Symp. Biocomput. 2010, 57–68. [Google Scholar] [CrossRef]
- McCaskill, J.S. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolym. Orig. Res. Biomol. 1990, 29, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Sabarinathan, R.; Tafer, H.; Seemann, S.E.; Hofacker, I.L.; Stadler, P.F.; Gorodkin, J. RNAsnp: Efficient detection of local RNA secondary structure changes induced by SNPs. Hum. Mutat. 2013, 34, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Hecker, N.; Christensen-Dalsgaard, M.; Seemann, S.E.; Havgaard, J.H.; Stadler, P.F.; Hofacker, I.L.; Nielsen, H.; Gorodkin, J. Optimizing RNA structures by sequence extensions using RNAcop. Nucleic Acids Res. 2015, 43, 8135–8145. [Google Scholar] [CrossRef]
- Will, S.; Joshi, T.; Hofacker, I.; Stadler, P.; Backofen, R. LocARNA-P: Accurate boundary prediction and improved detection of structural RNAs. RNA 2012, 18, 900–914. [Google Scholar] [CrossRef]
- Miladi, M.; Junge, A.; Costa, F.; Seemann, S.E.; Havgaard, J.H.; Gorodkin, J.; Backofen, R. RNAscClust: clustering RNA sequences using structure conservation and graph based motifs. Bioinformatics 2017, 33, 2089–2096. [Google Scholar] [CrossRef]
- Seemann, S.E.; Gorodkin, J.; Backofen, R. Unifying evolutionary and thermodynamic information for RNA folding of multiple alignments. Nucleic Acids Res. 2008, 36, 6355–6362. [Google Scholar] [CrossRef]
- Puton, T.; Kozlowski, L.P.; Rother, K.M.; Bujnicki, J.M. CompaRNA: A server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res. 2013, 41, 4307–4323. [Google Scholar] [CrossRef]
- Seemann, S.; Mirza, A.; Hansen, C.; Bang-Berthelsen, C.; Garde, C.; Christensen-Dalsgaard, M.; Torarinsson, E.; Yao, Z.; Workman, C.; Pociot, F.; et al. The identification and functional annotation of RNA structures conserved in vertebrates. Genome Res. 2017, 27, 1371–1383. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Bejerano, G.; Siepel, A.; Rosenbloom, K.; Lindblad-Toh, K.; Lander, E.S.; Kent, J.; Miller, W.; Haussler, D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput. Biol. 2006, 2, e33. [Google Scholar] [CrossRef] [PubMed]
- Washietl, S.; Hofacker, I.L.; Stadler, P.F. Fast and reliable prediction of noncoding RNAs. Proc. Natl. Acad. Sci. USA 2005, 102, 2454–2459. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Gesell, T.; Stadler, P.; Mattick, J. Widespread purifying selection on RNA structure in mammals. Nucleic Acids Res. 2013, 41, 8220–8236. [Google Scholar] [CrossRef] [PubMed]
- Gorodkin, J.; Hofacker, I.L.; Torarinsson, E.; Yao, Z.; Havgaard, J.H.; Ruzzo, W.L. De novo prediction of structured RNAs from genomic sequences. Trends Biotechnol. 2010, 28, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.P.; Daub, J.; Tate, J.; Moore, B.L.; Osuch, I.H.; Griffiths-Jones, S.; Finn, R.D.; Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 2010, 39, D141–D145. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef]
- Jühling, F.; Mörl, M.; Hartmann, R.K.; Sprinzl, M.; Stadler, P.F.; Pütz, J. tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009, 37, D159–D162. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- Lestrade, L.; Weber, M.J. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res. 2006, 34, D158–D162. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, M.; Kent, W.J.; Riemer, C.; Elnitski, L.; Smit, A.F.A.; Roskin, K.M.; Baertsch, R.; Rosenbloom, K.; Clawson, H.; Green, E.D.; et al. Aligning Multiple Genomic Sequences With the Threaded Blockset Aligner. Genome Res. 2004, 14, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Steffen, P.; Voß, B.; Rehmsmeier, M.; Reeder, J.; Giegerich, R. RNAshapes: An integrated RNA analysis package based on abstract shapes. Bioinformatics 2005, 22, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Kiryu, H.; Kin, T.; Asai, K. Rfold: An exact algorithm for computing local base pairing probabilities. Bioinformatics 2007, 24, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Kiryu, H.; Terai, G.; Imamura, O.; Yoneyama, H.; Suzuki, K.; Asai, K. A detailed investigation of accessibilities around target sites of siRNAs and miRNAs. Bioinformatics 2011, 27, 1788–1797. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, S.H.; Hofacker, I.L.; Stadler, P.F. Local RNA base pairing probabilities in large sequences. Bioinformatics 2005, 22, 614–615. [Google Scholar] [CrossRef]
- Lange, S.; Maticzka, D.; Mohl, M.; Gagnon, J.; Brown, C.; Backofen, R. Global or local? Predicting secondary structure and accessibility in mRNAs. Nucleic Acids Res. 2012, 40, 5215–5226. [Google Scholar] [CrossRef]
- Nawrocki, E.; Eddy, S. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Smith, M.; Seemann, S.; Quek, X.; Mattick, J. DotAligner: Identification and clustering of RNA structure motifs. Genome Biol. 2017, 18, 244. [Google Scholar] [CrossRef]





| RNA Family | Window = 100 | 150 | 200 |
|---|---|---|---|
| miRNA | 244 | 642 | 661 |
| tRNA | 185 | 166 | 127 |
| cisreg | 30 | 42 | 46 |
| snoRNA | 6 | 37 | 81 |
| ribozyme | 2 | 3 | 2 |
| snRNA | 1 | 3 | 6 |
| others | 1 | 6 | 9 |
| lncRNA | 0 | 0 | 1 |
| Total | 469 | 899 | 933 |
| Window | Family (n) | RNAbound | Dotu et al. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Single seq. | Multiple seq. | Single seq. | Multiple seq. | ||||||
| Left | Right | Left | Right | Left | Right | Left | Right | ||
| 100 | Single hairpin structures | ||||||||
| miRNA (244) | 0.0 | −2.0 | 4.0 | 1.0 | −2.0 | −3.0 | 1.0 | 0.0 | |
| cisreg (28) | 6.5 | −1.0 | 5.0 | −2.0 | −5.0 | −5.0 | −8.5 | −15.5 | |
| snoRNA (6) | 2.0 | −2.5 | 1.5 | −6.0 | −19.0 | −16.0 | −18.0 | −17.5 | |
| all (279) | 0.0 | −2.0 | 4.0 | 0.0 | −2.0 | −4.0 | 1.0 | −1.0 | |
| Branched structures | |||||||||
| tRNA (185) | −5.0 | −7.0 | −5.0 | −9.0 | 1.0 | −2.0 | 0.0 | −2.0 | |
| all (190) | −5.0 | −7.0 | −5.0 | −9.0 | 1.0 | −2.0 | 0.0 | −2.0 | |
| 150 | Single hairpin structures | ||||||||
| miRNA (642) | 10.0 | 6.0 | 11.0 | 9.0 | −5.0 | −6.0 | −1.0 | −1.5 | |
| cisreg (36) | 13.0 | 19.5 | 9.0 | 7.0 | −0.5 | −3.5 | −5.0 | −8.5 | |
| snoRNA (24) | 8.0 | 6.0 | 5.5 | −1.5 | −27.5 | −29.5 | 7.0 | 3.0 | |
| all (709) | 10.0 | 7.0 | 9.0 | 8.0 | −6.0 | −7.0 | −1.0 | −2.0 | |
| Branched structures | |||||||||
| tRNA (166) | 18.0 | 16.0 | −6.0 | −9.0 | 2.0 | −1.0 | 0.0 | −2.0 | |
| cisreg (6) | 11.5 | 13.5 | −4.5 | 3.0 | −0.5 | −21.5 | −4.5 | 1.0 | |
| snoRNA (13) | 0.0 | −8.0 | 0.0 | −12.0 | −12.0 | −71.0 | −14.0 | −82.0 | |
| all (190) | 14.5 | 14.0 | −6.0 | −9.5 | 1.0 | −2.0 | 0.0 | −3.0 | |
| 200 | Single hairpin structures | ||||||||
| miRNA (661) | 30.0 | 30.0 | 30.0 | 33.0 | −6.0 | −7.0 | −2.0 | 1.0 | |
| cisreg (37) | 35.0 | 35.0 | 33.0 | 29.0 | 5.0 | −4.0 | −5.0 | −16.0 | |
| snoRNA (20) | 24.5 | 8.5 | 12.0 | 5.0 | −52.0 | −4.5 | 7.0 | 3.0 | |
| all (727) | 30.0 | 29.0 | 29.0 | 32.0 | −6.0 | −7.0 | −1.0 | 0.0 | |
| Branched structures | |||||||||
| tRNA (127) | 38.0 | 39.0 | −6.0 | −9.0 | 2.0 | −2.0 | 0.0 | −3.0 | |
| cisreg (9) | 23.0 | 17.0 | 23.0 | 26.0 | −5.0 | 3.0 | 16.0 | 26.0 | |
| snoRNA (61) | 11.0 | −2.0 | −7.0 | −69.0 | −20.0 | −24.0 | −18.0 | −86.0 | |
| all (206) | 23.0 | 18.5 | −6.0 | −11.5 | −14.5 | −12.5 | −14.0 | −9.0 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabarinathan, R.; Anthon, C.; Gorodkin, J.; Seemann, S.E. Multiple Sequence Alignments Enhance Boundary Definition of RNA Structures. Genes 2018, 9, 604. https://doi.org/10.3390/genes9120604
Sabarinathan R, Anthon C, Gorodkin J, Seemann SE. Multiple Sequence Alignments Enhance Boundary Definition of RNA Structures. Genes. 2018; 9(12):604. https://doi.org/10.3390/genes9120604
Chicago/Turabian StyleSabarinathan, Radhakrishnan, Christian Anthon, Jan Gorodkin, and Stefan E. Seemann. 2018. "Multiple Sequence Alignments Enhance Boundary Definition of RNA Structures" Genes 9, no. 12: 604. https://doi.org/10.3390/genes9120604
APA StyleSabarinathan, R., Anthon, C., Gorodkin, J., & Seemann, S. E. (2018). Multiple Sequence Alignments Enhance Boundary Definition of RNA Structures. Genes, 9(12), 604. https://doi.org/10.3390/genes9120604