The Emerging Role of Cohesin in the DNA Damage Response


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
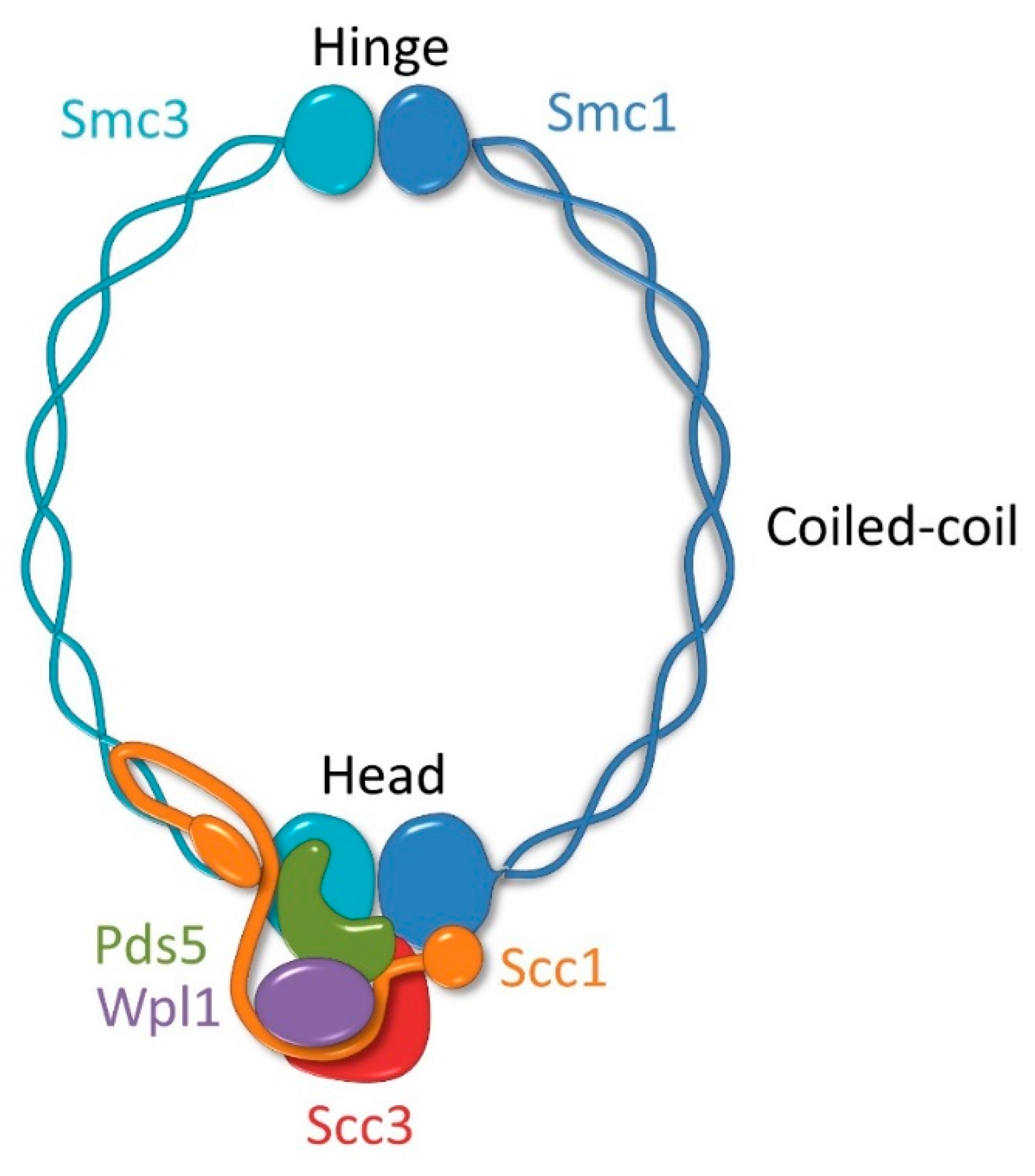
2. Molecular Architecture of the Cohesin Complex
3. Sister Chromatid Cohesion Process
3.1. Cohesin Loading
3.2. Cohesion Establishment and Maintenance
3.3. Cohesion Dissolution
4. DNA Damage Response Mechanisms
4.1. Signaling and Repair of DNA Double-Strand Breaks
4.1.1. DNA Damage Checkpoint Activation in Response to DNA Double-Strand Breaks
4.1.2. Repair of DNA Double-Strand Breaks by Homologous Recombination in Mitotic Cells
4.1.3. Nonhomologous End Joining
4.2. DNA Damage Tolerance Mechanisms
5. Mechanisms of Cohesin Recruitment to DNA Damage Sites
5.1. Targeting Yeast Cohesin to DNA Double-Strand Breaks
5.2. Targeting Human Cohesin to DNA Double-Strand Breaks
5.3. Cohesin Recruitment and Dynamics at Stalled Replication Forks
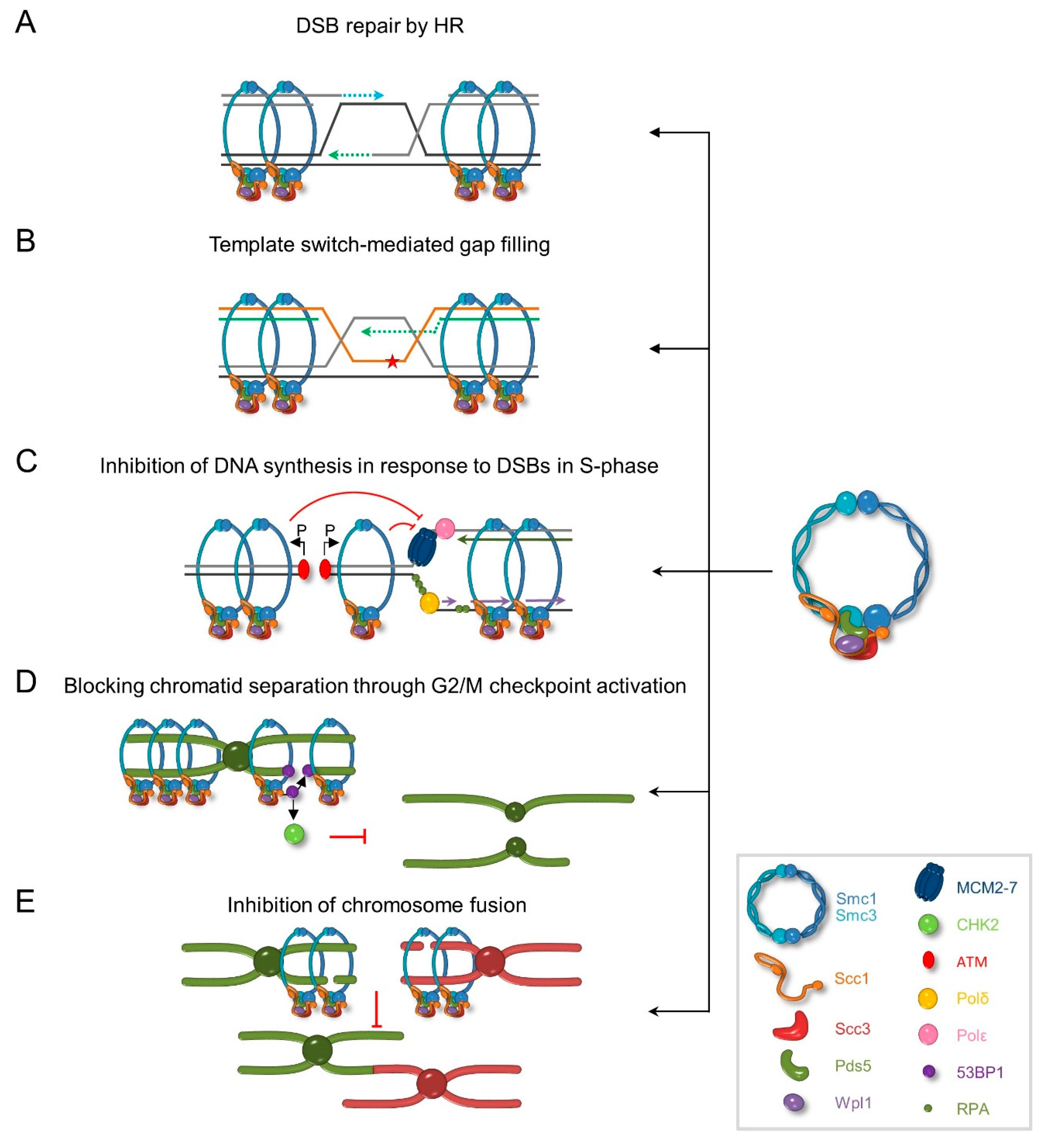
6. Role of Cohesin in DNA Damage Response
7. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Gómez-González, B.; Aguilera, A. DNA repair in mammalian cells: DNA double-strand break repair: How to fix a broken relationship. Cell. Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Szakal, B. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair 2016, 44, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.-B.S.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Guacci, V.; Koshland, D.; Strunnikov, A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 1997, 91, 47–57. [Google Scholar] [CrossRef]
- Michaelis, C.; Ciosk, R.; Nasmyth, K. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell 1997, 91, 35–45. [Google Scholar] [CrossRef]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Zuin, J.; Dixon, J.R.; van der Reijden, M.I.; Ye, Z.; Kolovos, P.; Brouwer, R.W.; van de Corput, M.P.; van de Werken, H.J.; Knoch, T.A.; van Ijcken, W.F.; et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Kojic, A.; Cuadrado, A.; De Koninck, M.; Giménez-Llorente, D.; Rodríguez-Corsino, M.; Gómez-López, G.; Le Dily, F.; Marti-Renom, M.A.; Losada, A. Distinct roles of cohesin-SA1 and cohesin-SA2 in 3D chromosome organization. Nat. Struct. Mol. Biol. 2018, 25, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Birkenbihl, R.P.; Subramani, S. Cloning and characterization of rad21 an essential gene of Schizosaccharomyces pombe involved in DNA double- strand-break repair. Nucleic Acids Res. 1992, 20, 6605–6611. [Google Scholar] [CrossRef] [PubMed]
- Marston, A.L. Chromosome segregation in budding yeast: Sister chromatid cohesion and related mechanisms. Genetics 2014, 196, 31–63. [Google Scholar] [CrossRef] [PubMed]
- Murayama, Y.; Uhlmann, F. DNA entry into and exit out of the cohesin ring by an interlocking gate mechanism. Cell 2015, 163, 1628–1640. [Google Scholar] [CrossRef] [PubMed]
- Gligoris, T.; Löwe, J. Structural insights into ring formation of cohesin and related Smc complexes. Trends Cell Biol. 2016, 26, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Haering, C.H.; Farcas, A.-M.; Arumugam, P.; Metson, J.; Nasmyth, K. The cohesin ring concatenates sister DNA molecules. Nature 2008, 454, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Gligoris, T.G.; Scheinost, J.C.; Burmann, F.; Petela, N.; Chan, K.-L.; Uluocak, P.; Beckouet, F.; Gruber, S.; Nasmyth, K.; Lowe, J. Closing the cohesin ring: Structure and function of its Smc3-kleisin interface. Science 2014, 346, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Kulemzina, I.; Schumacher, M.R.; Verma, V.; Reiter, J.; Metzler, J.; Failla, A.V.; Lanz, C.; Sreedharan, V.T.; Rätsch, G.; Ivanov, D. Cohesin rings devoid of Scc3 and Pds5 maintain their stable association with the DNA. PLoS Genet. 2012, 8, e1002856. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Zakian, S.; Hu, X.-W.; Singleton, M.R. Structural insights into the regulation of cohesion establishment by Wpl1. EMBO J. 2013, 32, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Haering, C.H.; Löwe, J.; Hochwagen, A.; Nasmyth, K. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol. Cell 2002, 9, 773–788. [Google Scholar] [CrossRef]
- Muir, K.W.; Kschonsak, M.; Li, Y.; Metz, J.; Haering, C.H.; Panne, D. Structure of the Pds5-Scc1 complex and implications for cohesin function. Cell Rep. 2016, 14, 2116–2126. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, F.; Lottspeich, F.; Nasmyth, K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 1999, 400, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Ciosk, R.; Shirayama, M.; Shevchenko, A.; Tanaka, T.; Toth, A.; Shevchenko, A.; Nasmyth, K. Cohesin’s Binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol. Cell 2000, 5, 243–254. [Google Scholar] [CrossRef]
- Darwiche, N.; Freeman, L.A.; Strunnikov, A. Characterization of the components of the putative mammalian sister chromatid cohesion complex. Gene 1999, 233, 39–47. [Google Scholar] [CrossRef]
- Sumara, I.; Vorlaufer, E.; Gieffers, C.; Peters, B.H.; Peters, J.-M. Characterization of vertebrate cohesin complexes and their regulation in prophase. J. Cell Biol. 2000, 151, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Krantz, I.D.; McCallum, J.; DeScipio, C.; Kaur, M.; Gillis, L.A.; Yaeger, D.; Jukofsky, L.; Wasserman, N.; Bottani, A.; Morris, C.A.; et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 2004, 36, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Watrin, E.; Schleiffer, A.; Tanaka, K.; Eisenhaber, F.; Nasmyth, K.; Peters, J.-M. Human Scc4 is required for cohesin binding to chromatin, sister-chromatid cohesion, and mitotic progression. Curr. Biol. 2006, 16, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Seitan, V.C.; Banks, P.; Laval, S.; Majid, N.A.; Dorsett, D.; Rana, A.; Smith, J.; Bateman, A.; Krpic, S.; Hostert, A.; et al. Metazoan Scc4 homologs link sister chromatid cohesion to cell and axon migration guidance. PLoS Biol. 2006, 4, e242. [Google Scholar] [CrossRef] [PubMed]
- Murayama, Y.; Uhlmann, F. Biochemical reconstitution of topological DNA binding by the cohesin ring. Nature 2014, 505, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.C.H.; Murayama, Y.; Muñoz, S.; Jones, A.W.; Wade, B.O.; Purkiss, A.G.; Hu, X.-W.; Borg, A.; Snijders, A.P.; Uhlmann, F.; et al. Structure of the cohesin loader Scc2. Nat. Commun. 2017, 8, 13952. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, S.M.; Makrantoni, V.; Kerr, A.; Marston, A.L.; Harrison, S.C. Structural evidence for Scc4-dependent localization of cohesin loading. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.C.H.; Murayama, Y.; Muñoz, S.; Costa, A.; Uhlmann, F.; Singleton, M.R. Structural studies reveal the functional modularity of the Scc2-Scc4 cohesin loader. Cell Rep. 2015, 12, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Braunholz, D.; Hullings, M.; Gil-Rodríguez, M.C.; Fincher, C.T.; Mallozzi, M.B.; Loy, E.; Albrecht, M.; Kaur, M.; Limon, J.; Rampuria, A.; et al. Isolated NIBPL missense mutations that cause Cornelia de Lange syndrome alter MAU2 interaction. Eur. J. Hum. Genet. 2012, 20, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Borek, D.M.; Otwinowski, Z.; Tomchick, D.R.; Yu, H. Crystal structure of the cohesin loader Scc2 and insight into cohesinopathy. Proc. Natl. Acad. Sci. USA 2016, 113, 12444–12449. [Google Scholar] [CrossRef] [PubMed]
- Petela, N.J.; Gligoris, T.G.; Metson, J.; Lee, B.-G.; Voulgaris, M.; Hu, B.S.; Chapard, C.; Chen, W.; Rajendra, E.; Srinivisan, M.; et al. Scc2 is a potent activator of cohesin’s ATPase that promotes loading by binding Scc1 without Pds5. Mol. Cell 2018, 70, 1134–1148. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, P.; Gruber, S.; Tanaka, K.; Haering, C.H.; Mechtler, K.; Nasmyth, K. ATP hydrolysis is required for cohesin’s association with chromosomes. Curr. Biol. 2003, 13, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Gruber, S.; Arumugam, P.; Katou, Y.; Kuglitsch, D.; Helmhart, W.; Shirahige, K.; Nasmyth, K. Evidence that loading of cohesin onto chromosomes involves opening of its SMC hinge. Cell 2006, 127, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Murayama, Y.; Samora, C.P.; Kurokawa, Y.; Iwasaki, H.; Uhlmann, F. Establishment of DNA-DNA interactions by the cohesin ring. Cell 2018, 172, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Zuin, J.; Franke, V.; van IJcken, W.F.J.; van der Sloot, A.; Krantz, I.D.; van der Reijden, M.I.J.A.; Nakato, R.; Lenhard, B.; Wendt, K.S. A Cohesin-independent role for NIPBL at promoters provides insights in CdLS. PLoS Genet. 2014, 10, e1004153. [Google Scholar] [CrossRef] [PubMed]
- Lengronne, A.; Katou, Y.; Mori, S.; Yokobayashi, S.; Kelly, G.P.; Itoh, T.; Watanabe, Y.; Shirahige, K.; Uhlmann, F. Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature 2004, 430, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Serra, L.; Kelly, G.; Patel, H.; Stewart, A.; Uhlmann, F. The Scc2–Scc4 complex acts in sister chromatid cohesion and transcriptional regulation by maintaining nucleosome-free regions. Nat. Genet. 2014, 46, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Kanchwala, M.; Xing, C.; Yu, H. MCM2–7-dependent cohesin loading during S phase promotes sister-chromatid cohesion. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, F.; Nasmyth, K. Cohesion between sister chromatids must be established during DNA replication. Curr. Biol. 1998, 8, 1095–1102. [Google Scholar] [CrossRef]
- Tittel-Elmer, M.; Lengronne, A.; Davidson, M.B.; Bacal, J.; François, P.; Hohl, M.; Petrini, J.H.J.; Pasero, P.; Cobb, J.A. Cohesin association to replication sites depends on Rad50 and promotes fork restart. Mol. Cell 2012, 48, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, S.M.; Makrantoni, V.; Harrison, S.C.; Marston, A.L. The kinetochore receptor for the cohesin loading complex. Cell 2017, 171, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Fernius, J.; Nerusheva, O.O.; Galander, S.; de Alves, F.L.; Rappsilber, J.; Marston, A.L. Cohesin-dependent association of Scc2/4 with the centromere initiates pericentromeric cohesion establishment. Curr. Biol. 2013, 23, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Woodman, J.; Fara, T.; Dzieciatkowska, M.; Trejo, M.; Luong, N.; Hansen, K.C.; Megee, P.C. Cell cycle-specific cleavage of Scc2 regulates its cohesin deposition activity. Proc. Natl. Acad. Sci. USA 2014, 111, 7060–7065. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.; Mazza, D.; Nasmyth, K.; Uphoff, S. Scc2/Nipbl hops between chromosomal cohesin rings after loading. eLife 2017, 6, e30000. [Google Scholar] [CrossRef] [PubMed]
- Litwin, I.; Bakowski, T.; Maciaszczyk-Dziubinska, E.; Wysocki, R. The LSH/HELLS homolog Irc5 contributes to cohesin association with chromatin in yeast. Nucleic Acids Res. 2017, 45, 6404–6416. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, M.-A.; Bochar, D.A.; Schmiesing, J.A.; Dong, Y.; Barak, O.G.; Speicher, D.W.; Yokomori, K.; Shiekhattar, R. A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature 2002, 418, 994–998. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.; Seah, C.; Moulin, J.; Isaac, C.; Dick, F.; Bérubé, N.G. Loss of ATRX leads to chromosome cohesion and congression defects. J. Cell Biol. 2008, 180, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.; Demattei, M.-V.; Episkopou, H.; Augé-Gouillou, C.; Decottignies, A.; Grandin, N.; Charbonneau, M. Genetic inactivation of ATRX leads to a decrease in the amount of telomeric cohesin and level of telomere transcription in human glioma cells. Mol. Cell. Biol. 2015, 35, 2818–2830. [Google Scholar] [CrossRef] [PubMed]
- Ocampo-Hafalla, M.; Muñoz, S.; Samora, C.P.; Uhlmann, F. Evidence for cohesin sliding along budding yeast chromosomes. Open Biol. 2016, 6, 150178. [Google Scholar] [CrossRef] [PubMed]
- Davidson, I.F.; Goetz, D.; Zaczek, M.P.; Molodtsov, M.I.; Huis in ’t Veld, P.J.; Weissmann, F.; Litos, G.; Cisneros, D.A.; Ocampo-Hafalla, M.; Ladurner, R.; et al. Rapid movement and transcriptional re-localization of human cohesin on DNA. EMBO J. 2016, 35, 2671–2685. [Google Scholar] [CrossRef] [PubMed]
- Vian, L.; Pękowska, A.; Rao, S.S.P.; Kieffer-Kwon, K.R.; Jung, S.; Baranello, L.; Huang, S.C.; El Khattabi, L.; Dose, M.; Pruett, N.; et al. The energetics and physiological impact of cohesin extrusion. Cell 2018, 173, 1165–1178. [Google Scholar] [CrossRef] [PubMed]
- Gerlich, D.; Koch, B.; Dupeux, F.; Peters, J.-M.; Ellenberg, J. Live-cell imaging reveals a stable cohesin-chromatin interaction after but not before DNA replication. Curr. Biol. 2006, 16, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Bernard, P.; Schmidt, C.K.; Vaur, S.; Dheur, S.; Drogat, J.; Genier, S.; Ekwall, K.; Uhlmann, F.; Javerzat, J.-P. Cell-cycle regulation of cohesin stability along fission yeast chromosomes. EMBO J. 2008, 27, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Gause, M.; Misulovin, Z.; Bilyeu, A.; Dorsett, D. Dosage-sensitive regulation of cohesin chromosome binding and dynamics by Nipped-B, Pds5, and Wapl. Mol. Cell. Biol. 2010, 30, 4940–4951. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-L.; Roig, M.B.; Hu, B.; Beckouët, F.; Metson, J.; Nasmyth, K. Cohesin’s DNA exit gate is distinct from its entrance gate and is regulated by acetylation. Cell 2012, 150, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA controls establishment of sister chromatid cohesion during S phase. Mol. Cell 2006, 23, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Lengronne, A.; McIntyre, J.; Katou, Y.; Kanoh, Y.; Hopfner, K.-P.; Shirahige, K.; Uhlmann, F. Establishment of sister chromatid cohesion at the S. cerevisiae replication fork. Mol. Cell 2006, 23, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shi, X.; Li, Y.; Kim, B.-J.; Jia, J.; Huang, Z.; Yang, T.; Fu, X.; Jung, S.Y.; Wang, Y.; et al. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol. Cell 2008, 31, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Alomer, R.M.; da Silva, E.M.L.; Chen, J.; Piekarz, K.M.; McDonald, K.; Sansam, C.G.; Sansam, C.L.; Rankin, S. Esco1 and Esco2 regulate distinct cohesin functions during cell cycle progression. Proc. Natl. Acad. Sci. USA 2017, 114, 9906–9911. [Google Scholar] [CrossRef] [PubMed]
- Çamdere, G.; Guacci, V.; Stricklin, J.; Koshland, D. The ATPases of cohesin interface with regulators to modulate cohesin-mediated DNA tethering. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Elbatsh, A.M.O.; Haarhuis, J.H.I.; Petela, N.; Chapard, C.; Fish, A.; Celie, P.H.; Stadnik, M.; Ristic, D.; Wyman, C.; Medema, R.H.; et al. Cohesin releases DNA through asymmetric ATPase-driven ring opening. Mol. Cell 2016, 61, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Boone, C.; Brown, G.W. Genetic dissection of parallel sister-chromatid cohesion pathways. Genetics 2007, 176, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Samora, C.P.; Saksouk, J.; Goswami, P.; Wade, B.O.; Singleton, M.R.; Bates, P.A.; Lengronne, A.; Costa, A.; Uhlmann, F. Ctf4 Links DNA Replication with sister chromatid cohesion establishment by recruiting the Chl1 helicase to the replisome. Mol. Cell 2016, 63, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Borges, V.; Smith, D.J.; Whitehouse, I.; Uhlmann, F. An Eco1-independent sister chromatid cohesion establishment pathway in S. cerevisiae. Chromosoma 2013, 122, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Fumasoni, M.; Zwicky, K.; Vanoli, F.; Lopes, M.; Branzei, D. Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polα/Primase/Ctf4 complex. Mol. Cell 2015, 57, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-L.; Gligoris, T.; Upcher, W.; Kato, Y.; Shirahige, K.; Nasmyth, K.; Beckouet, F. Pds5 promotes and protects cohesin acetylation. Proc. Natl. Acad. Sci. USA 2013, 110, 13020–13025. [Google Scholar] [CrossRef] [PubMed]
- Roig, M.B.; Löwe, J.; Chan, K.-L.; Beckouët, F.; Metson, J.; Nasmyth, K. Structure and function of cohesin’s Scc3/SA regulatory subunit. FEBS Lett. 2014, 588, 3692–3702. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, L.M.; Lavoie, B.D. Pds5 prevents the polySUMO-dependent separation of sister chromatids. Curr. Biol. 2014, 24, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, T.; Ladurner, R.; Schmitz, J.; Kreidl, E.; Schleiffer, A.; Bhaskara, V.; Bando, M.; Shirahige, K.; Hyman, A.A.; Mechtler, K.; et al. Sororin mediates sister chromatid cohesion by antagonizing Wapl. Cell 2010, 143, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Borges, V.; Lehane, C.; Lopez-Serra, L.; Flynn, H.; Skehel, M.; Ben-Shahar, T.R.; Uhlmann, F. Hos1 deacetylates Smc3 to close the cohesin acetylation cycle. Mol. Cell 2010, 39, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yue, Z.; Tanaka, T.U. Smc3 Deacetylation by Hos1 facilitates efficient dissolution of sister chromatid cohesion during early anaphase. Mol. Cell 2017, 68, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Waizenegger, I.C.; Hauf, S.; Meinke, A.; Peters, J.-M. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 2000, 103, 399–410. [Google Scholar] [CrossRef]
- Gandhi, R.; Gillespie, P.J.; Hirano, T. Human Wapl is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Curr. Biol. 2006, 16, 2406–2417. [Google Scholar] [CrossRef] [PubMed]
- Pâques, F.; Haber, J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999, 63, 349–404. [Google Scholar] [PubMed]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Löbrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef] [PubMed]
- Clikeman, J.A.; Khalsa, G.J.; Barton, S.L.; Nickoloff, J.A. Homologous recombinational repair of double-strand breaks in yeast is enhanced by MAT heterozygosity through yKU-dependent and -independent mechanisms. Genetics 2001, 157, 579–589. [Google Scholar] [PubMed]
- Hartwell, L.; Weinert, T. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Lisby, M.; Barlow, J.H.; Burgess, R.C.; Rothstein, R. Choreography of the DNA damage response: Spatiotemporal relationships among checkpoint and repair proteins. Cell 2004, 118, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Seeber, A.; Hegnauer, A.M.; Hustedt, N.; Deshpande, I.; Poli, J.; Eglinger, J.; Pasero, P.; Gut, H.; Shinohara, M.; Hopfner, K.-P.; et al. RPA mediates recruitment of MRX to forks and double-strand breaks to hold sister chromatids together. Mol. Cell 2016, 64, 951–966. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Chahwan, C.; Bailis, J.; Hunter, T.; Russell, P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 2005, 25, 5363–5379. [Google Scholar] [CrossRef] [PubMed]
- Nakada, D. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 2003, 17, 1957–1962. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Arbel-Eden, A.; Pilch, D.; Ira, G.; Bonner, W.M.; Petrini, J.H.; Haber, J.E.; Lichten, M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 2004, 14, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Vialard, J.E. The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J. 1998, 17, 5679–5688. [Google Scholar] [CrossRef] [PubMed]
- Hammet, A.; Magill, C.; Heierhorst, J.; Jackson, S.P. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 2007, 8, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.F.; Duong, J.K.; Sun, Z.; Morrow, J.S.; Pradhan, D.; Stern, D.F. Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol. Cell 2002, 9, 1055–1065. [Google Scholar] [CrossRef]
- Blankley, R.T. A domain of Rad9 specifically required for activation of Chk1 in budding yeast. J. Cell Sci. 2004, 117, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Hsiao, J.; Fay, D.S.; Stern, D.F. Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 1998, 281, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.S.; Green, C.M.; Lowndes, N.F. Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol. Cell 2001, 8, 129–136. [Google Scholar] [CrossRef]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Gobbini, E.; Cassani, C.; Villa, M.; Bonetti, D.; Longhese, M. Functions and regulation of the MRX complex at DNA double-strand breaks. Microb. Cell 2016, 3, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Wold, M.S. Replication protein A: A Heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Mantiero, D.; Clerici, M.; Lucchini, G.; Longhese, M.P. Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep. 2007, 8, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, B.; Zou, L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Paciotti, V.; Clerici, M.; Lucchini, G.; Longhese, M.P. The checkpoint protein Ddc2, functionally related to S. pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev. 2000, 14, 2046–2059. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Rouse, J.; Jackson, S.P. Lcd1p recruits Mec1p to DNA lesions in vitro and in vivo. Mol. Cell 2002, 9, 857–869. [Google Scholar] [CrossRef]
- Deshpande, I.; Seeber, A.; Shimada, K.; Keusch, J.J.; Gut, H.; Gasser, S.M. Structural basis of Mec1-Ddc2-RPA assembly and activation on single-stranded DNA at sites of damage. Mol. Cell 2017, 68, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.B.; Albuquerque, C.P.; Chen, S.-H.; Zhou, H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 10364–10369. [Google Scholar] [CrossRef] [PubMed]
- Sung, P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J. Biol. Chem. 1997, 272, 28194–28197. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Zaitseva, E.M.; Kowalczykowski, S.C. A single-stranded DNA-binding protein is needed for efficient presynaptic complex formation by the Saccharomyces cerevisiae Rad51 protein. J. Biol. Chem. 1997, 272, 7940–7945. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yang, H.; Pavletich, N.P. Mechanism of homologous recombination from the RecA–ssDNA/dsDNA structures. Nature 2008, 453, 489. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Terakawa, T.; Qi, Z.; Steinfeld, J.B.; Redding, S.; Kwon, Y.; Gaines, W.A.; Zhao, W.; Sung, P.; Greene, E.C. Base triplet stepping by the Rad51/RecA family of recombinases. Science 2015, 349, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Kadyk, L.C.; Hartwell, L.H. Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 1992, 132, 387–402. [Google Scholar] [PubMed]
- Wright, W.D.; Heyer, W.-D. Rad54 functions as a heteroduplex DNA pump modulated by its DNA substrates and Rad51 during D loop formation. Mol. Cell 2014, 53, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Fasching, C.L.; Cejka, P.; Kowalczykowski, S.C.; Heyer, W.-D. Top3-Rmi1 dissolve Rad51-mediated D loops by a topoisomerase-based mechanism. Mol. Cell 2015, 57, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ede, C.; Wright, W.D.; Gore, S.K.; Jenkins, S.S.; Freudenthal, B.D.; Washington, M.T.; Veaute, X.; Heyer, W.-D. Srs2 promotes synthesis-dependent strand annealing by disrupting DNA polymerase δ-extending D-loops. eLife 2017, 6, pii–e22195. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Satory, D.; Dray, E.; Papusha, A.; Scheller, J.; Kramer, W.; Krejci, L.; Klein, H.; Haber, J.E.; Sung, P.; et al. Yeast Mph1 helicase dissociates Rad51-made D-loops: Implications for crossover control in mitotic recombination. Genes Dev. 2009, 23, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Sica, R.A.; Kowalczykowski, S.C. Rad52 promotes second-end DNA capture in double-stranded break repair to form complement-stabilized joint molecules. Proc. Natl. Acad. Sci. USA 2009, 106, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Bizard, A.H.; Hickson, I.D. The dissolution of double holliday junctions. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Matos, J.; West, S.C. Holliday junction resolution: Regulation in space and time. DNA Repair 2014, 19, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Dynan, W.S.; Yoo, S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998, 26, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 2010, 29, 3358–3369. [Google Scholar] [CrossRef] [PubMed]
- DeFazio, L.G. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J. 2002, 21, 3192–3200. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.M.; Jackson, P. The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell 1993, 72, 131–142. [Google Scholar] [CrossRef]
- Yoo, S. Geometry of a complex formed by double strand break repair proteins at a single DNA end: Recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res. 1999, 27, 4679–4686. [Google Scholar] [CrossRef] [PubMed]
- Malu, S.; De Ioannes, P.; Kozlov, M.; Greene, M.; Francis, D.; Hanna, M.; Pena, J.; Escalante, C.R.; Kurosawa, A.; Erdjument-Bromage, H.; et al. Artemis C-terminal region facilitates V(D)J recombination through its interactions with DNA Ligase IV and DNA-PKcs. J. Exp. Med. 2012, 209, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Clements, P.M.; Breslin, C.; Deeks, E.D.; Byrd, P.J.; Ju, L.; Bieganowski, P.; Brenner, C.; Moreira, M.-C.; Taylor, A.M.R.; Caldecott, K.W. The ataxia–oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA Repair 2004, 3, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.A.; Agyei, R.; Galicia, S.; Metalnikov, P.; O’Donnell, P.; Starostine, A.; Weinfeld, M.; Durocher, D. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J. 2004, 23, 3874–3885. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, K.N.; Nick McElhinny, S.A.; Mitchell, B.S.; Ramsden, D.A. Association of DNA polymerase μ (pol μ) with Ku and ligase IV: Role for pol μ in end-joining double-strand break repair. Mol. Cell. Biol. 2002, 22, 5194–5202. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, K.; Shevelev, I.V.; Maga, G.; Hübscher, U. De novo DNA synthesis by human DNA polymerase λ, DNA polymerase μ and terminal deoxyribonucleotidyl transferase. J. Mol. Biol. 2004, 339, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Herreros, F.; Romero-Granados, R.; Zeng, Z.; Álvarez-Quilón, A.; Quintero, C.; Ju, L.; Umans, L.; Vermeire, L.; Huylebroeck, D.; Caldecott, K.W.; et al. TDP2–dependent non-homologous end-joining protects against topoisomerase II–induced DNA breaks and genome instability in cells and in vivo. PLoS Genet. 2013, 9, e1003226. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Strande, N.; Burkhalter, M.D.; Strom, C.; Havener, J.M.; Hasty, P.; Ramsden, D.A. Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends. Nature 2010, 464, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [PubMed]
- Sishc, B.J.; Davis, A.J. The role of the core non-homologous end joining factors in carcinogenesis and cancer. Cancers 2017, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.A.; Huttner, D.; Daigaku, Y.; Chen, S.; Ulrich, H.D. Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein A. Mol. Cell 2008, 29, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Stelter, P.; Ulrich, H.D. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 2003, 425, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Tateishi, S.; Kawasuji, M.; Tsurimoto, T.; Inoue, H.; Yamaizumi, M. Rad18 guides polη to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–3896. [Google Scholar] [CrossRef] [PubMed]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase η with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Terai, K.; Abbas, T.; Jazaeri, A.A.; Dutta, A. CRL4Cdt2 E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol. Cell 2010, 37, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chea, J.; Meng, X.; Zhou, Y.; Lee, E.Y.C.; Lee, M.Y.W.T. PCNA is ubiquitinated by RNF8. Cell Cycle 2008, 7, 3399–3404. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.D. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 2000, 19, 3388–3397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lawrence, C.W. The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc. Natl. Acad. Sci. USA 2005, 102, 15954–15959. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, M.; Zwicky, K.; Follonier, C.; Foiani, M.; Lopes, M.; Branzei, D. Visualization of recombination-mediated damage bypass by template switching. Nat. Struct. Mol. Biol. 2014, 21, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Unk, I.; Hajdu, I.; Fatyol, K.; Szakal, B.; Blastyak, A.; Bermudez, V.; Hurwitz, J.; Prakash, L.; Prakash, S.; Haracska, L. Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. USA 2006, 103, 18107–18112. [Google Scholar] [CrossRef] [PubMed]
- Unk, I.; Hajdu, I.; Fatyol, K.; Hurwitz, J.; Yoon, J.-H.; Prakash, L.; Prakash, S.; Haracska, L. Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc. Natl. Acad. Sci. USA 2008, 105, 3768–3773. [Google Scholar] [CrossRef] [PubMed]
- Karras, G.I.; Jentsch, S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 2010, 141, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Daigaku, Y.; Davies, A.A.; Ulrich, H.D. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 2010, 465, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Gangavarapu, V.; Prakash, S.; Prakash, L. Requirement of RAD52 group genes for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007, 27, 7758–7764. [Google Scholar] [CrossRef] [PubMed]
- Vanoli, F.; Fumasoni, M.; Szakal, B.; Maloisel, L.; Branzei, D. Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet. 2010, 6, e1001205. [Google Scholar] [CrossRef] [PubMed]
- Liberi, G. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 2005, 19, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Gritenaite, D.; Princz, L.N.; Szakal, B.; Bantele, S.C.S.; Wendeler, L.; Schilbach, S.; Habermann, B.H.; Matos, J.; Lisby, M.; Branzei, D.; et al. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014, 28, 1604–1619. [Google Scholar] [CrossRef] [PubMed]
- Szakal, B.; Branzei, D. Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 2013, 32, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Vanoli, F.; Foiani, M. SUMOylation regulates Rad18-mediated template switch. Nature 2008, 456, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Huici, V.; Szakal, B.; Urulangodi, M.; Psakhye, I.; Castellucci, F.; Menolfi, D.; Rajakumara, E.; Fumasoni, M.; Bermejo, R.; Jentsch, S.; et al. DNA bending facilitates the error-free DNA damage tolerance pathway and upholds genome integrity. EMBO J. 2014, 33, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Bazán, M.Á.; Gallo-Fernández, M.; Saugar, I.; Jiménez-Martín, A.; Vázquez, M.V.; Tercero, J.A. Rad5 plays a major role in the cellular response to DNA damage during chromosome replication. Cell Rep. 2014, 9, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Piening, B.D.; Paulovich, A.G. The preference for error-free or error-prone postreplication repair in Saccharomyces cerevisiae exposed to low-dose methyl methanesulfonate is cell cycle dependent. Mol. Cell. Biol. 2013, 33, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.M.; Lambert, S. Replication stress-induced genome instability: The dark side of replication maintenance by homologous recombination. J. Mol. Biol. 2013, 425, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Carr, A.M. Replication stress and genome rearrangements: Lessons from yeast models. Curr. Opin. Genet. Dev. 2013, 23, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Ünal, E.; Arbel-Eden, A.; Sattler, U.; Shroff, R.; Lichten, M.; Haber, J.E.; Koshland, D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol. Cell 2004, 16, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Ström, L.; Lindroos, H.B.; Shirahige, K.; Sjögren, C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell 2004, 16, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Ström, L.; Karlsson, C.; Lindroos, H.B.; Wedahl, S.; Katou, Y.; Shirahige, K.; Sjögren, C. Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science 2007, 317, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Unal, E.; Heidinger-Pauli, J.M.; Koshland, D. DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7). Science 2007, 317, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Conde, F.; Refolio, E.; Cordon-Preciado, V.; Cortes-Ledesma, F.; Aragon, L.; Aguilera, A.; San-Segundo, P.A. The Dot1 histone methyltransferase and the Rad9 checkpoint adaptor contribute to cohesin-dependent double-strand break repair by sister chromatid recombination in Saccharomyces cerevisiae. Genetics 2009, 182, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Qiu, J.; Ratnakumar, K.; Laurent, B.C. RSC functions as an early double-strand-break sensor in the cell’s response to DNA damage. Curr. Biol. 2007, 17, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Oum, J.-H.; Seong, C.; Kwon, Y.; Ji, J.-H.; Sid, A.; Ramakrishnan, S.; Ira, G.; Malkova, A.; Sung, P.; Lee, S.E.; et al. RSC facilitates Rad59-dependent homologous recombination between sister chromatids by promoting cohesin loading at DNA double-strand breaks. Mol. Cell. Biol. 2011, 31, 3924–3937. [Google Scholar] [CrossRef] [PubMed]
- Heidinger-Pauli, J.M.; Ünal, E.; Guacci, V.; Koshland, D. The kleisin subunit of cohesin dictates damage-induced cohesion. Mol. Cell 2008, 31, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Heidinger-Pauli, J.M.; Ünal, E.; Koshland, D. Distinct targets of the Eco1 acetyltransferase modulate cohesion in S phase and in response to DNA damage. Mol. Cell 2009, 34, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Almedawar, S.; Colomina, N.; Bermúdez-López, M.; Pociño-Merino, I.; Torres-Rosell, J. A SUMO-dependent step during establishment of sister chromatid cohesion. Curr. Biol. 2012, 22, 1576–1581. [Google Scholar] [CrossRef] [PubMed]
- McAleenan, A.; Cordon-Preciado, V.; Clemente-Blanco, A.; Liu, I.-C.; Sen, N.; Leonard, J.; Jarmuz, A.; Aragón, L. SUMOylation of the α-kleisin subunit of cohesin is required for DNA damage-induced cohesion. Curr. Biol. 2012, 22, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Enervald, E.; Lindgren, E.; Katou, Y.; Shirahige, K.; Ström, L. Importance of Polη for damage-induced cohesion reveals differential regulation of cohesion establishment at the break site and genome-wide. PLoS Genet. 2013, 9, e1003158. [Google Scholar] [CrossRef] [PubMed]
- McAleenan, A.; Clemente-Blanco, A.; Cordon-Preciado, V.; Sen, N.; Esteras, M.; Jarmuz, A.; Aragón, L. Post-replicative repair involves separase-dependent removal of the kleisin subunit of cohesin. Nature 2012, 493, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Caron, P.; Aymard, F.; Iacovoni, J.S.; Briois, S.; Canitrot, Y.; Bugler, B.; Massip, L.; Losada, A.; Legube, G. Cohesin protects genes against γH2AX induced by DNA double-strand breaks. PLoS Genet. 2012, 8, e1002460. [Google Scholar] [CrossRef] [PubMed]
- Bot, C.; Pfeiffer, A.; Giordano, F.; Edara, D.M.; Dantuma, N.P.; Ström, L. Independent mechanisms recruit the cohesin loader protein NIPBL to sites of DNA damage. J. Cell Sci. 2017, 130, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-J.; Li, Y.; Zhang, J.; Xi, Y.; Li, Y.; Yang, T.; Jung, S.Y.; Pan, X.; Chen, R.; Li, W.; et al. Genome-wide reinforcement of cohesin binding at pre-existing cohesin sites in response to ionizing radiation in human cells. J. Biol. Chem. 2010, 285, 22784–22792. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Ball, A.R.; Pham, H.X.; Zeng, W.; Chen, H.-Y.; Schmiesing, J.A.; Kim, J.-S.; Berns, M.; Yokomori, K. Distinct functions of human cohesin-SA1 and cohesin-SA2 in double-strand break repair. Mol. Cell. Biol. 2014, 34, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Oka, Y.; Suzuki, K.; Yamauchi, M.; Mitsutake, N.; Yamashita, S. Recruitment of the cohesin loading factor NIPBL to DNA double-strand breaks depends on MDC1, RNF168 and HP1γ in human cells. Biochem. Biophys. Res. Commun. 2011, 411, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Krasieva, T.B.; LaMorte, V.; Taylor, A.M.R.; Yokomori, K. Specific recruitment of human cohesin to laser-induced DNA damage. J. Biol. Chem. 2002, 277, 45149–45153. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, Y.; Mu, J.-J.; Zhang, J.; Tonaka, T.; Hamamori, Y.; Jung, S.Y.; Wang, Y.; Qin, J. Regulation of intra-S phase checkpoint by ionizing radiation (IR)-dependent and IR-independent phosphorylation of SMC3. J. Biol. Chem. 2008, 283, 19176–19183. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Kong, X.; Ji, Z.; Zeng, W.; Potts, P.R.; Yokomori, K.; Yu, H. Scc1 sumoylation by Mms21 promotes sister chromatid recombination through counteracting Wapl. Genes Dev. 2012, 26, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Litwin, I.; Bakowski, T.; Szakal, B.; Pilarczyk, E.; Maciaszczyk-Dziubinska, E.; Branzei, D.; Wysocki, R. Error-free DNA damage tolerance pathway is facilitated by the Irc5 translocase through cohesin. EMBO J. 2018, 37, e98732. [Google Scholar] [CrossRef] [PubMed]
- Frattini, C.; Villa-Hernández, S.; Pellicanò, G.; Jossen, R.; Katou, Y.; Shirahige, K.; Bermejo, R. Cohesin ubiquitylation and mobilization facilitate stalled replication fork dynamics. Mol. Cell 2017, 68, 758–772. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, R. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004, 18, 1423–1438. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-T.; Xu, B.; Kastan, M.B. Involvement of the cohesin protein, Smc1, in ATM-dependent and independent responses to DNA damage. Genes Dev. 2002, 16, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Boeing, S.; Williamson, L.; Encheva, V.; Gori, I.; Saunders, R.E.; Instrell, R.; Aygün, O.; Rodriguez-Martinez, M.; Weems, J.C.; Kelly, G.P.; et al. Multiomic analysis of the UV-induced DNA damage response. Cell Rep. 2016, 15, 1597–1610. [Google Scholar] [CrossRef] [PubMed]
- Bauerschmidt, C.; Arrichiello, C.; Burdak-Rothkamm, S.; Woodcock, M.; Hill, M.A.; Stevens, D.L.; Rothkamm, K. Cohesin promotes the repair of ionizing radiation-induced DNA double-strand breaks in replicated chromatin. Nucleic Acids Res. 2010, 38, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Brough, R.; Bajrami, I.; Vatcheva, R.; Natrajan, R.; Reis-Filho, J.S.; Lord, C.J.; Ashworth, A. APRIN is a cell cycle specific BRCA2-interacting protein required for genome integrity and a predictor of outcome after chemotherapy in breast cancer: APRIN interacts with BRCA2 and is required for genomic integrity. EMBO J. 2012, 31, 1160–1176. [Google Scholar] [CrossRef] [PubMed]
- Heidinger-Pauli, J.M.; Mert, O.; Davenport, C.; Guacci, V.; Koshland, D. Systematic reduction of cohesin differentially affects chromosome segregation, condensation, and DNA repair. Curr. Biol. 2010, 20, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, C.; Nasmyth, K. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr. Biol. 2001, 11, 991–995. [Google Scholar] [CrossRef]
- Countryman, P.; Fan, Y.; Gorthi, A.; Pan, H.; Strickland, J.; Kaur, P.; Wang, X.; Lin, J.; Lei, X.; White, C.; et al. Cohesin SA2 is a sequence-independent DNA-binding protein that recognizes DNA replication and repair intermediates. J. Biol. Chem. 2018, 293, 1054–1069. [Google Scholar] [CrossRef] [PubMed]
- Couturier, A.M.; Fleury, H.; Patenaude, A.-M.; Bentley, V.L.; Rodrigue, A.; Coulombe, Y.; Niraj, J.; Pauty, J.; Berman, J.N.; Dellaire, G.; et al. Roles for APRIN (PDS5B) in homologous recombination and in ovarian cancer prediction. Nucleic Acids Res. 2016, 44, 10879–10897. [Google Scholar] [CrossRef] [PubMed]
- Covo, S.; Chiou, E.; Gordenin, D.A.; Resnick, M.A. Suppression of allelic recombination and aneuploidy by cohesin is independent of Chk1 in Saccharomyces cerevisiae. PLoS ONE 2014, 9, e113435. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Bachant, J.; Wang, H.; Hu, F.; Tetzlaff, M.; Liu, D.; Elledge, S.J. Control of the DNA damage checkpoint by chkl and rad53 protein kinases through distinct mechanisms. Science 1999, 286, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Fasullo, M.; Dong, Z.; Sun, M.; Zeng, L. Saccharomyces cerevisiae RAD53 (CHK2) but not CHK1 is required for double-strand break-initiated SCE and DNA damage-associated SCE after exposure to X rays and chemical agents. DNA Repair 2005, 4, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Ledesma, F.; Aguilera, A. Double-strand breaks arising by replication through a nick are repaired by cohesin-dependent sister-chromatid exchange. EMBO Rep. 2006, 7, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Covo, S.; Westmoreland, J.W.; Gordenin, D.A.; Resnick, M.A. Cohesin is limiting for the suppression of DNA damage–induced recombination between homologous chromosomes. PLoS Genet. 2010, 6, e1001006. [Google Scholar] [CrossRef] [PubMed]
- Dion, V.; Gasser, S.M. Chromatin movement in the maintenance of genome stability. Cell 2013, 152, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Agmon, N.; Liefshitz, B.; Zimmer, C.; Fabre, E.; Kupiec, M. Effect of nuclear architecture on the efficiency of double-strand break repair. Nat. Cell Biol. 2013, 15, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Dion, V.; Kalck, V.; Seeber, A.; Schleker, T.; Gasser, S.M. Cohesin and the nucleolus constrain the mobility of spontaneous repair foci. EMBO Rep. 2013, 14, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Gelot, C.; Guirouilh-Barbat, J.; Le Guen, T.; Dardillac, E.; Chailleux, C.; Canitrot, Y.; Lopez, B.S. The cohesin complex prevents the end joining of distant DNA double-strand ends. Mol. Cell 2016, 61, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, P.T.; Wang, Y.; Zhao, S.; Patel, N.; Lee, E.Y.; Qin, J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002, 16, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Watrin, E.; Peters, J.-M. The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. EMBO J. 2009, 28, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Losada, A. Cohesin in cancer: Chromosome segregation and beyond. Nat. Rev. Cancer 2014, 14, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Remeseiro, S.; Cuadrado, A.; Losada, A. Cohesin in development and disease. Development 2013, 140, 3715–3718. [Google Scholar] [CrossRef] [PubMed]
- Guacci, V.; Koshland, D. Cohesin-independent segregation of sister chromatids in budding yeast. Mol. Biol. Cell 2012, 23, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Merkenschlager, M.; Nora, E.P. CTCF and cohesin in genome folding and transcriptional gene regulation. Annu. Rev. Genomics Hum. Genet. 2016, 17, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Mannini, L.; Menga, S.; Tonelli, A.; Zanotti, S.; Bassi, M.T.; Magnani, C.; Musio, A. SMC1A codon 496 mutations affect the cellular response to genotoxic treatments. Am. J. Med. Genet. A 2012, 158A, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Vrouwe, M.G.; Elghalbzouri-Maghrani, E.; Meijers, M.; Schouten, P.; Godthelp, B.C.; Bhuiyan, Z.A.; Redeker, E.J.; Mannens, M.M.; Mullenders, L.H.F.; Pastink, A.; et al. Increased DNA damage sensitivity of Cornelia de Lange syndrome cells: Evidence for impaired recombinational repair. Hum. Mol. Genet. 2007, 16, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Revenkova, E.; Focarelli, M.L.; Susani, L.; Paulis, M.; Bassi, M.T.; Mannini, L.; Frattini, A.; Delia, D.; Krantz, I.; Vezzoni, P.; et al. Cornelia de Lange syndrome mutations in SMC1A or SMC3 affect binding to DNA. Hum. Mol. Genet. 2009, 18, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Enervald, E.; Du, L.; Visnes, T.; Björkman, A.; Lindgren, E.; Wincent, J.; Borck, G.; Colleaux, L.; Cormier-Daire, V.; van Gent, D.C.; et al. A regulatory role for the cohesin loader NIPBL in nonhomologous end joining during immunoglobulin class switch recombination. J. Exp. Med. 2013, 210, 2503–2513. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litwin, I.; Pilarczyk, E.; Wysocki, R. The Emerging Role of Cohesin in the DNA Damage Response. Genes 2018, 9, 581. https://doi.org/10.3390/genes9120581
Litwin I, Pilarczyk E, Wysocki R. The Emerging Role of Cohesin in the DNA Damage Response. Genes. 2018; 9(12):581. https://doi.org/10.3390/genes9120581
Chicago/Turabian StyleLitwin, Ireneusz, Ewa Pilarczyk, and Robert Wysocki. 2018. "The Emerging Role of Cohesin in the DNA Damage Response" Genes 9, no. 12: 581. https://doi.org/10.3390/genes9120581
APA StyleLitwin, I., Pilarczyk, E., & Wysocki, R. (2018). The Emerging Role of Cohesin in the DNA Damage Response. Genes, 9(12), 581. https://doi.org/10.3390/genes9120581