Systems Analysis of Transcriptomic and Proteomic Profiles Identifies Novel Regulation of Fibrotic Programs by miRNAs in Pulmonary Fibrosis Fibroblasts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA-seq and miRNA-seq Analysis
2.3. Real-Time Reverse Transcription-Polymerase Chain Reaction
2.4. Gene Ontology and Signature Analysis
2.5. miRNA Mimic Transfection and Nanostring Gene Expression Analysis
2.6. Extracellular matrix Proteomic Profiling
3. Results
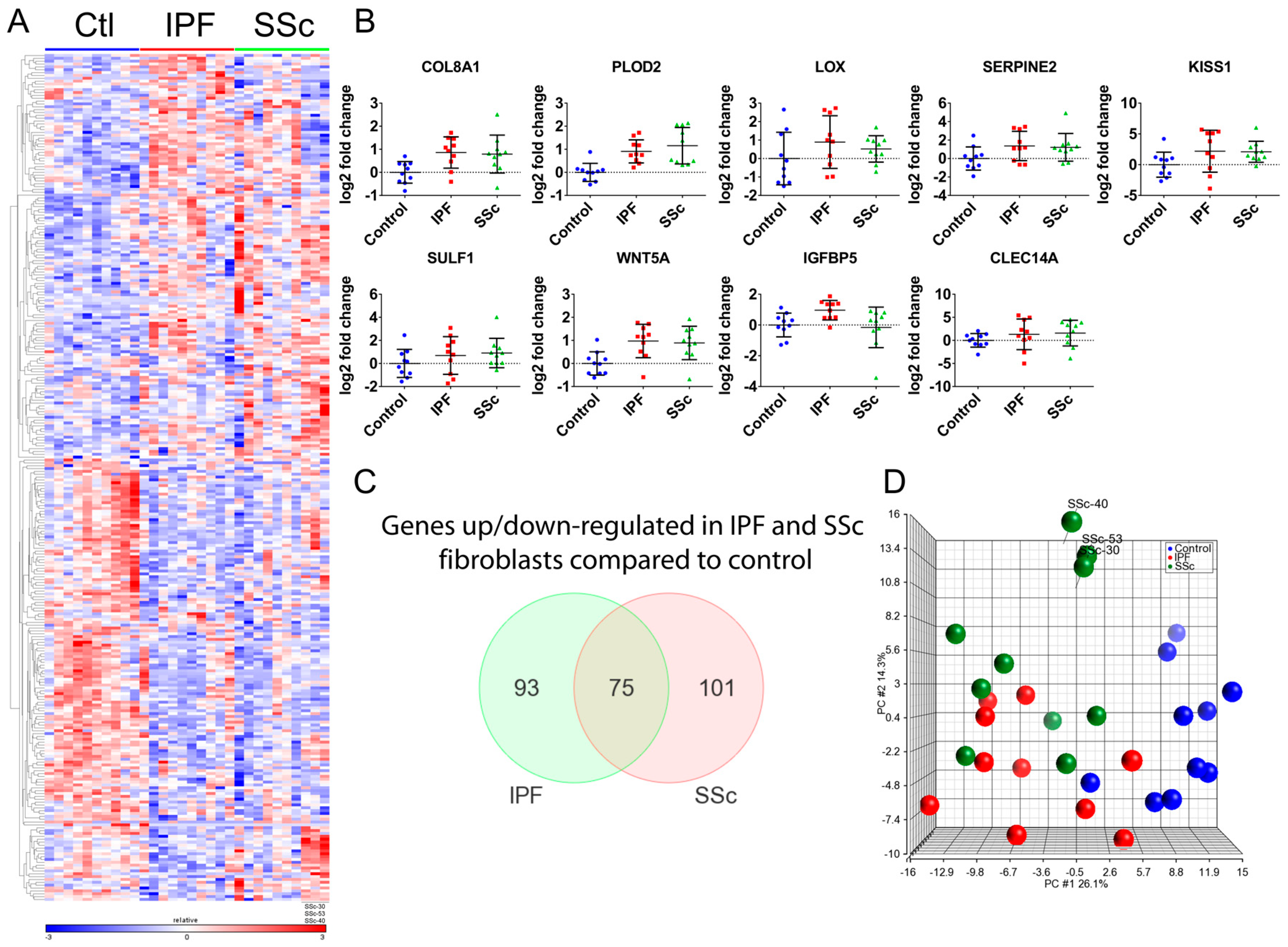
3.1. Transcriptional Profiling Identified Similar Dysregulated Gene Expression Programs in Idiopathic Pulmonary Fibrosis and Systemic Sclerosis Lung Fibroblasts
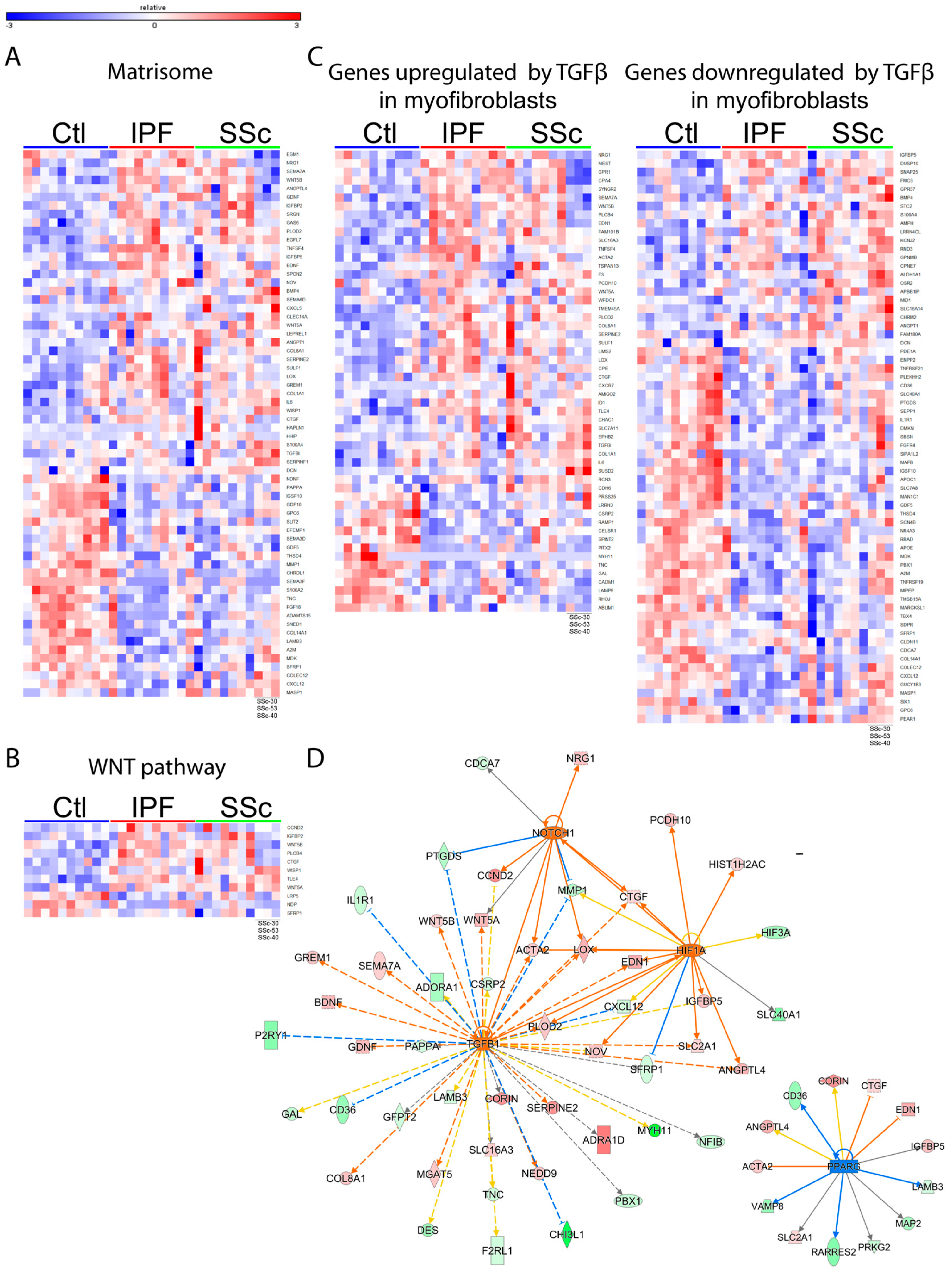
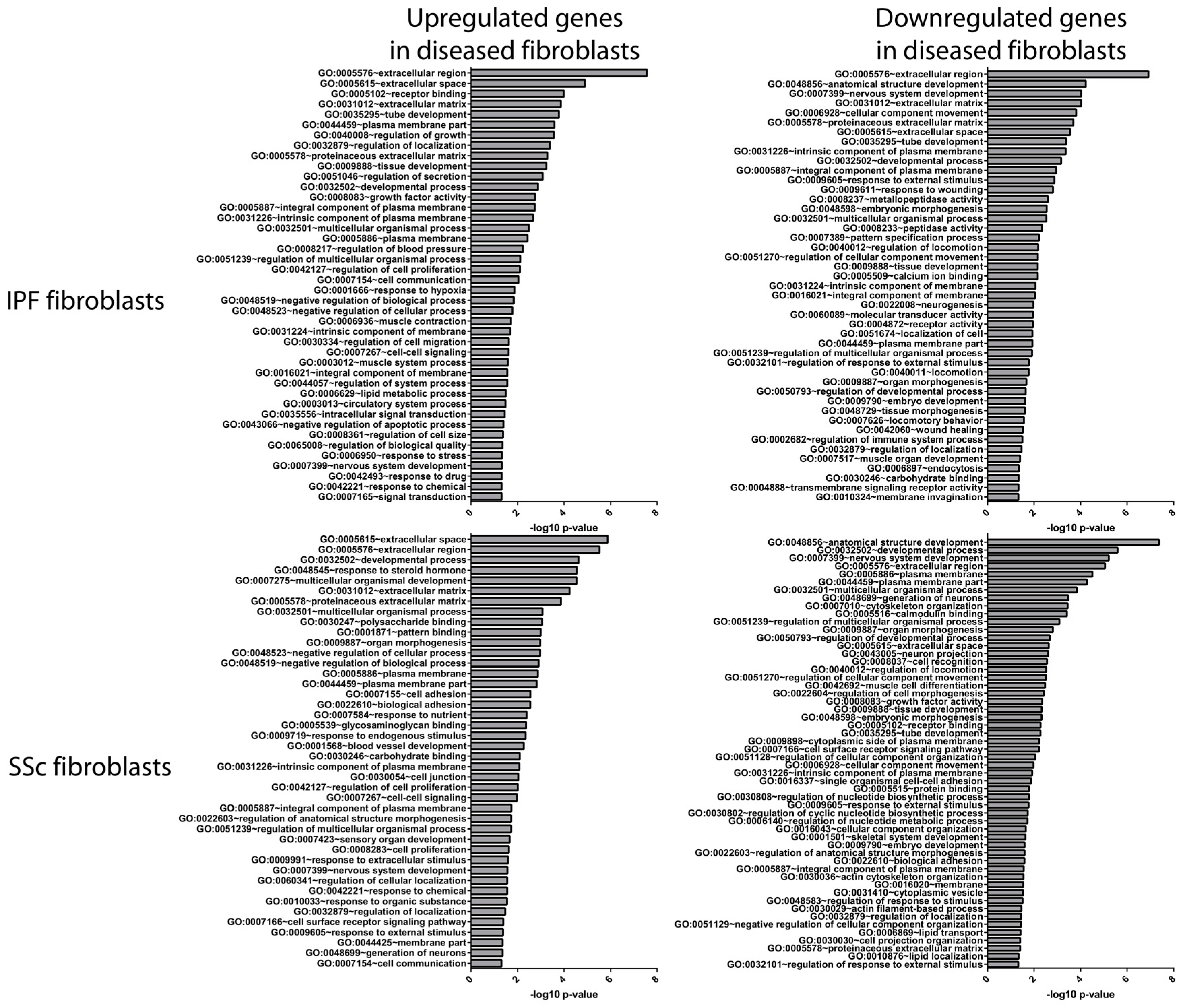
3.2. Idiopathic Pulmonary Fibrosis and Systemic Sclerosis Lung Fibroblast Disease Signatures are Associated with Pro-Fibrotic Pathways and Extracellular Matrix
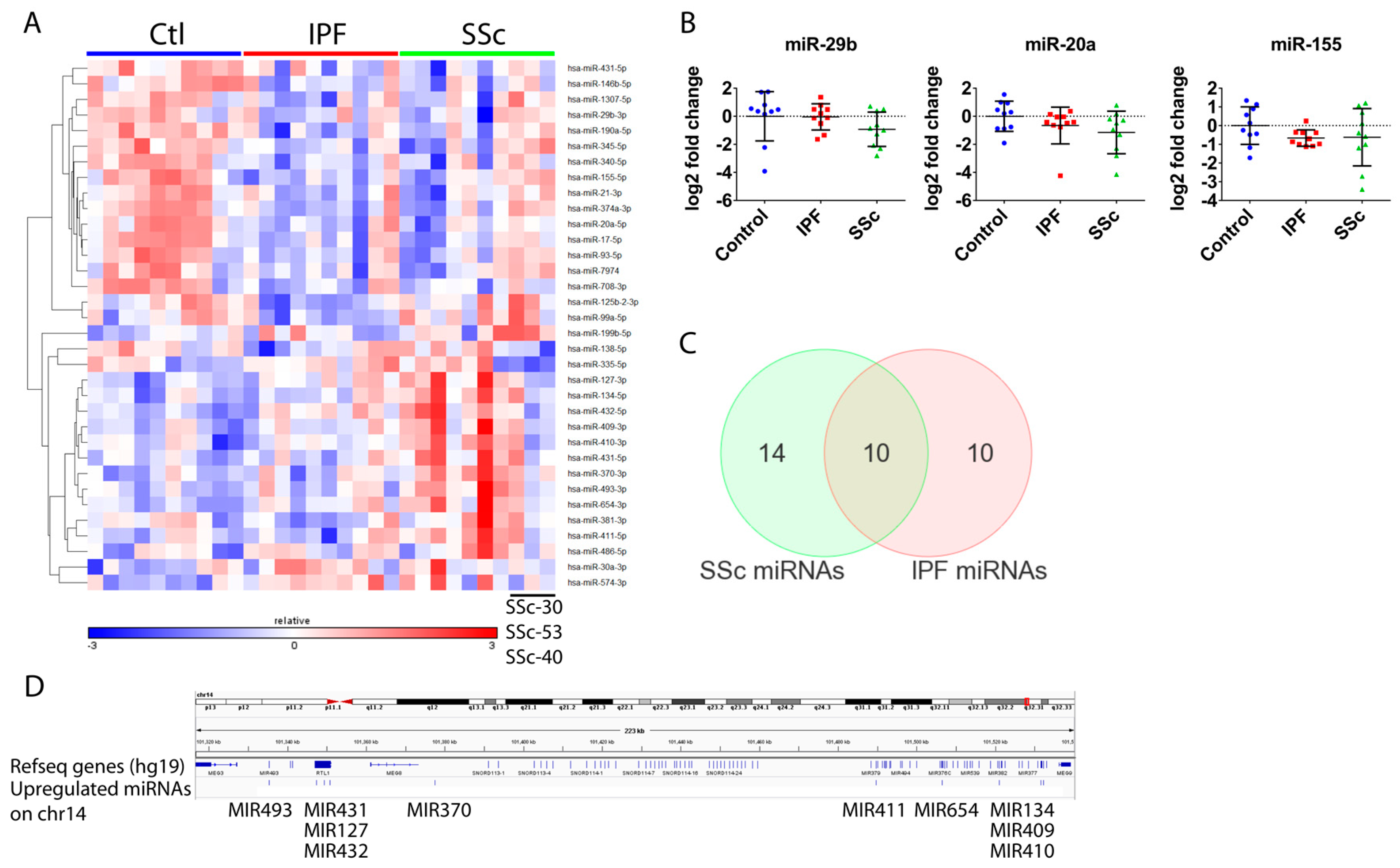
3.3. Global microRNA Profiling Identified Similar Fibrotic microRNA Signatures in Idiopathic Pulmonary Fibrosis and Systemic Sclerosis Lung Fibroblasts
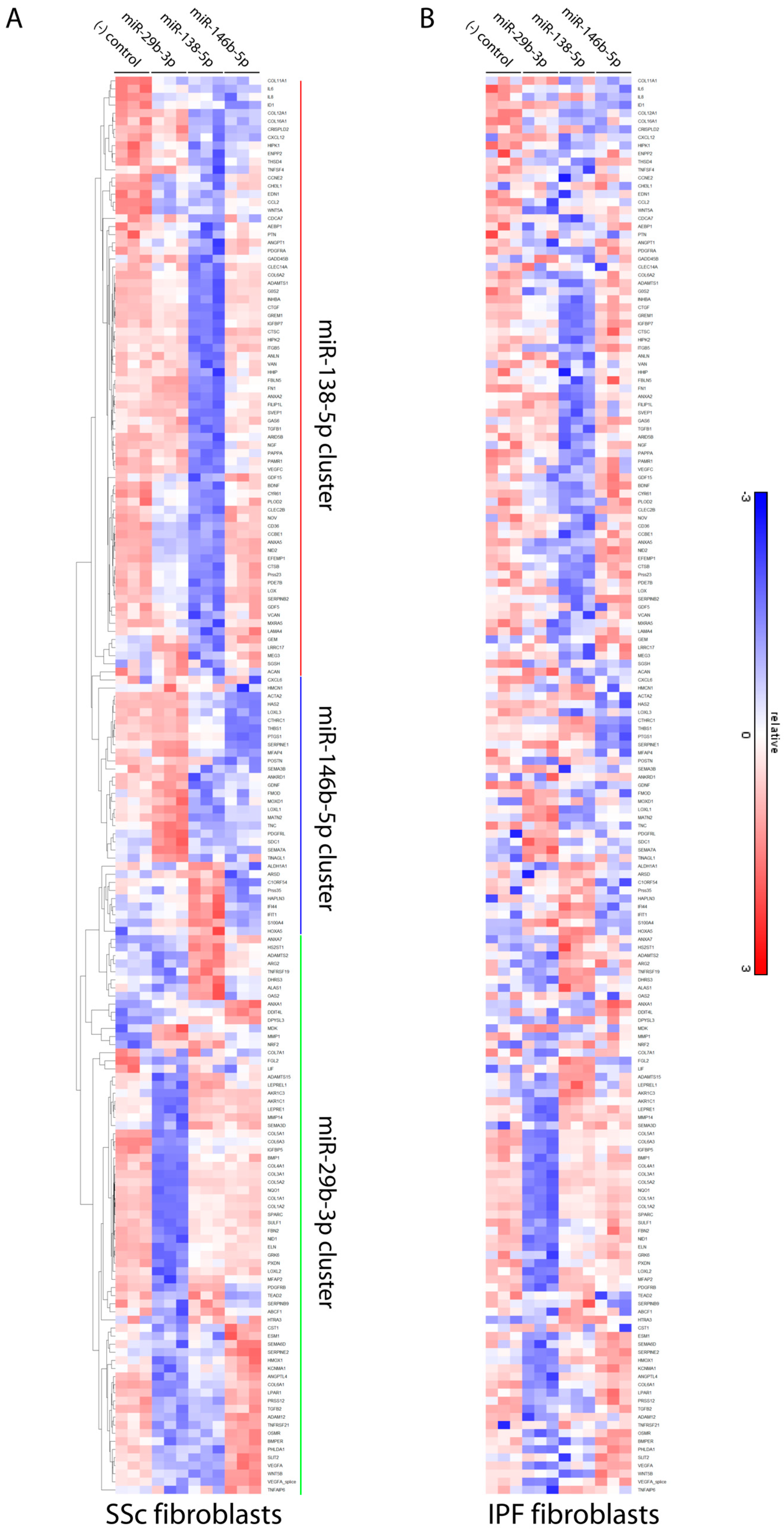
3.4. Differentially Expressed microRNAs in Idiopathic Pulmonary Fibrosis and Systemic Sclerosis Fibroblasts Regulate Expression of Fibrosis-Associated Genes
3.5. Proteomic Profiling Characterized a Distinct Fibrotic Matrisome Deposited by Idiopathic Pulmonary Fibrosis and Systemic Sclerosis Lung Fibroblasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Herzog, E.L.; Mathur, A.; Tager, A.M.; Feghali-Bostwick, C.; Schneider, F.; Varga, J. Review: interstitial lung disease associated with systemic sclerosis and idiopathic pulmonary fibrosis: how similar and distinct? Arthritis Rheumatol. 2014, 66, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Emblom-Callahan, M.C.; Chhina, M.K.; Shlobin, O.A.; Ahmad, S.; Reese, E.S.; Iyer, E.P.; Cox, D.N.; Brenner, R.; Burton, N.A.; Grant, G.M.; et al. Genomic phenotype of non-cultured pulmonary fibroblasts in idiopathic pulmonary fibrosis. Genomics 2010, 96, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.; Shi, H.; Jordan, R.M.; Lyons-Weiler, J.; Pilewski, J.M.; Feghali-Bostwick, C.A. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 2011, 63, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Kabuyama, Y.; Oshima, K.; Kitamura, T.; Homma, M.; Yamaki, J.; Munakata, M.; Homma, Y. Involvement of selenoprotein P in the regulation of redox balance and myofibroblast viability in idiopathic pulmonary fibrosis. Genes Cells 2007, 12, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, G.E.; Stock, C.J.; Shi-Wen, X.; Leoni, P.; Sestini, P.; Howat, S.L.; Bou-Gharios, G.; Nicholson, A.G.; Denton, C.P.; Grutters, J.C.; et al. Microarray profiling reveals suppressed interferon stimulated gene program in fibroblasts from scleroderma-associated interstitial lung disease. Respir. Res. 2013, 14, 80. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Sridhar, S.; Tyagi, G.; Phillips, J.E.; Garrido, R.; Harris, P.; Burns, L.; Renteria, L.; Woods, J.; Chen, L.; et al. Bleomycin induces molecular changes directly relevant to idiopathic pulmonary fibrosis: a model for “active” disease. PLoS ONE 2013, 8, e59348. [Google Scholar] [CrossRef] [PubMed]
- Kass, D.J.; Kaminski, N. Evolving genomic approaches to idiopathic pulmonary fibrosis: moving beyond genes. Clin. Transl. Sci. 2011, 4, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Christmann, R.B.; Sampaio-Barros, P.; Stifano, G.; Borges, C.L.; de Carvalho, C.R.; Kairalla, R.; Parra, E.R.; Spira, A.; Simms, R.; Capellozzi, V.L.; et al. Association of Interferon- and transforming growth factor beta-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol. 2014, 66, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Rossi, D.; Peterson, M.; Smith, K.; Sikström, K.; White, E.S.; Connett, J.E.; Henke, C.A.; Larsson, O.; Bitterman, P.B. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J. Clin. Invest. 2014, 124, 1622–1635. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Kupershmidt, I.; Su, Q.J.; Grewal, A.; Sundaresh, S.; Halperin, I.; Flynn, J.; Shekar, M.; Wang, H.; Park, J.; Cui, W.; et al. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Didangelos, A.; Yin, X.; Mandal, K.; Baumert, M.; Jahangiri, M.; Mayr, M. Proteomics characterization of extracellular space components in the human aorta. Mol. Cell Proteomics 2010, 9, 2048–2062. [Google Scholar] [CrossRef] [PubMed]
- Decaris, M.L.; Gatmaitan, M.; FlorCruz, S.; Luo, F.; Li, K.; Holmes, W.E.; Hellerstein, M.K.; Turner, S.M.; Emson, C.L. Proteomic analysis of altered extracellular matrix turnover in bleomycin-induced pulmonary fibrosis. Mol. Cell Proteomics 2014, 13, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteomics 2012, 11, M111 014647. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Huang, C.; Lin, X.; Li, J. MicroRNA-29 family, a crucial therapeutic target for fibrosis diseases. Biochimie 2013, 95, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Yu, G.; Latimer, P.A.; Stack, C.; Robinson, K.; Dalby, C.M.; Kaminski, N.; van Rooij, E. MicroRNA mimicry blocks pulmonary fibrosis. EMBO Mol. Med. 2014, 6, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Meng, X.M.; Huang, X.R.; Chung, A.C.; Feng, Y.L.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. miR-29 inhibits bleomycin-induced pulmonary fibrosis in mice. Mol. Ther. 2012, 20, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lu, J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Chung, A.C.; Huang, X.R.; Meng, X.M.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 2011, 22, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, X.R.; Wei, L.H.; Chung, A.C.; Yu, C.M.; Lan, H.Y. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol. Ther. 2014, 22, 974–985. [Google Scholar] [CrossRef] [PubMed]
- van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed]
- Roderburg, C.; Urban, G.W.; Bettermann, K.; Vucur, M.; Zimmermann, H.; Schmidt, S.; Janssen, J.; Koppe, C.; Knolle, P.; Castoldi, M.; et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 2011, 53, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, Y.; Ogawa, T.; Yoshizato, K.; Ikeda, K.; Kawada, N. Suppression of hepatic stellate cell activation by microRNA-29b. Biochem. Biophys. Res. Commun. 2011, 412, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, L.; Wang, Y.; Zhang, M.; Li, L.; Zhu, D.; Li, X.; Gu, H.; Zhang, C.Y.; Zen, K. Protective role of estrogen-induced miRNA-29 expression in carbon tetrachloride-induced mouse liver injury. J. Biol. Chem. 2012, 287, 14851–14862. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.; Stanczyk, J.; Jungel, A.; Akhmetshina, A.; Trenkmann, M.; Brock, M.; Kowal-Bielecka, O.; Gay, R.E.; Michel, B.A.; Distler, J.H.; et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010, 62, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Cushing, L.; Kuang, P.; Lu, J. The role of miR-29 in pulmonary fibrosis. Biochem. Cell Biol. 2015, 93, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Pandit, K.V.; Milosevic, J. MicroRNA regulatory networks in idiopathic pulmonary fibrosis. Biochem. Cell Biol. 2015, 93, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Pandit, K.V.; Corcoran, D.; Yousef, H.; Yarlagadda, M.; Tzouvelekis, A.; Gibson, K.F.; Konishi, K.; Yousem, S.A.; Singh, M.; Handley, D.; et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.Y.; Fan, P.J.; Lei, S.R.; Qi, M.; Yang, X.H. MiR-138/peroxisome proliferator-activated receptor beta signaling regulates human hypertrophic scar fibroblast proliferation and movement in vitro. J. Dermatol. 2015, 42, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.; Xia, X.; Wu, H.H.; Tu, C.Q.; Pan, X.M. PDGF-regulated miRNA-138 inhibits the osteogenic differentiation of mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2014, 448, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Eskildsen, T.; Taipaleenmaki, H.; Stenvang, J.; Abdallah, B.M.; Ditzel, N.; Nossent, A.Y.; Bak, M.; Kauppinen, S.; Kassem, M. MicroRNA-138 regulates osteogenic differentiation of human stromal (mesenchymal) stem cells in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 6139–6144. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, C.; Chen, Z.; Jin, Y.; Wang, Y.; Kolokythas, A.; Dai, Y.; Zhou, X. MicroRNA-138 suppresses epithelial-mesenchymal transition in squamous cell carcinoma cell lines. Biochem. J. 2011, 440, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Geraldo, M.V.; Yamashita, A.S.; Kimura, E.T. MicroRNA miR-146b-5p regulates signal transduction of TGF-beta by repressing SMAD4 in thyroid cancer. Oncogene 2012, 31, 1910–1922. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Huang, C.; Sun, X.; Long, X.R.; Lv, X.W.; Li, J. MicroRNA-146a modulates TGF-beta1-induced hepatic stellate cell proliferation by targeting SMAD4. Cell Signal 2012, 24, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lu, C.L.; Cui, L.P.; Hu, Y.L.; Yu, Q.; Jiang, Y.; Ma, T.; Jiao, D.K.; Wang, D.; Jia, C.Y. MicroRNA-146a modulates TGF-beta1-induced phenotypic differentiation in human dermal fibroblasts by targeting SMAD4. Arch. Dermatol. Res. 2012, 304, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Morishita, Y.; Imai, T.; Yoshizawa, H.; Watanabe, M.; Ishibashi, K.; Muto, S.; Nagata, D. Delivery of microRNA-146a with polyethylenimine nanoparticles inhibits renal fibrosis in vivo. Int. J. Nanomedicine 2015, 10, 3475–3488. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, J.; Pandit, K.; Magister, M.; Rabinovich, E.; Ellwanger, D.C.; Yu, G.; Vuga, L.J.; Weksler, B.; Benos, P.V.; Gibson, K.F.; et al. Profibrotic role of miR-154 in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 879–887. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mullenbrock, S.; Liu, F.; Szak, S.; Hronowski, X.; Gao, B.; Juhasz, P.; Sun, C.; Liu, M.; McLaughlin, H.; Xiao, Q.; et al. Systems Analysis of Transcriptomic and Proteomic Profiles Identifies Novel Regulation of Fibrotic Programs by miRNAs in Pulmonary Fibrosis Fibroblasts. Genes 2018, 9, 588. https://doi.org/10.3390/genes9120588
Mullenbrock S, Liu F, Szak S, Hronowski X, Gao B, Juhasz P, Sun C, Liu M, McLaughlin H, Xiao Q, et al. Systems Analysis of Transcriptomic and Proteomic Profiles Identifies Novel Regulation of Fibrotic Programs by miRNAs in Pulmonary Fibrosis Fibroblasts. Genes. 2018; 9(12):588. https://doi.org/10.3390/genes9120588
Chicago/Turabian StyleMullenbrock, Steven, Fei Liu, Suzanne Szak, Xiaoping Hronowski, Benbo Gao, Peter Juhasz, Chao Sun, Mei Liu, Helen McLaughlin, Qiurong Xiao, and et al. 2018. "Systems Analysis of Transcriptomic and Proteomic Profiles Identifies Novel Regulation of Fibrotic Programs by miRNAs in Pulmonary Fibrosis Fibroblasts" Genes 9, no. 12: 588. https://doi.org/10.3390/genes9120588
APA StyleMullenbrock, S., Liu, F., Szak, S., Hronowski, X., Gao, B., Juhasz, P., Sun, C., Liu, M., McLaughlin, H., Xiao, Q., Feghali-Bostwick, C., & Zheng, T. S. (2018). Systems Analysis of Transcriptomic and Proteomic Profiles Identifies Novel Regulation of Fibrotic Programs by miRNAs in Pulmonary Fibrosis Fibroblasts. Genes, 9(12), 588. https://doi.org/10.3390/genes9120588