Selective Pressures on Human Cancer Genes along the Evolution of Mammals


Abstract
1. Introduction
2. Materials and Methods
2.1. Cancer Genes
2.2. Sequence Data Collection
2.3. Multiple Sequence Alignment
2.4. Estimation of Phylogenetic Trees
2.5. Codon-Based Selection Models
2.6. Gene Ontology Enrichment Analysis
2.7. Pathogenic Germline Mutations
2.8. Comparison of dN/dS Ratios across COSMIC Categories
3. Results
3.1. Long-Term Selective Pressures on Human Cancer Genes
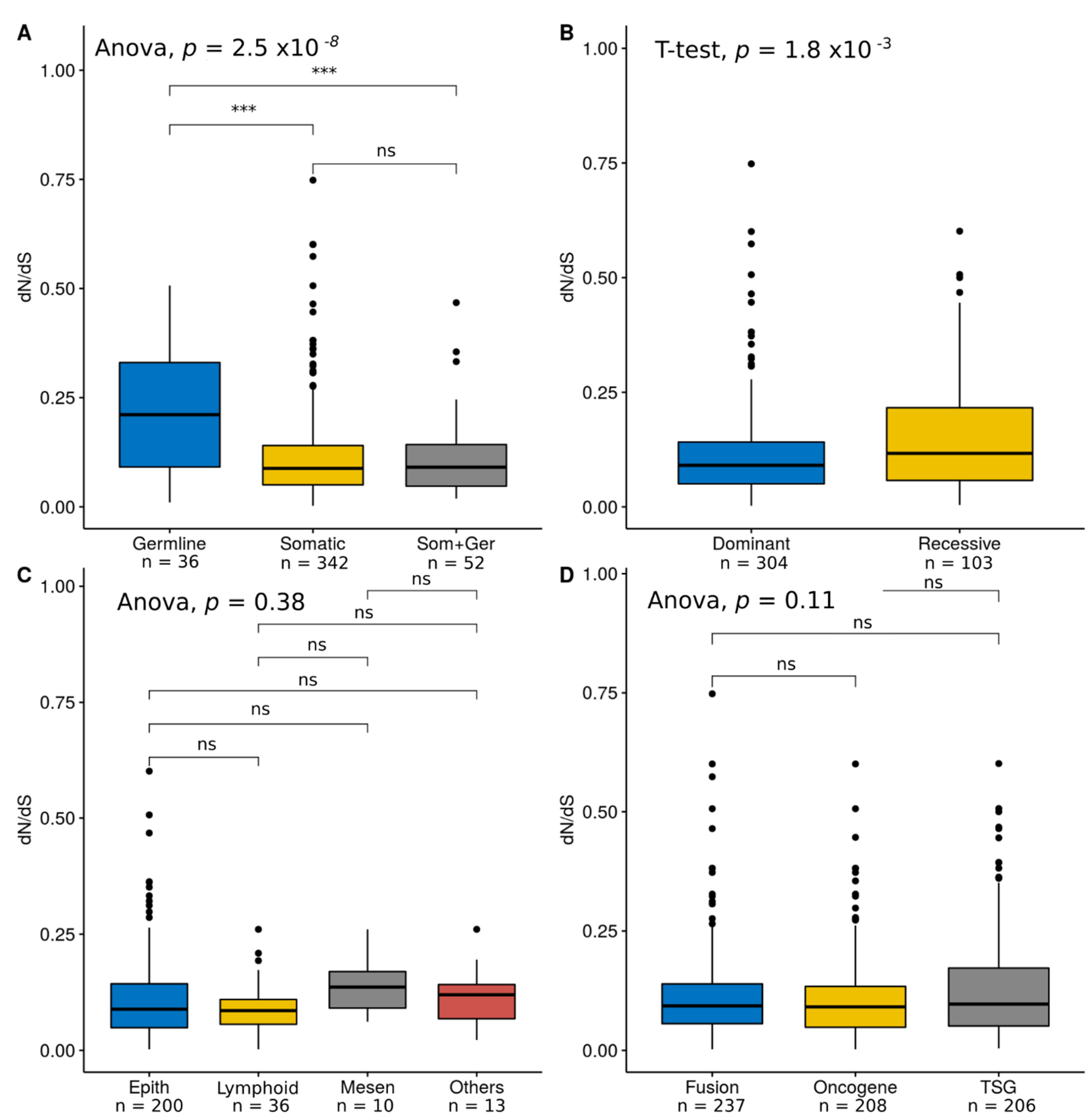
3.2. Comparison of Selection Estimates across Functional Categories
3.3. Functional Enrichment of Positively Selected Cancer Genes
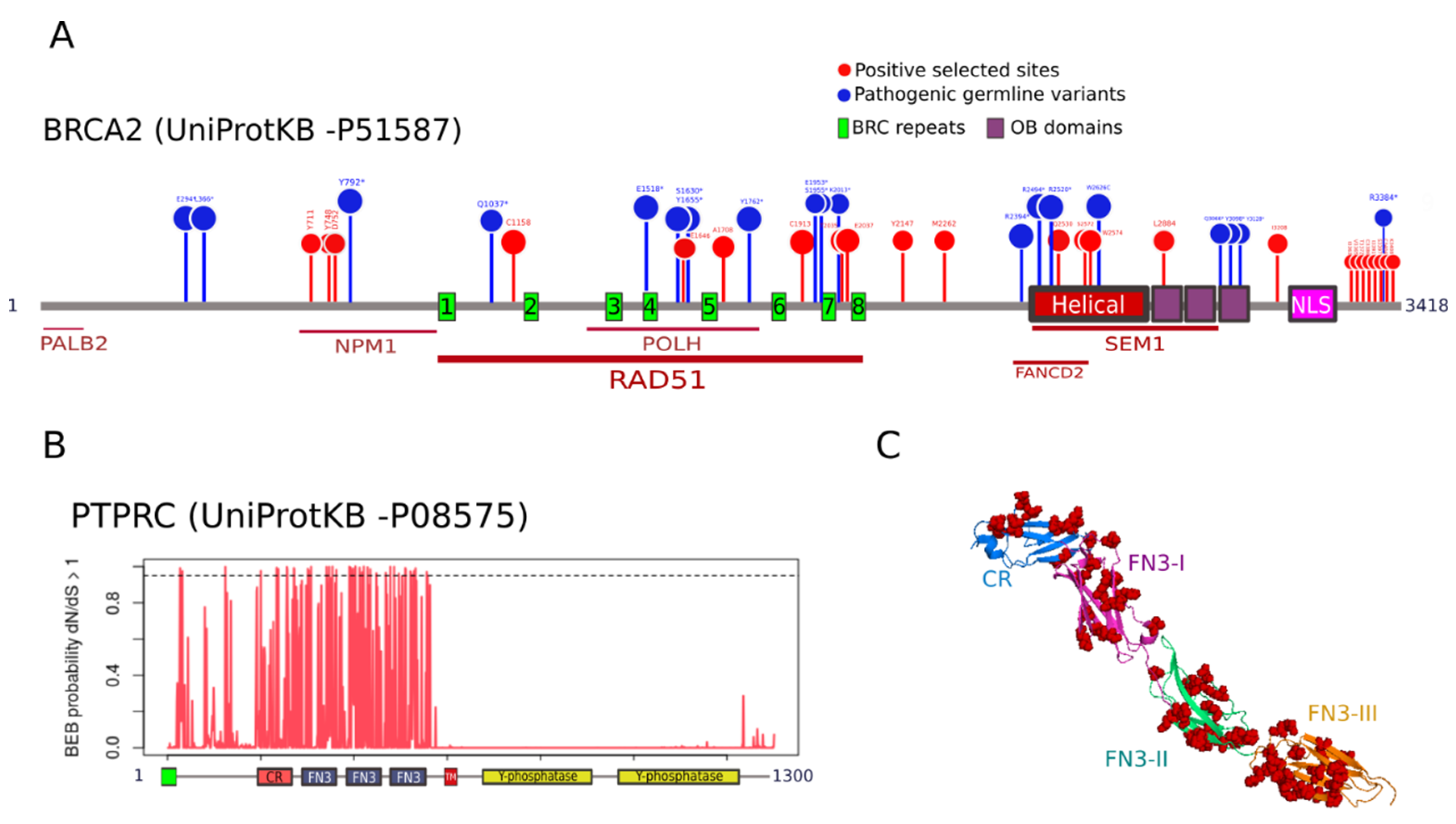
3.4. Functional Relevance of Positively Selected Sites in Cancer Genes
4. Discussion
4.1. Cancer Genes Show Relatively Low dN/dS Values
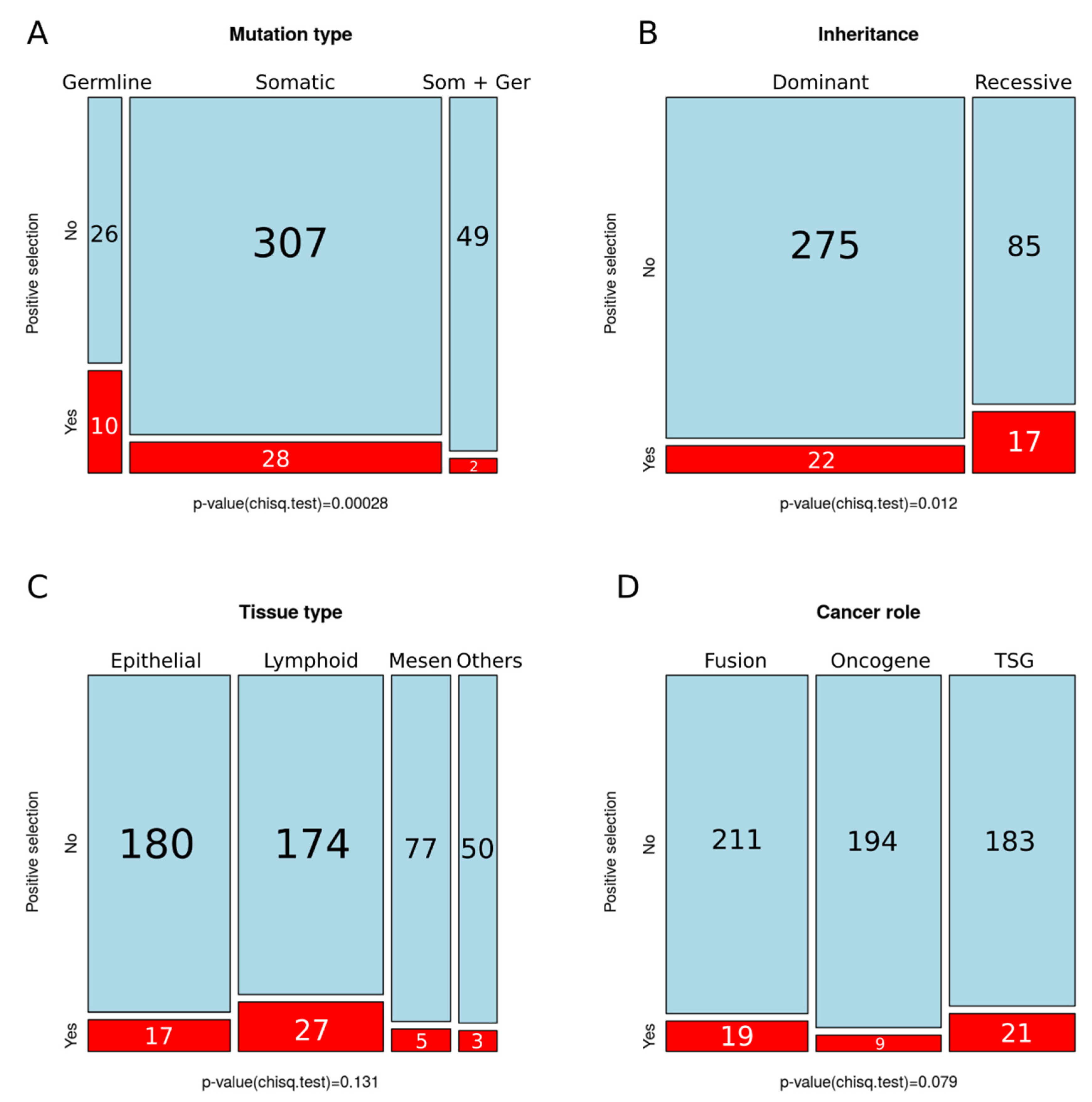
4.2. Positive Selection on Human Cancer Genes is Associated with Hereditary Cancer and Recessive Mutations
4.3. Lack of Variation in Selection across Tissues or Cancer Gene Role
4.4. Signalling Pathways and Biological Functions of Cancer Genes under Positive Selection
4.5. Functional Relevance of Residues under Positive Selection in Cancer Genes
Data Availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bodmer, W.; Tomilson, I. Rare genetic variants and the risk of cancer. Curr. Opin. Genet. Dev. 2010, 20, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal patterns of selection in cancer and somatic tissues. bioRxiv 2017. [Google Scholar] [CrossRef] [PubMed]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.G.; Glanowski, S.; Nielsen, R.; Thomas, P.D.; Kejariwal, A.; Todd, M.A.; Tanenbaum, D.M.; Civello, D.; Lu, F.; Murphy, B.; et al. Inferring nonneutral evolution from human-chimp-mouse orthologous gene trios. Science 2003, 302, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Bustamante, C.; Clark, A.G.; Glanowski, S.; Sackton, T.B.; Hubisz, M.J.; Fledel-Alon, A.; Tanenbaum, D.M.; Civello, D.; White, T.J.; et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005, 3, e170. [Google Scholar] [CrossRef]
- Kosiol, C.; Vinař, T.; da Fonseca, R.R.; Hubisz, M.J.; Bustamante, C.D.; Nielsen, R.; Siepel, A. Patterns of positive selection in six mammalian genomes. PLoS Genet. 2008, 4, e1000144. [Google Scholar] [CrossRef]
- Da Fonseca, R.R.; Kosiol, C.; Vinař, T.; Siepel, A.; Nielsen, R. Positive selection on apoptosis related genes. FEBS Lett. 2010, 584, 469–476. [Google Scholar] [CrossRef]
- Kleene, K.C. Sexual selection, genetic conflict, selfish genes, and the atypical patterns of gene expression in spermatogenic cells. Dev. Biol. 2005, 277, 16–26. [Google Scholar] [CrossRef]
- Crespi, B.J.; Summers, K. Positive selection in the evolution of cancer. Biol. Rev. Camb. Philos. Soc. 2006, 81, 407–424. [Google Scholar] [CrossRef]
- Kang, L.; Michalak, P. The evolution of cancer-related genes in hominoids. J. Mol. Evol. 2015, 80, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [PubMed]
- Ranwez, V.; Harispe, S.; Delsuc, F.; Douzery, E.J.P. MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS ONE 2011, 6, e22594. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, F.; Oliver, J.L.; Marín, A.; Medina, J.R. The general stochastic model of nucleotide substitution. J. Theor. Biol. 1990, 142, 485–501. [Google Scholar] [CrossRef]
- Yang, Z. Maximum-likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Mol. Biol. Evol. 1993, 10, 1396–1401. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Gharib, W.H.; Robinson-Rechavi, M. The branch-site test of positive selection is surprisingly robust but lacks power under synonymous substitution saturation and variation in GC. Mol. Biol. Evol. 2013, 30, 1675–1686. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R.; Goldman, N.; Pedersen, A. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 2000, 155, 431–449. [Google Scholar] [PubMed]
- Wong, W.S.W.; Yang, Z.; Goldman, N.; Nielsen, R. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 2004, 168, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-wide identification of episodic selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Frost, S.D.W.; Muse, S. V HyPhy: hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, S.W.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 2009, 10, 48. [Google Scholar] [CrossRef]
- Huang, K.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic germline variants in 10,389 adult cancers. Cell 2018, 173, 355–370. [Google Scholar] [CrossRef]
- Team, R. core R: A language and Environment for statistical computing. Available online: http://www.r-project.org (accessed on 5 May 2018).
- Bogliolo, M.; Surrallés, J. The Fanconi Anemia/BRCA pathway: FANCD2 at the crossroad between repair and checkpoint responses to DNA damage. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Wang, H.-F.; Takenaka, K.; Nakanishi, A.; Miki, Y. BRCA2 and nucleophosmin coregulate centrosome amplification and form a complex with the ρ effector kinase ROCK2. Cancer Res. 2011, 71, 68–77. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef]
- Buisson, R.; Niraj, J.; Pauty, J.; Maity, R.; Zhao, W.; Coulombe, Y.; Sung, P.; Masson, J.-Y. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase η in recombination-associated DNA synthesis at blocked replication forks. Cell Rep. 2014, 6, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Dayhoff, M.; Schwartz, R.; Orcutt, B. Atlas of Protein Sequence and Structure; Dayhoff, M., Ed.; National b.: Washington, DC, USA, 1978. [Google Scholar]
- Marston, N.J.; Richards, W.J.; Hughes, D.; Bertwistle, D.; Marshall, C.J.; Ashworth, A. Interaction between the product of the breast cancer susceptibility gene BRCA2 and DSS1, a protein functionally conserved from yeast to mammals. Mol. Cell. Biol. 1999, 19, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Wilson, J.B.; Medhurst, A.L.; Hejna, J.; Witt, E.; Ananth, S.; Davies, A.; Masson, J.-Y.; Moses, R.; West, S.C.; et al. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum. Mol. Genet. 2004, 13, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Zeng, G.; Chen, L.; Wang, G.; Wang, X.; Cao, X.; Lu, M.; Liu, X.; Qian, G.; Xiao, Y.; et al. Identification of key genes and pathways in human clear cell renal cell carcinoma (ccRCC) by co-expression analysis. Int. J. Biol. Sci. 2018, 14, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Chang, V.T.; Fernandes, R.A.; Ganzinger, K.A.; Lee, S.F.; Siebold, C.; McColl, J.; Jönsson, P.; Palayret, M.; Harlos, K.; Coles, C.H.; et al. Initiation of T cell signaling by CD45 segregation at “close contacts”. Nat. Immunol. 2016, 17, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Romiguier, J.; Ranwez, V.; Douzery, E.J.P.; Galtier, N. Genomic evidence for large, long-lived ancestors to placental mammals. Mol. Biol. Evol. 2013, 30, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Figuet, E.; Nabholz, B.; Bonneau, M.; Mas Carrio, E.; Nadachowska-Brzyska, K.; Ellegren, H.; Galtier, N. Life history traits, protein evolution, and the nearly neutral theory in amniotes. Mol. Biol. Evol. 2016, 33, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.D.; Fledel-Alon, A.; Williamson, S.; Nielsen, R.; Todd Hubisz, M.; Glanowski, S.; Tanenbaum, D.M.; White, T.J.; Sninsky, J.J.; Hernandez, R.D.; et al. Natural selection on protein-coding genes in the human genome. Nature 2005, 437, 1153–1157. [Google Scholar] [CrossRef]
- Thomas, M.A.; Weston, B.; Joseph, M.; Wu, W.; Nekrutenko, A.; Tonellato, P.J. Evolutionary dynamics of oncogenes and tumor suppressor genes: Higher intensities of purifying selection than other genes. Mol. Biol. Evol. 2003, 20, 964–968. [Google Scholar] [CrossRef]
- Blekhman, R.; Man, O.; Herrmann, L.; Boyko, A.R.; Indap, A.; Kosiol, C.; Bustamante, C.D.; Teshima, K.M.; Przeworski, M. Natural selection on genes that underlie human disease susceptibility. Curr. Biol. 2008, 18, 883–889. [Google Scholar] [CrossRef]
- Eyre-Walker, A.; Keightley, P.D.; Smith, N.G.C.; Gaffney, D. Quantifying the slightly deleterious mutation model of molecular evolution. Mol. Biol. Evol. 2002, 19, 2142–2149. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Charlesworth, B.; Rudan, I.; Carothers, A.; Campbell, H. A polygenic basis for late-onset disease. Trends Genet. 2003, 19, 97–106. [Google Scholar] [CrossRef]
- Spataro, N.; Rodríguez, J.A.; Navarro, A.; Bosch, E. Properties of human disease genes and the role of genes linked to Mendelian disorders in complex disease aetiology. Hum. Mol. Genet. 2017, 26, ddw405. [Google Scholar] [CrossRef] [PubMed]
- Furney, S.J.; Albà, M.M.; López-Bigas, N. Differences in the evolutionary history of disease genes affected by dominant or recessive mutations. BMC Genomics 2006, 7, 165. [Google Scholar] [CrossRef]
- O’Connell, M.J. Selection and the cell cycle: Positive darwinian selection in a well-known DNA damage response pathway. J. Mol. Evol. 2010, 71, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Zhou, H.; Tang, K. Recent coselection in human populations revealed by protein–protein interaction network. Genome Biol. Evol. 2015, 7, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Tollis, M.; Schiffman, J.D.; Boddy, A.M. Evolution of cancer suppression as revealed by mammalian comparative genomics. Curr. Opin. Genet. Dev. 2017, 42, 40–47. [Google Scholar] [CrossRef]
- Morgan, C.C.; Mc Cartney, A.M.; Donoghue, M.T.A.; Loughran, N.B.; Spillane, C.; Teeling, E.C.; O’Connell, M.J. Molecular adaptation of telomere associated genes in mammals. BMC Evol. Biol. 2013, 13. [Google Scholar] [CrossRef]
- Zhang, G.; Cowled, C.; Shi, Z.; Huang, Z.; Bishop-Lilly, K.A.; Fang, X.; Wynne, J.W.; Xiong, Z.; Baker, M.L.; Zhao, W.; et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science 2013, 339, 456–460. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Function | dN/dS |
|---|---|---|
| IL2 | T cell proliferation and regulation of the immune response. | 0.748 |
| FAS | Apoptosis | 0.601 |
| FCRL4 | B cell receptor signaling | 0.601 |
| NUTM2A | Unknown | 0.574 |
| PALB2 | Tumor necrosis factor, apoptosis | 0.507 |
| PDCD1LG2 | T cell proliferation; immune response | 0.506 |
| FANCG | Fanconi Anemia (FA) group; DNA repair | 0.500 |
| BRCA2 | Double-strand break repair and/or homologous recombination | 0.468 |
| CD274 | T cell effector regulation; attenuation of anti-tumor immunity | 0.465 |
| FANCC | F.A. group; DNA repair | 0.445 |
| CASP8 | Protease inhibitor; apoptosis | 0.363 |
| PTPRC | Protein phosphatase; receptor; immune response | 0.361 |
| FANCD2 | F.A. group; DNA repair | 0.348 |
| BARD1 | Control of the cell cycle in response to DNA damage | 0.333 |
| ERCC5 | DNA repair | 0.321 |
| NCOA4 | Androgen receptor signaling | 0.312 |
| NIN | Centrosome localization | 0.307 |
| BRIP1 | Double-strand break repair and/or homologous recombination | 0.286 |
| COL1A1 | Collagen component | 0.276 |
| CD79B | B cell differentiation and activation | 0.275 |
| BLM | Basic helix-loop transcription factor | 0.264 |
| CD79A | B cell differentiation and activation | 0.253 |
| PMS2 | DNA binding protein | 0.225 |
| KTN1 | Kinesin-driven vesicle motility; cadherin binding | 0.211 |
| PRF1 | Apoptosis; immune response | 0.197 |
| SET | Chaperone; phosphatase inhibitor | 0.180 |
| ARHGEF12 | Regulation of RhoA GTPase | 0.178 |
| CHEK2 | Checkpoint-mediated cell cycle arrest, activation of DNA repair and apoptosis | 0.175 |
| PTPRB | Protein phosphatase; receptor; angiogenesis | 0.156 |
| SS18 | Chromatin-binding protein; transcription regulation | 0.153 |
| FLT3 | Regulation of apoptotic process | 0.144 |
| COL2A1 | Collagen component | 0.132 |
| MLLT6 | Nucleic acid binding; zinc finger transcription factor | 0.130 |
| KDM6A | Transcription factor; chromatin remodeling | 0.120 |
| POU2AF1 | Transcriptional coactivator; immune response | 0.106 |
| MED12 | Nucleic acid binding; transcription cofactor | 0.097 |
| RBM15 | RNA binding protein | 0.088 |
| RABEP1 | Membrane fusion; apoptosis | 0.086 |
| BRAF | Transduction of mitogenic signals; apoptosis | 0.079 |
| PICALM | Vesicle coat protein | 0.068 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vicens, A.; Posada, D. Selective Pressures on Human Cancer Genes along the Evolution of Mammals. Genes 2018, 9, 582. https://doi.org/10.3390/genes9120582
Vicens A, Posada D. Selective Pressures on Human Cancer Genes along the Evolution of Mammals. Genes. 2018; 9(12):582. https://doi.org/10.3390/genes9120582
Chicago/Turabian StyleVicens, Alberto, and David Posada. 2018. "Selective Pressures on Human Cancer Genes along the Evolution of Mammals" Genes 9, no. 12: 582. https://doi.org/10.3390/genes9120582
APA StyleVicens, A., & Posada, D. (2018). Selective Pressures on Human Cancer Genes along the Evolution of Mammals. Genes, 9(12), 582. https://doi.org/10.3390/genes9120582