Molecular and Cellular Functions of the Warsaw Breakage Syndrome DNA Helicase DDX11


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Discovery of Human DDX11
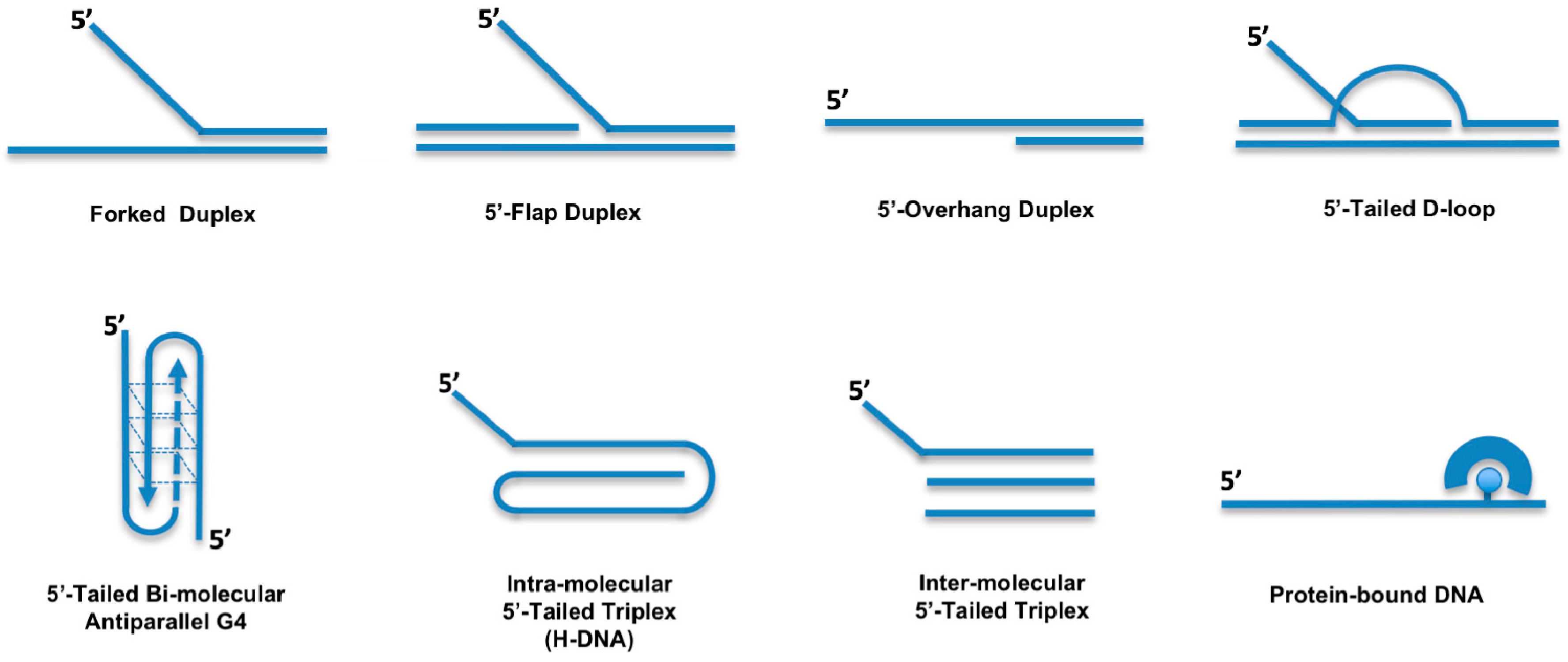
3. Enzymatic Properties of Human DDX11
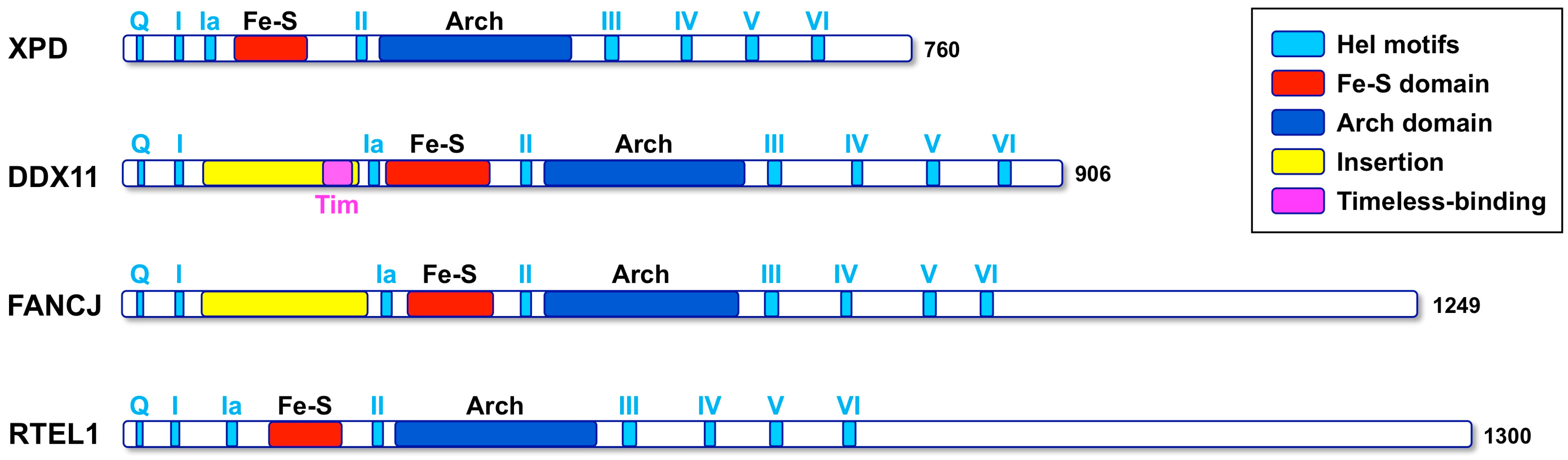
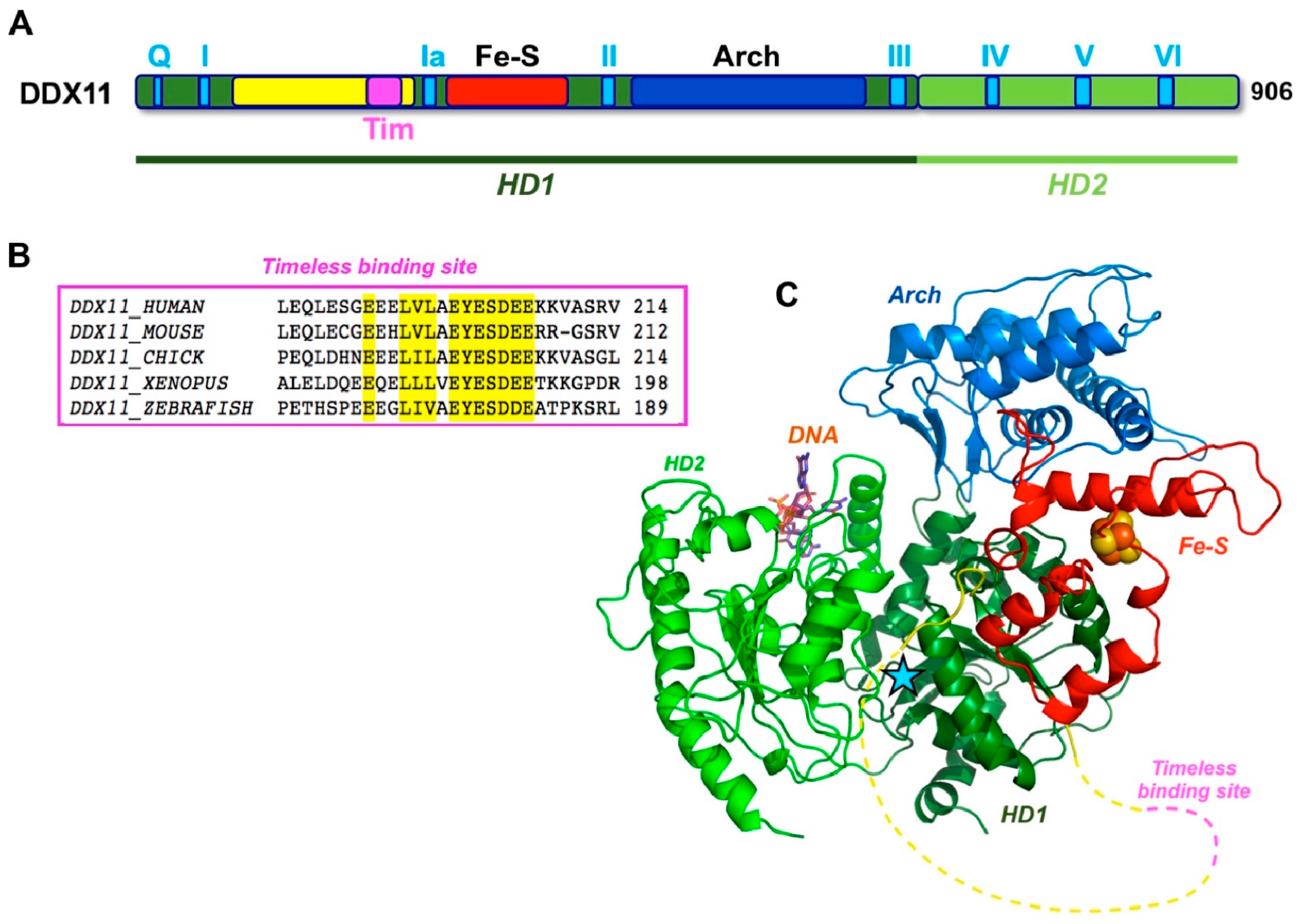
4. Structural Features of Human DDX11
5. Cellular Localization of DDX11
6. Role of DDX11 in Sister Chromatid Cohesion
7. DDX11 Functions in DNA Repair and Replication Fork Stabilization
8. DDX11 and Chromatin Structure
9. DDX11 and Cancer
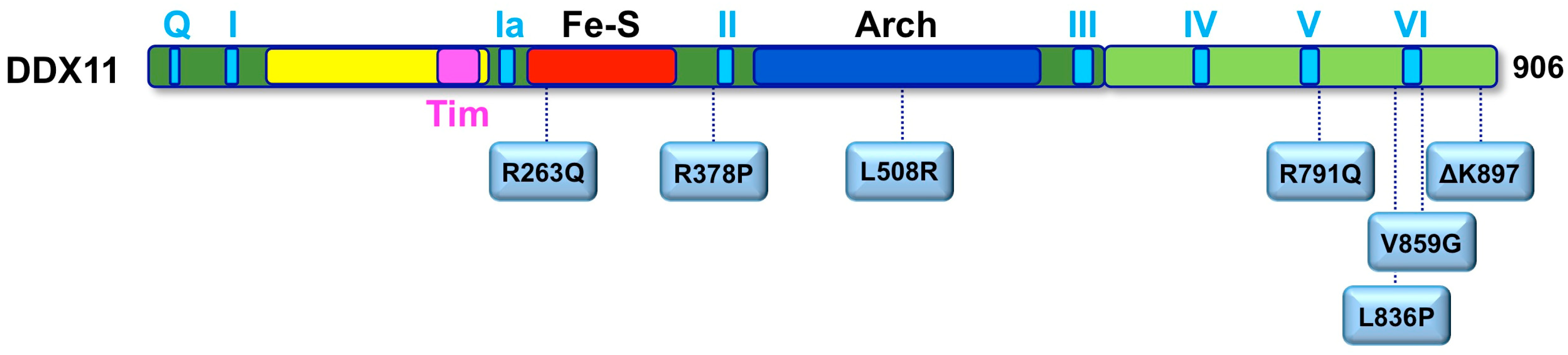
10. Warsaw Breakage Syndrome, a Cohesinopathy
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, Y.; Brosh, R.M., Jr. DNA helicase and helicase-nuclease enzymes with a conserved iron-sulfur cluster. Nucleic Acids Res. 2012, 40, 4247–4260. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M., Jr. DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer 2013, 13, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Khan, I.; Banerjee, T.; Sommers, J.A.; Wu, Y.; Brosh, R.M., Jr. Molecular functions and cellular roles of the ChlR1 (DDX11) helicase defective in the rare cohesinopathy Warsaw breakage syndrome. Cell. Mol. Life Sci. 2014, 71, 2625–2639. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Werner, S. The human homologue of the yeast CHL1 gene is a novel keratinocyte growth factor-regulated gene. J. Biol. Chem. 1996, 271, 24337–24340. [Google Scholar] [CrossRef] [PubMed]
- Amann, J.; Valentine, M.; Kidd, V.J.; Lahti, J.M. Localization of chi1-related helicase genes to human chromosome regions 12p11 and 12p13: Similarity between parts of these genes and conserved human telomeric-associated DNA. Genomics 1996, 32, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Amann, J.; Kidd, V.J.; Lahti, J.M. Characterization of putative human homologues of the yeast chromosome transmission fidelity gene, CHL1. J. Biol. Chem. 1997, 272, 3823–3832. [Google Scholar] [CrossRef] [PubMed]
- Gerring, S.L.; Spencer, F.; Hieter, P. The CHL1 (CTF1) gene product of Saccharomyces cerevisiae is important for chromosome transmission and normal cell cycle progression in G2/M. EMBO J. 1990, 9, 4347–4358. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Casamassimi, A.; Roberto, R.; Gianfrancesco, F.; Matarazzo, M.R.; D’Urso, M.; D’Esposito, M.; Rocchi, M.; Ciccodicola, A. DDX11L: A novel transcript family emerging from human subtelomeric regions. BMC Genomics 2009, 10, 250. [Google Scholar] [CrossRef] [PubMed]
- Hirota, Y.; Lahti, J.M. Characterization of the enzymatic activity of hChlR1, a novel human DNA helicase. Nucleic Acids Res. 2000, 28, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Farina, A.; Shin, J.H.; Kim, D.H.; Bermudez, V.P.; Kelman, Z.; Seo, Y.S.; Hurwitz, J. Studies with the human cohesin establishment factor, ChlR1. Association of ChlR1 with Ctf18-RFC and Fen1. J. Biol. Chem. 2008, 283, 20925–20936. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sommers, J.A.; Khan, I.; de Winter, J.P.; Brosh, R.M., Jr. Biochemical characterization of Warsaw breakage syndrome helicase. J. Biol. Chem. 2012, 287, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Suhasini, A.N.; Sommers, J.A.; Yu, S.; Wu, Y.; Xu, T.; Kelman, Z.; Kaplan, D.L.; Brosh, R.M., Jr. DNA repair and replication fork helicases are differentially affected by alkyl phosphotriester lesion. J. Biol. Chem. 2012, 287, 19188–19198. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Sommers, J.A.; George, F.; Kuper, J.; Hamon, F.; Shin-Ya, K.; Teulade-Fichou, M.P.; Kisker, C.; Brosh, R.M., Jr. Specialization among iron–sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. J. Biol. Chem. 2013, 288, 28217–28229. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Suhasini, A.N.; Banerjee, T.; Sommers, J.A.; Kaplan, D.L.; Kuper, J.; Kisker, C.; Brosh, R.M., Jr. Impact of age-associated cyclopurine lesions on DNA repair helicases. PLoS ONE 2014, 11, e113293. [Google Scholar] [CrossRef] [PubMed]
- León-Ortiz, A.M.; Svendsen, J.; Boulton, S.J. Metabolism of DNA secondary structures at the eukaryotic replication fork. DNA Repair 2014, 19, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Jaruga, P.; Dizdaroglu, M. 8,5′-Cyclopurine-2′-deoxynucleosides in DNA: Mechanisms of formation, measurement, repair and biological effects. DNA Repair 2008, 7, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, I.; Bender, C.; Romieu, A.; Cadet, J.; Wood, R.D.; et al. Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl. Acad. Sci. USA 2000, 97, 3832–3837. [Google Scholar] [CrossRef] [PubMed]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sommers, J.A.; Loiland, J.A.; Kitao, H.; Kuper, J.; Kisker, C.; Brosh, R.M., Jr. The Q motif of Fanconi anemia group J protein (FANCJ) DNA helicase regulates its dimerization, DNA binding, and DNA repair function. J. Biol. Chem. 2012, 287, 21699–216716. [Google Scholar] [CrossRef] [PubMed]
- Sommers, J.A.; Rawtani, N.; Gupta, R.; Bugreev, D.V.; Mazin, A.V.; Cantor, S.B.; Brosh, R.M., Jr. FANCJ uses its motor ATPase to destabilize protein-DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. J. Biol. Chem. 2009, 284, 7505–7517. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Hundseth, K.; Ding, H.; Vidhyasagar, V.; Inoue, A.; Nguyen, C.H.; Zain, R.; Lee, J.S.; Wu, Y. A distinct triplex DNA unwinding activity of ChlR1 helicase. J. Biol. Chem. 2015, 290, 5174–5189. [Google Scholar] [CrossRef] [PubMed]
- Duca, M.; Vekhoff, P.; Oussedik, K.; Halby, L.; Arimondo, P.B. The triple helix: 50 years later, the outcome. Nucleic Acids Res. 2008, 36, 5123–5138. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, K.M.; Glazer, P.M. Triplex-forming oligonucleotides: Principles and applications. Q. Rev. Biophys. 2002, 35, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Burkholder, G.D.; Latimer, L.J.; Lee, J.S. Immunofluorescent staining of mammalian nuclei and chromosomes with a monoclonal antibody to triplex DNA. Chromosoma 1988, 97, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Agazie, Y.M.; Lee, J.S.; Burkholder, G.D. Characterization of a new monoclonal antibody to triplex DNA and immunofluorescent staining of mammalian chromosomes. J. Biol. Chem. 1994, 269, 7019–7023. [Google Scholar] [PubMed]
- Wu, Y.; Rawtani, N.; Thazhathveetil, A.K.; Kenny, M.K.; Seidman, M.M.; Brosh, R.M., Jr. Human replication protein A melts a DNA triple helix structure in a potent and specific manner. Biochemistry 2008, 47, 5068–5077. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M., Jr.; Majumdar, A.; Desai, S.; Hickson, I.D.; Bohr, V.A.; Seidman, M.M. Unwinding of a DNA triple helix by the Werner and Bloom syndrome helicases. J. Biol. Chem. 2001, 276, 3024–3030. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Bacolla, A.; Del Mundo, I.M.; Zhao, J.; Wang, G.; Vasquez, K.M. DHX9 helicase is involved in preventing genomic instability induced by alternatively structured DNA in human cells. Nucleic Acids Res. 2013, 41, 10345–10357. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Fuss, J.O.; Cheng, Q.J.; Arvai, A.S.; Hammel, M.; Roberts, V.A.; Cooper, P.K.; Tainer, J.A. XPD helicase structures and activities: Insights into the cancer and aging phenotypes from XPD mutations. Cell 2008, 133, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Rudolf, J.; Johnson, K.A.; McMahon, S.A.; Oke, M.; Carter, L.; McRobbie, A.M.; Brown, S.E.; Naismith, J.H.; White, M.F. Structure of the DNA repair helicase XPD. Cell 2008, 133, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Wolski, S.C.; Kuper, J.; Hänzelmann, P.; Truglio, J.J.; Croteau, D.L.; Van Houten, B.; Kisker, C. Crystal structure of the FeS cluster-containing nucleotide excision repair helicase XPD. PLoS Biol. 2008, 6, e149. [Google Scholar] [CrossRef] [PubMed]
- Kuper, J.; Wolski, S.C.; Michels, G.; Kisker, C. Functional and structural studies of the nucleotide excision repair helicase XPD suggest a polarity for DNA translocation. EMBO J. 2012, 31, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu-Aruxandei, D.; Petrovic-Stojanovska, B.; Penedo, J.C.; White, M.F.; Naismith, J.H. Mechanism of DNA loading by the DNA repair helicase XPD. Nucleic Acids Res. 2016, 44, 2806–2815. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; Thompson, J.D.; Higgins, D.G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Xu, J. RaptorX: Exploiting structure information for protein alignment by statistical inference. Proteins 2011, 79, 161–171. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yan, C.; Fang, J.; Inouye, C.; Tjian, R.; Ivanov, I.; Nogales, E. Near-atomic resolution visualization of human transcription promoter opening. Nature 2016, 533, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Parish, J.L.; Rosa, J.; Wang, X.; Lahti, J.M.; Doxsey, S.J.; Androphy, E. The DNA helicase ChlR1 is required for sister chromatid cohesion in mammalian cells. J. Cell. Sci. 2006, 119, 4857–4865. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.E. Bisexual mating behavior in a diploid of Saccharomyces cerevisiae: Evidence for genetically controlled non-random chromosome loss during vegetative growth. Genetics 1974, 78, 843–858. [Google Scholar] [PubMed]
- Holloway, S.L. CHL1 is a nuclear protein with an essential ATP binding site that exhibits a size-dependent effect on chromosome segregation. Nucleic Acids Res. 2000, 28, 3056–3064. [Google Scholar] [CrossRef]
- Ansbach, A.B.; Noguchi, C.; Klansek, I.W.; Heidlebaugh, M.; Nakamura, T.M.; Noguchi, E. RFCCtf18 and the Swi1-Swi3 complex function in separate and redundant pathways required for the stabilization of replication forks to facilitate sister chromatid cohesion in Schizosaccharomyces pombe. Mol. Biol. Cell. 2008, 19, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.L.; Pot, I.; Chang, M.; Xu, H.; Aneliunas, V.; Kwok, T.; Newitt, R.; Aebersold, R.; Boone, C.; Brown, G.W.; Hieter, P. Identification of protein complexes required for efficient sister chromatid cohesion. Mol. Biol. Cell. 2004, 15, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Petronczki, M.; Chwalla, B.; Siomos, M.F.; Yokobayashi, S.; Helmhart, W.; Deutschbauer, A.M.; Davis, R.W.; Watanabe, Y.; Nasmyth, K. Sister-chromatid cohesion mediated by the alternative RF-CCtf18/Dcc1/Ctf8, the helicase Chl1 and the polymerase-α-associated protein Ctf4 is essential for chromatid disjunction during meiosis II. J. Cell. Sci. 2004, 117, 3547–3559. [Google Scholar] [CrossRef] [PubMed]
- Skibbens, R.V. Chl1p, a DNA helicase-like protein in budding yeast, functions in sister-chromatid cohesion. Genetics 2004, 166, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Rudra, S.; Skibbens, R.V. Sister chromatid cohesion establishment occurs in concert with lagging strand synthesis. Cell Cycle 2012, 11, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Li, T.; Roby, S.K.; Valentine, M.B.; Inoue, M.; Boyd, K.; Kidd, V.J.; Lahti, J.M. Loss of ChlR1 helicase in mouse causes lethality due to the accumulation of aneuploid cells generated by cohesion defects and placental malformation. Cell Cycle 2007, 6, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Leman, A.R.; Noguchi, C.; Lee, C.Y.; Noguchi, E. Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J. Cell. Sci. 2010, 123, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Samora, C.P.; Saksouk, J.; Goswami, P.; Wade, B.O.; Singleton, M.R.; Bates, P.A.; Lengronne, A.; Costa, A.; Uhlmann, F. Ctf4 links DNA replication with sister chromatid cohesion establishment by recruiting the Chl1 helicase to the replisome. Mol. Cell 2016, 63, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.C.; Zhou, J.C.; Perera, R.L.; van Deursen, F.; Evrin, C.; Ivanova, M.E.; Kilkenny, M.L.; Renault, L.; Kjaer, S.; Matak-Vinković, D.; Labib, K.; Costa, A.; Pellegrini, L. A Ctf4 trimer couples the CMG helicase to DNA polymerase α in the eukaryotic replisome. Nature 2014, 510, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Calì, F.; Bharti, S.K.; Di Perna, R.; Brosh, R.M., Jr.; Pisani, F.M. Tim/Timeless, a member of the replication fork protection complex, operates with the Warsaw breakage syndrome DNA helicase DDX11 in the same fork recovery pathway. Nucleic Acids Res. 2016, 44, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Cortone, G.; Zheng, G.; Pensieri, P.; Chiappetta, V.; Tatè, R.; Malacaria, E.; Pichierri, P.; Yu, H.; Pisani, F.M. Interaction of the Warsaw breakage syndrome DNA helicase DDX11 with the replication fork-protection factor Timeless promotes sister chromatid cohesion. PLoS Genet. 2018, 14, e1007622. [Google Scholar] [CrossRef] [PubMed]
- Aze, A.; Zhou, J.C.; Costa, A.; Costanzo, V. DNA replication and homologous recombination factors: Acting together to maintain genome stability. Chromosoma 2013, 5, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Kawasumi, R.; Arakawa, H.; Hori, T.; Shirahige, K.; Losada, A.; Fukagawa, T.; Branzei, D. Chromatin determinants of the inner-centromere rely on replication factors with functions that impart cohesion. Oncotarget 2016, 7, 67934–67947. [Google Scholar] [CrossRef] [PubMed]
- Marchese, F.P.; Grossi, E.; Marín-Béjar, O.; Bharti, S.K.; Raimondi, I.; González, J.; Martínez-Herrera, D.J.; Athie, A.; Amadoz, A.; Brosh, R.M., Jr.; et al. Long noncoding RNA regulates sister chromatid cohesion. Mol. Cell 2016, 63, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Murayama, Y.; Samora, C.P.; Kurokawa, Y.; Iwasaki, H.; Uhlmann, F. Establishment of DNA-DNA Interactions by the Cohesin Ring. Cell 2018, 172, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Kanchwala, M.; Xing, C.; Yu, H. MCM2-7-dependent cohesin loading during S phase promotes sister-chromatid cohesion. Elife 2018, 7, e33920. [Google Scholar] [CrossRef] [PubMed]
- Laha, S.; Das, S.P.; Hajra, S.; Sau, S.; Sinha, P. The budding yeast protein Chl1p is required to preserve genome integrity upon DNA damage in S-phase. Nucleic Acids Res. 2006, 34, 5880–5891. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, H.; Ohuchi, T.; Ui, A.; Tada, S.; Enomoto, T.; Seki, M. Ctf18 is required for homologous recombination-mediated double-strand break repair. Nucleic Acids Res. 2007, 35, 4989–5000. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Inoue, A.; Lee, S.W.; Beishline, K.; Lahti, J.M.; Noguchi, E. Roles of Chlr1 DNA helicase in replication fork recovery from DNA damage. Exp. Cell Res. 2013, 319, 2244–2253. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Ooka, M.; Kawasumi, R.; Miyata, K.; Takata, M.; Hirota, K.; Branzei, D. Warsaw breakage syndrome DDX11 helicase acts jointly with RAD17 in the repair of bulky lesions and replication through abasic sites. Proc. Natl. Acad. Sci. USA 2018, 115, 8412–8417. [Google Scholar] [CrossRef] [PubMed]
- Litman, R.; Peng, M.; Jin, Z.; Zhang, F.; Zhang, J.; Powell, S.; Andreassen, P.R.; Cantor, S.B. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 2005, 8, 255–265. [Google Scholar] [CrossRef] [PubMed]
- van der Lelij, P.; Chrzanowska, K.H.; Godthelp, B.C.; Rooimans, M.A.; Oostra, A.B.; Stumm, M.; Zdzienicka, M.Z.; Joenje, H.; de Winter, J.P. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am. J. Hum. Genet. 2010, 86, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Bacolla, A.; Chakraborty, P.; Grosse, F.; Vasquez, K.M. Human DHX9 helicase unwinds triple-helical DNA structures. Biochemistry 2010, 49, 6992–6999. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Mortusewicz, O.; Ma, H.T.; Herr, P.; Poon, R.R.; Helleday, T.; Qian, C. Timeless interacts with PARP-1 to promote homologous recombination repair. Mol. Cell 2015, 60, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Young, L.M.; Marzio, A.; Perez-Duran, P.; Reid, D.A.; Meredith, D.N.; Roberti, D.; Star, A.; Rothenberg, E.; Ueberheide, B.; Pagano, M. TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the DNA damage response. Cell Rep. 2015, 13, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Stoepker, C.; Faramarz, A.; Rooimans, M.A.; van Mil, S.E.; Balk, J.A.; Velleuer, E.; Ameziane, N.; Te Riele, H.; de Winter, J.P. DNA helicases FANCM and DDX11 are determinants of PARP inhibitor sensitivity. DNA Repair 2015, 26, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Hyle, J.; Lechner, M.S.; Lahti, J.M. Mammalian ChlR1 has a role in heterochromatin organization. Exp. Cell Res. 2011, 317, 2522–2535. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chen, H.; Deng, Z.; Hu, B.; Luo, H.; Zeng, X.; Han, L.; Cai, G.; Ma, L. The Warsaw breakage syndrome-related protein DDX11 is required for ribosomal RNA synthesis and embryonic development. Hum. Mol. Genet. 2015, 24, 4901–4915. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, C.; Wang, X.; Becker, D. The DEAD/DEAH box helicase, DDX11, is essential for the survival of advanced melanomas. Mol. Cancer 2012, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, H.; Jallepalli, P.V.; Rago, C.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Inactivation of hCDC4 can cause chromosomal instability. Nature 2004, 428, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.D.; McManus, K.; Yuen, K.W.; Reis, M.; Parmigiani, G.; Shen, D.; Barrett, I.; Nouhi, Y.; Spencer, F.; Markowitz, S.; et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 3443–3448. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Benavente, B.; García, J.L.; Rodríguez, M.S.; Pineda-Lucena, A.; Piechaczyk, M.; Font de Mora, J.; Farràs, R. GSK3-SCFFBXW7 targets JunB for degradation in G2 to preserve chromatid cohesion before anaphase. Oncogene 2013, 32, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- de Lange, J.; Faramarz, A.; Oostra, A.B.; de Menezes, R.X.; van der Meulen, I.H.; Rooimans, M.A.; Rockx, D.A.; Brakenhoff, R.H.; van Beusechem, V.W.; King, R.W.; de Winter, J.P.; Wolthuis, R.M. Defective sister chromatid cohesion is synthetically lethal with impaired APC/C function. Nat. Commun. 2015, 6, 8399. [Google Scholar] [CrossRef] [PubMed]
- Parish, J.L.; Bean, A.M.; Park, R.B.; Androphy, E.J. ChlR1 is required for loading papillomavirus E2 onto mitotic chromosomes and viral genome maintenance. Mol. Cell 2006, 24, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Feeney, K.M.; Saade, A.; Okrasa, K.; Parish, J.L. In vivo analysis of the cell cycle dependent association of the bovine papillomavirus E2 protein and ChlR1. Virology 2011, 414, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.; McFarlane-Majeed, L.; Campos-León, K.; Roberts, S.; Parish, J.L. The cellular DNA helicase ChlR1 regulates chromatin and nuclear matrix attachment of the human papillomavirus 16 E2 protein and high-copy-number viral genome establishment. J. Virol. 2016, 91, e01853-16. [Google Scholar] [CrossRef] [PubMed]
- Dheekollu, J.; Lieberman, P.M. The replisome pausing factor Timeless is required for episomal maintenance of latent Epstein-Barr virus. J. Virol. 2011, 85, 5853–5863. [Google Scholar] [CrossRef] [PubMed]
- Dheekollu, J.; Chen, H.S.; Kaye, K.M.; Lieberman, P.M. Timeless-dependent DNA replication-coupled recombination promotes Kaposi’s Sarcoma-associated herpesvirus episome maintenance and terminal repeat stability. J. Virol. 2013, 87, 3699–3709. [Google Scholar] [CrossRef] [PubMed]
- Capo-Chichi, J.M.; Bharti, S.K.; Sommers, J.A.; Yammine, T.; Chouery, E.; Patry, L.; Rouleau, G.A.; Samuels, M.E.; Hamdan, F.F.; Michaud, J.L.; et al. Identification and biochemical characterization of a novel mutation in DDX11 causing Warsaw breakage syndrome. Hum. Mutat. 2013, 34, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.; Fryer, A.E.; Greenslade, M. Warsaw Breakage Syndrome—A further report, emphasising cutaneous findings. Eur. J. Med. Genet. 2015, 58, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Eppley, S.; Hopkin, R.J.; Mendelsohn, B.; Slavotinek, A.M. Clinical Report: Warsaw breakage syndrome with small radii and fibulae. Am. J. Med. Genet. A 2017, 173, 3075–3081. [Google Scholar] [CrossRef] [PubMed]
- Alkhunaizi, E.; Shaheen, R.; Bharti, S.K.; Joseph-George, A.M.; Chong, K.; Abdel-Salam, G.M.H.; Alowain, M.; Blaser, S.I.; Papsin, B.C.; Butt, M.; et al. Warsaw breakage syndrome: Further clinical and genetic delineation. Am. J. Med. Genet 2018. [Google Scholar] [CrossRef] [PubMed]
- Vega, H.; Waisfisz, Q.; Gordillo, M.; Sakai, N.; Yanagihara, I.; Yamada, M.; van Gosliga, D.; Kayserili, H.; Xu, C.; Ozono, K.; et al. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat. Genet. 2005, 37, 468–470. [Google Scholar] [CrossRef] [PubMed]
- Skibbens, R.V.; Corson, L.B.; Koshland, D.; Hieter, P. Ctf7p is essential for sister chromatid cohesion and links mitotic chromosome structure to the DNA replication machinery. Genes Dev. 1999, 13, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Tóth, A.; Ciosk, R.; Uhlmann, F.; Galova, M.; Schleiffer, A.; Nasmyth, K. Yeast cohesin complex requires a conserved protein, Eco1p(Ctf7), to establish cohesion between sister chromatids during DNA replication. Genes Dev. 1999, 13, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Krantz, I.D. Cohesin and human disease. Annu. Rev. Genomics Hum. Genet. 2008, 9, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Krantz, I.D. Cohesin embraces new phenotypes. Nat. Genet. 2014, 46, 1157–1158. [Google Scholar] [CrossRef] [PubMed]
- Chetaille, P.; Preuss, C.; Burkhard, S.; Côté, J.M.; Houde, C.; Castilloux, J.; Piché, J.; Gosset, N.; Leclerc, S.; Wünnemann, F.; et al. Mutations in SGOL1 cause a novel cohesinopathy affecting heart and gut rhythm. Nat. Genet. 2014, 46, 1245–1249. [Google Scholar] [CrossRef] [PubMed]
- van der Lelij, P.; Oostra, A.B.; Rooimans, M.A.; Joenje, H.; de Winter, J.P. Diagnostic overlap between Fanconi anemia and the cohesinopathies: Roberts syndrome and Warsaw breakage syndrome. Anemia 2010, 565268. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Kang, K.M.; Jung, S.Y.; Choi, H.K.; Seo, J.H.; Chae, J.H.; Cho, E.J.; Youn, H.D.; Qin, J.; Kim, S.T. Esco2 is a novel corepressor that associates with various chromatin modifying enzymes. Biochem. Biophys. Res. Commun. 2008, 372, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Rudra, S.; Skibbens, R.V. Chl1 DNA helicase regulates Scc2 deposition specifically during DNA replication in Saccharomyces cerevisiae. PLoS ONE 2013, 8, e75435. [Google Scholar] [CrossRef] [PubMed]
- Skibbens, R.V.; Colquhoun, J.M.; Green, M.J.; Molnar, C.A.; Sin, D.N.; Sullivan, B.J.; Tanzosh, E.E. Cohesinopathies of a feather flock together. PLoS Genet. 2013, 9, e1004036. [Google Scholar] [CrossRef] [PubMed]
- Gerton, J.L. Translational mechanisms at work in the cohesinopathies. Nucleus 2012, 3, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Lu, S.; Gerton, J.L. Roberts syndrome: A deficit in acetylated cohesin leads to nucleolar dysfunction. Rare Dis. 2014, 2, e27743. [Google Scholar] [CrossRef] [PubMed]
- Bose, T.; Lee, K.K.; Lu, S.; Xu, B.; Harris, B.; Slaughter, B.; Unruh, J.; Garrett, A.; McDowell, W.; Box, A.; et al. Cohesin proteins promote ribosomal RNA production and protein translation in yeast and human cells. PLoS Genet. 2012, 8, e1002749. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Lee, K.K.; Harris, B.; Xiong, B.; Bose, T.; Saraf, A.; Hattem, G.; Florens, L.; Seidel, C.; Gerton, J.L. The cohesin acetyltransferase Eco1 coordinates rDNA replication and transcription. EMBO Rep. 2014, 15, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Harris, B.; Bose, T.; Lee, K.K.; Wang, F.; Lu, S.; Ross, R.T.; Zhang, Y.; French, S.L.; Beyer, A.L.; Slaughter, B.D.; et al. Cohesion promotes nucleolar structure and function. Mol. Biol. Cell. 2014, 25, 337–346. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisani, F.M.; Napolitano, E.; Napolitano, L.M.R.; Onesti, S. Molecular and Cellular Functions of the Warsaw Breakage Syndrome DNA Helicase DDX11. Genes 2018, 9, 564. https://doi.org/10.3390/genes9110564
Pisani FM, Napolitano E, Napolitano LMR, Onesti S. Molecular and Cellular Functions of the Warsaw Breakage Syndrome DNA Helicase DDX11. Genes. 2018; 9(11):564. https://doi.org/10.3390/genes9110564
Chicago/Turabian StylePisani, Francesca M., Ettore Napolitano, Luisa M. R. Napolitano, and Silvia Onesti. 2018. "Molecular and Cellular Functions of the Warsaw Breakage Syndrome DNA Helicase DDX11" Genes 9, no. 11: 564. https://doi.org/10.3390/genes9110564
APA StylePisani, F. M., Napolitano, E., Napolitano, L. M. R., & Onesti, S. (2018). Molecular and Cellular Functions of the Warsaw Breakage Syndrome DNA Helicase DDX11. Genes, 9(11), 564. https://doi.org/10.3390/genes9110564