Exploration of Survival Traits, Probiotic Determinants, Host Interactions, and Functional Evolution of Bifidobacterial Genomes Using Comparative Genomics


Abstract
1. Introduction
2. Materials and Methods
2.1. Retrieval of Genome Sequences
2.2. Pan-Genome Analyses
2.3. Selection Pressure Analyses
2.4. Genome-Size Variation and Mobilome Analyses
2.5. Functional Analyses
2.6. Interspecific Interactions between Human and Bifidobacteria
2.7. Interactome Analysis of Bifidobacterial Core Proteins
3. Results
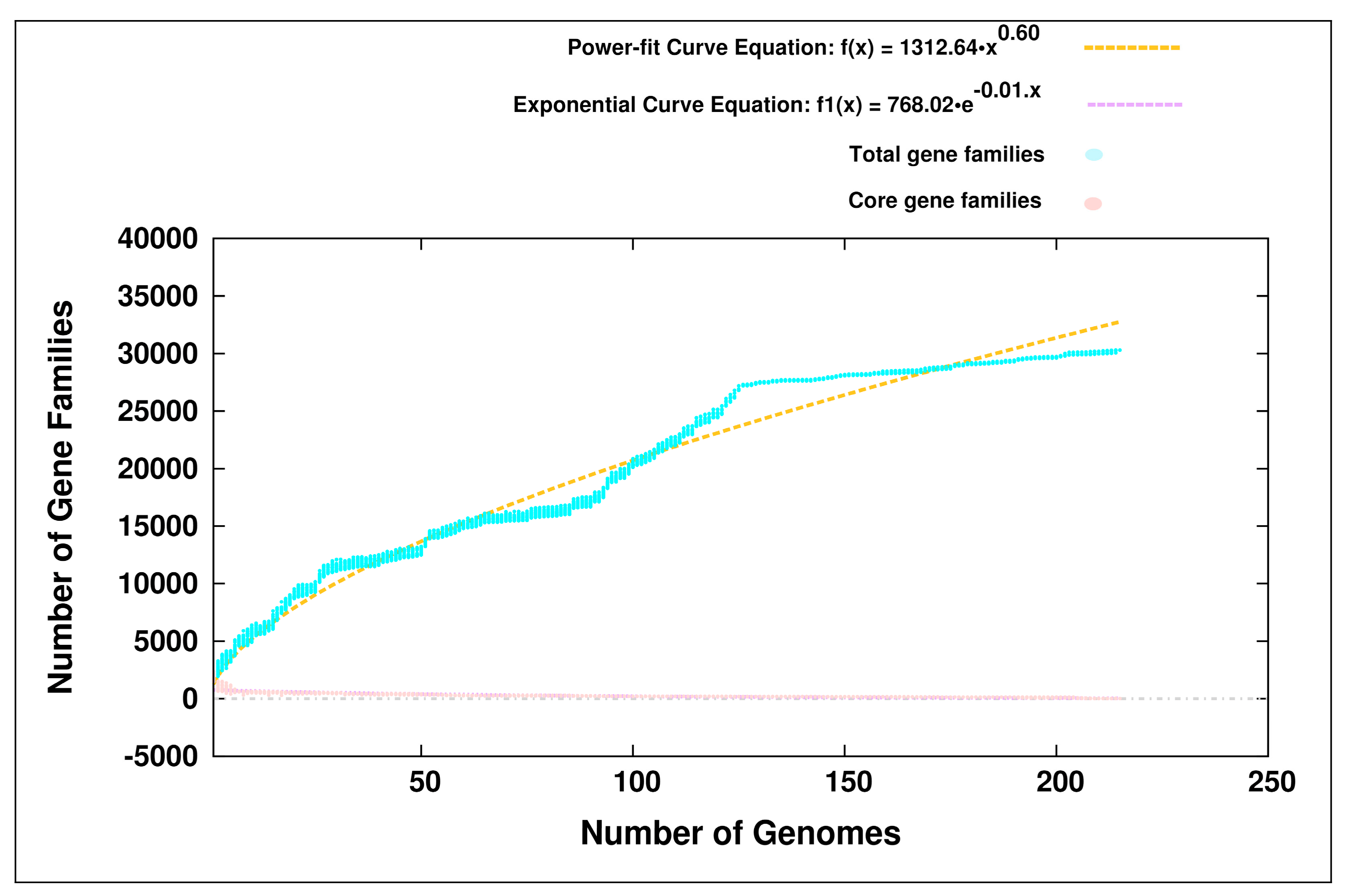
3.1. Pan-Genome Analyses
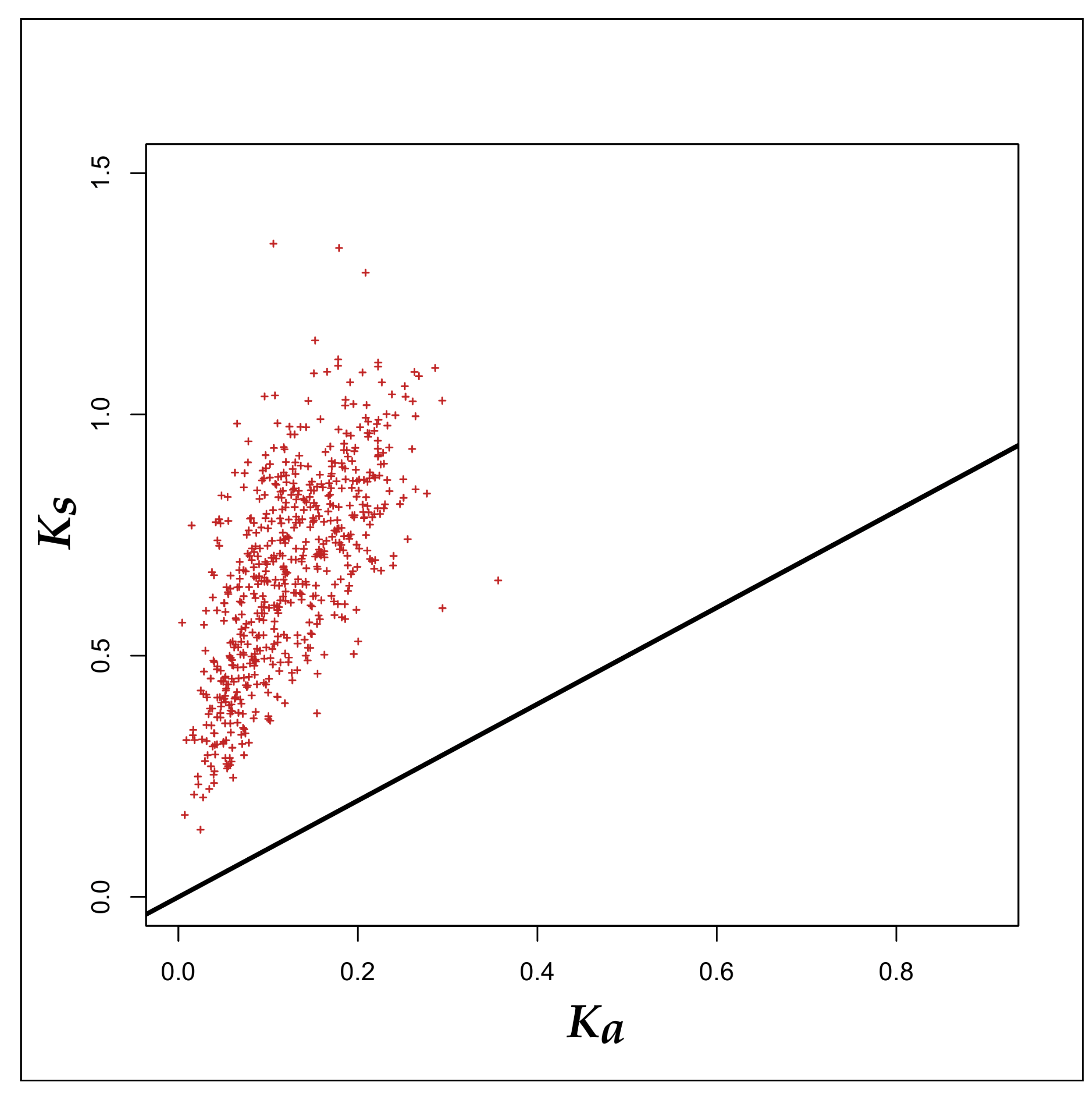
3.2. Selection Pressure Analyses
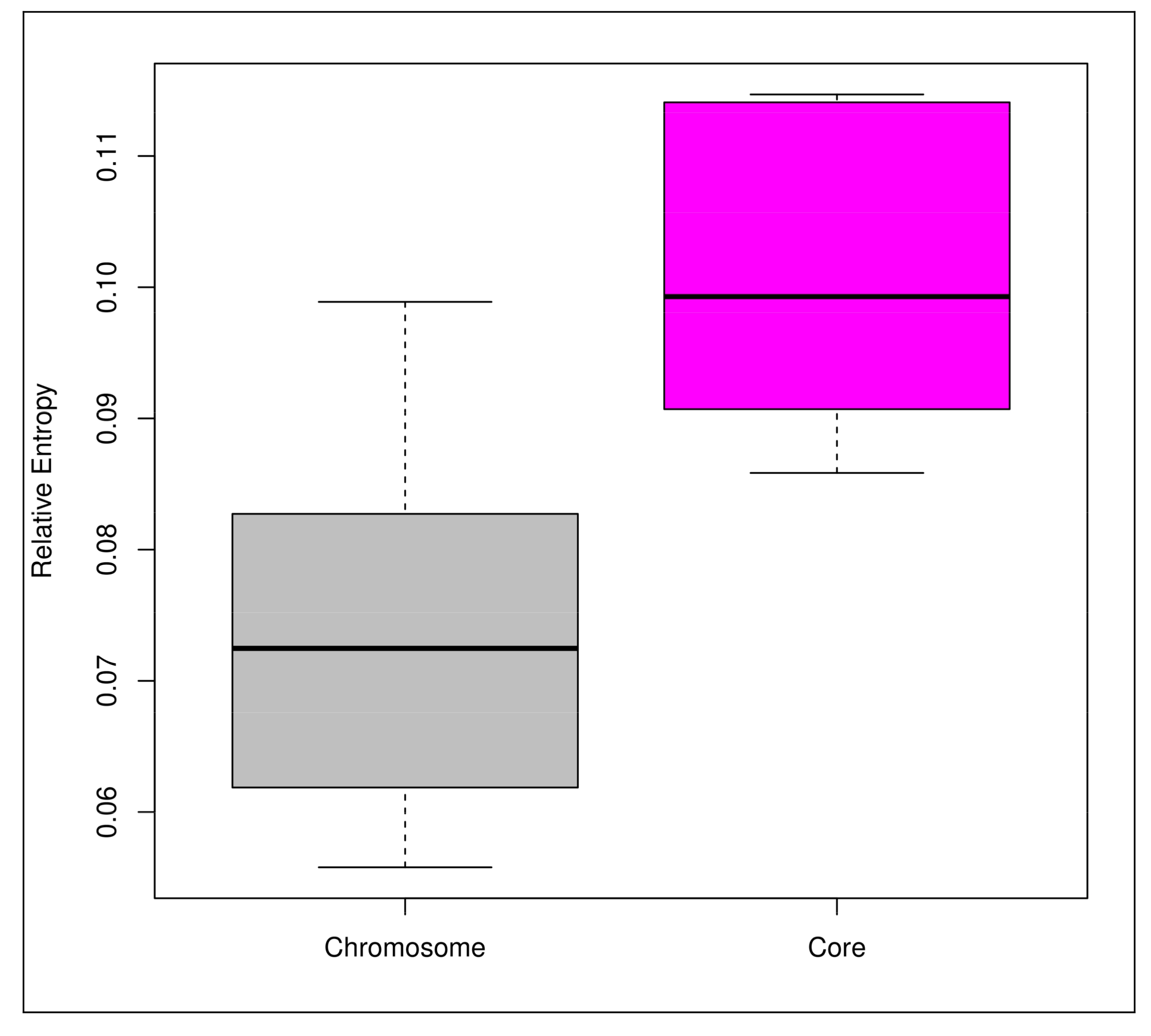
3.3. Mobilome Analyses
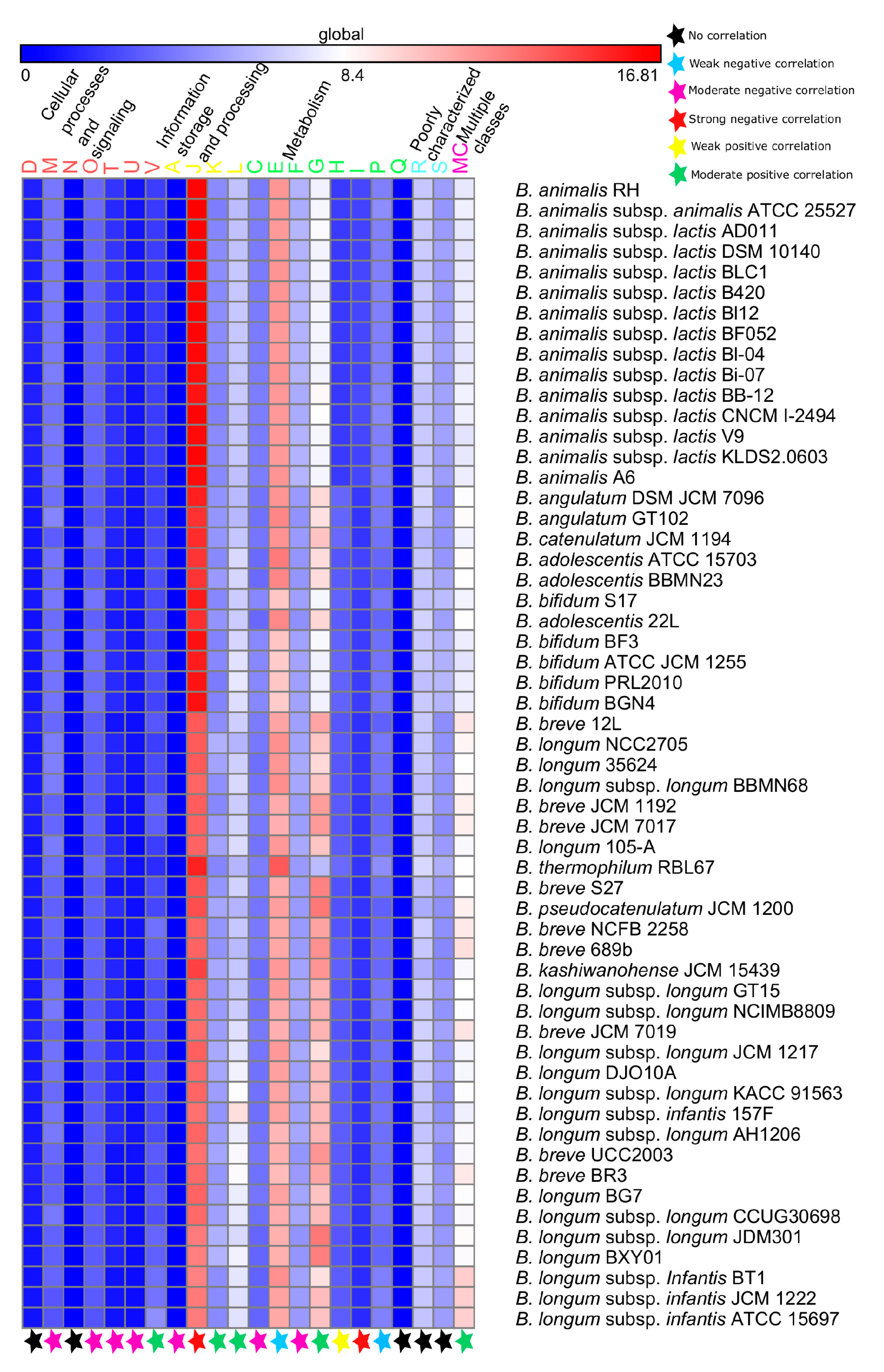
3.4. Functional Analyses
3.4.1. Probiotic-Traits
3.4.2. Survival-Strategies
3.5. In Silico Protein-Protein Interaction Analyses
4. Discussion
4.1. Open Pan-Genomes of the Genus Bifidobacterium and its Probiotic and Human-Gut Strains
4.2. Survival- and Probiotic-Traits of the Probiotic and Human-Gut Strains of Bifidobacteria
4.3. Conserved Protein-Protein Interactions of the Human Host and the Probiotic and Human-Gut Strains of Bifidobacteria
4.4. Functional Evolution Versus Genome-Size Variations among the Probiotic and Human-Gut Strains of Bifidobacteria
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. Genbank. Nucleic Acids Res. 2012, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Arboleya, S.; Watkins, C.; Stanton, C.; Ross, R.P. Gut bifidobacteria populations in human health and aging. Front. Microbiol. 2016, 7, 1204. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Mandal, S. Bifidobacteria—Insight into clinical outcomes and mechanisms of its probiotic action. Microbiol. Res. 2016, 192, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Pinto-Sánchez, M.I.; Smecuol, E.C.; Temprano, M.P.; Sugai, E.; González, A.; Moreno, M.L.; Huang, X.; Bercik, P.; Cabanne, A.; Vázquez, H. Bifidobacterium infantis NLS super strain reduces the expression of α-defensin-5, a marker of innate immunity, in the mucosa of active celiac disease patients. J. Clin. Gastroenterol. 2017, 51, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, B.; Champomier-Verges, M.-C.; del Carmen Collado, M.; Anglade, P.; Baraige, F.; Sanz, Y.; Clara, G.; Margolles, A.; Zagorec, M. Low-pH adaptation and the acid tolerance response of Bifidobacterium longum biotype longum. Appl. Environ. Microbiol. 2007, 73, 6450–6459. [Google Scholar] [CrossRef] [PubMed]
- Bruni, N.; Capucchio, M.T.; Biasibetti, E.; Pessione, E.; Cirrincione, S.; Giraudo, L.; Corona, A.; Dosio, F. Antimicrobial activity of lactoferrin-related peptides and applications in human and veterinary medicine. Molecules 2016, 21, 752. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, K.; Yamamoto, Y.; Sugiyama, M.; Takaki, T.; Urashima, T.; Fukiya, S.; Yokota, A.; Okada, N.; Mukai, T. Bifidobacterium bifidum extracellular sialidase enhances adhesion to the mucosal surface and supports carbohydrate assimilation. MBio 2017, 8, e00928-17. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Lugli, G.A.; Duranti, S.; Turroni, F.; Bottacini, F.; Mangifesta, M.; Sanchez, B.; Viappiani, A.; Mancabelli, L.; Taminiau, B. Genome encyclopaedia of type strains of the genus Bifidobacterium. Appl. Environ. Microbiol. 2014, 80, 6290–6302. [Google Scholar] [CrossRef] [PubMed]
- Bottacini, F.; Medini, D.; Pavesi, A.; Turroni, F.; Foroni, E.; Riley, D.; Giubellini, V.; Tettelin, H.; van Sinderen, D.; Ventura, M. Comparative genomics of the genus Bifidobacterium. Microbiology 2010, 156, 3243–3254. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Milani, C.; Turroni, F.; Duranti, S.; Ferrario, C.; Viappiani, A.; Mancabelli, L.; Mangifesta, M.; Taminiau, B.; Delcenserie, V. Investigation of the evolutionary development of the genus Bifidobacterium by comparative genomics. Appl. Environ. Microbiol. 2014, 80, 6383–6394. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, W.; Guo, C.; Yang, X.; Liu, W.; Wu, Y.; Song, Y.; Kwok, L.Y.; Cui, Y.; Menghe, B. Comparative genomic analysis of 45 type strains of the genus Bifidobacterium: A snapshot of its genetic diversity and evolution. PLoS ONE 2015, 10, e0117912. [Google Scholar] [CrossRef] [PubMed]
- Satti, M.; Tanizawa, Y.; Endo, A.; Arita, M. Comparative analysis of probiotic bacteria based on a new definition of core genome. J. Bioinform. Comput. Biol. 2018, 16, 1840012. [Google Scholar] [CrossRef] [PubMed]
- Bottacini, F.; Motherway, M.O.C.; Kuczynski, J.; O’Connell, K.J.; Serafini, F.; Duranti, S.; Milani, C.; Turroni, F.; Lugli, G.A.; Zomer, A. Comparative genomics of the Bifidobacterium breve taxon. BMC Genom. 2014, 15, 170. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, A.; Bottacini, F.; Motherway, M.C.; Van Sinderen, D. Pangenome analysis of Bifidobacterium longum and site-directed mutagenesis through by-pass of restriction-modification systems. BMC Genom. 2015, 16, 832. [Google Scholar] [CrossRef] [PubMed]
- Duranti, S.; Milani, C.; Lugli, G.A.; Turroni, F.; Mancabelli, L.; Sanchez, B.; Ferrario, C.; Viappiani, A.; Mangifesta, M.; Mancino, W. Insights from genomes of representatives of the human gut commensal Bifidobacterium bifidum. Appl. Environ. Microbiol. 2015, 17, 2515–2531. [Google Scholar]
- Milani, C.; Duranti, S.; Lugli, G.A.; Bottacini, F.; Strati, F.; Arioli, S.; Foroni, E.; Turroni, F.; van Sinderen, D.; Ventura, M. Comparative genomics of Bifidobacterium animalis subsp. lactis reveals a strict monophyletic bifidobacterial taxon. Appl. Environ. Microbiol. 2013, 79, 4304–4315. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zhang, C.; Wu, H.; Wang, R.; Shen, J.; Wang, L.; Zhao, Y.; Pang, X.; Zhang, X.; Zhao, L. Genomic microdiversity of Bifidobacterium pseudocatenulatum underlying differential strain-level responses to dietary carbohydrate intervention. MBio 2017, 8, e02348-16. [Google Scholar] [CrossRef] [PubMed]
- Duranti, S.; Milani, C.; Lugli, G.A.; Mancabelli, L.; Turroni, F.; Ferrario, C.; Mangifesta, M.; Viappiani, A.; Sánchez, B.; Margolles, A. Evaluation of genetic diversity among strains of the human gut commensal Bifidobacterium adolescentis. Sci. Rep. 2016, 6, 23971. [Google Scholar] [CrossRef] [PubMed]
- Maury, M.M.; Tsai, Y.-H.; Charlier, C.; Touchon, M.; Chenal-Francisque, V.; Leclercq, A.; Criscuolo, A.; Gaultier, C.; Roussel, S.; Brisabois, A. Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nat. Genet. 2016, 48, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Qin, Y.; Wang, Y.; Li, H.; Shang, N.; Li, P. Complete genome sequence of Bifidobacterium animalis RH, a probiotic bacterium producing exopolysaccharides. J. Biotechnol. 2014, 189, 86–87. [Google Scholar] [CrossRef] [PubMed]
- Note, G. Evaluation of selective media for enumeration of Lactobacillus acidophilus and Bifidobacterium animalis present in probiotic formulations. Med. Sci. 2016, 20, 104–109. [Google Scholar]
- Stahl, B.; Barrangou, R. Complete genome sequences of probiotic strains Bifidobacterium animalis subsp. lactis B420 and Bi-07. J. Bacteriol. 2012, 194, 4131–4132. [Google Scholar] [CrossRef] [PubMed]
- Charnchai, P.; Jantama, S.S.; Jantama, K. Genome analysis of food-processing stressful-resistant probiotic Bifidobacterium animalis subsp. lactis BF052, and its potential application in fermented soymilk. FEMS Microbiol. Lett. 2017, 364. [Google Scholar] [CrossRef] [PubMed]
- Chervaux, C.; Grimaldi, C.; Bolotin, A.; Quinquis, B.; Legrain-Raspaud, S.; van Hylckama Vlieg, J.E.; Denariaz, G.; Smokvina, T. Genome sequence of the probiotic strain Bifidobacterium animalis subsp. lactis CNCM I-2494. J. Bacteriol. 2011, 193, 5560–5561. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Sun, Y.; Huo, G.-C.; Yang, L.; Liu, F.; Li, A.; Meng, X.-C. Complete genome sequence of Bifidobacterium animalis subsp. lactis KLDS 2.0603, a probiotic strain with digestive tract resistance and adhesion to the intestinal epithelial cells. J. Biotechnol. 2016, 220, 49–50. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.; Zhao, L.; Ren, F.; Liu, S.; Zhang, M.; Guo, H. Complete genome sequence of Bifidobacterium animalis subsp. lactis A6, a probiotic strain with high acid resistance ability. J. Biotechnol. 2015, 200, 8–9. [Google Scholar] [CrossRef] [PubMed]
- El-Alry, M.; Shenana, M.; El-Nagar, G.; Atallah, A. Growth and survival of some probiotic bacteria under manufacturing conditions of yoghurt and ras cheese. Egypt. J. Dairy Sci. 2012, 40, 123–133. [Google Scholar]
- Liu, S.; Zhao, L.; Ren, F.; Sun, E.; Zhang, M.; Guo, H. Complete genome sequence of Bifidobacterium adolesentis BBMN23, a probiotic strain from healthy centenarian. J. Biotechnol. 2015, 198, 44–45. [Google Scholar] [CrossRef] [PubMed]
- Duranti, S.; Turroni, F.; Lugli, G.A.; Milani, C.; Viappiani, A.; Mangifesta, M.; Gioiosa, L.; Palanza, P.; van Sinderen, D.; Ventura, M. Genomic characterization and transcriptional studies of the starch-utilizing Bifidobacterium adolescentis 22L. Appl. Environ. Microbiol. 2014, 80, 6080–6090. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-H.; Chung, M.-J.; Seo, J.-G. A multistrain probiotic formulation attenuates skin symptoms of atopic dermatitis in a mouse model through the generation of CD4+ Foxp3+ T cells. Food Nutr. Res. 2016, 60, 32550. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-G.; Xu, H.-B.; Xu, F.; Zeng, Z.-L.; Wei, H. Efficacy of oral Bifidobacterium bifidum ATCC 29521 on microflora and antioxidant in mice. Can. J. Microbiol. 2015, 62, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.; Park, M.S.; Ji, G.E.; You, H.J. Review on Bifidobacterium bifidum BGN4: Functionality and nutraceutical applications as a probiotic microorganism. Int. J. Mol. Sci. 2016, 17, 1544. [Google Scholar] [CrossRef] [PubMed]
- Altmann, F.; Kosma, P.; O’Callaghan, A.; Leahy, S.; Bottacini, F.; Molloy, E.; Plattner, S.; Schiavi, E.; Gleinser, M.; Groeger, D. Genome analysis and characterisation of the exopolysaccharide produced by Bifidobacterium longum subsp. longum 35624™. PLoS ONE 2016, 11, e0162983. [Google Scholar] [CrossRef] [PubMed]
- Tahoun, A.; Masutani, H.; El-Sharkawy, H.; Gillespie, T.; Honda, R.P.; Kuwata, K.; Inagaki, M.; Yabe, T.; Nomura, I.; Suzuki, T. Capsular polysaccharide inhibits adhesion of Bifidobacterium longum 105-A to enterocyte-like Caco-2 cells and phagocytosis by macrophages. Gut Pathog. 2017, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Coakley, M.; Ross, R.; Nordgren, M.; Fitzgerald, G.; Devery, R.; Stanton, C. Conjugated linoleic acid biosynthesis by human-derived Bifidobacterium species. J. Appl. Microbiol. 2003, 94, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Zakharevich, N.V.; Averina, O.V.; Klimina, K.M.; Kudryavtseva, A.V.; Kasianov, A.S.; Makeev, V.J.; Danilenko, V.N. Complete genome sequence of Bifidobacterium longum GT15: Identification and characterization of unique and global regulatory genes. Microb. Ecol. 2015, 70, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Gómez, M.X.; Martínez, I.; Bottacini, F.; O’Callaghan, A.; Ventura, M.; van Sinderen, D.; Hillmann, B.; Vangay, P.; Knights, D.; Hutkins, R.W. Stable engraftment of Bifidobacterium longum AH1206 in the human gut depends on individualized features of the resident microbiome. Cell. Host Microbe 2016, 20, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.-J.; Yoon, J.-K.; Kwon, S.-K.; Chung, M.-J.; Seo, J.-G.; Kim, J.F. Complete genome sequence of the probiotic bacterium Bifidobacterium breve KCTC 12201BP isolated from a healthy infant. J. Biotechnol. 2015, 214, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.-K.; Kwak, M.-J.; Seo, J.-G.; Chung, M.J.; Kim, J.F. Complete genome sequence of Bifidobacterium longum KCTC 12200BP, a probiotic strain promoting the intestinal health. J. Biotechnol. 2015, 214, 169–170. [Google Scholar] [CrossRef] [PubMed]
- Rodes, L.; Saha, S.; Tomaro-Duchesneau, C.; Prakash, S. Microencapsulated Bifidobacterium longum subsp. infantis ATCC 15697 favorably modulates gut microbiota and reduces circulating endotoxins in F344 rats. BioMed Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Bohlin, J.; van Passel, M.W.; Snipen, L.; Kristoffersen, A.B.; Ussery, D.; Hardy, S.P. Relative entropy differences in bacterial chromosomes, plasmids, phages and genomic islands. BMC Genom. 2012, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Peterson, D.; Tamura, K. MEGA-CC: Computing core of molecular evolutionary genetics analysis program for automated and iterative data analysis. Bioinformatics 2012, 28, 2685–2686. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar] [PubMed]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. Kaks_calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Leimbach, A. Bac-Genomics-Scripts. Available online: https://github.com/aleimba/bac-genomics-scripts (accessed on 9 March 2018).
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Hsiao, W.W.; Brinkman, F.S. Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinform. 2008, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Waack, S.; Keller, O.; Asper, R.; Brodag, T.; Damm, C.; Fricke, W.F.; Surovcik, K.; Meinicke, P.; Merkl, R. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinform. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, W.; Wan, I.; Jones, S.J.; Brinkman, F.S. IslandPath: Aiding detection of genomic islands in prokaryotes. Bioinformatics 2003, 19, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Varani, A.M.; Siguier, P.; Gourbeyre, E.; Charneau, V.; Chandler, M. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 2011, 12, R30. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Gould, J. GENE-E. Available online: https://www.broadinstitute.org/cancer/software/GENE-E/index.html (accessed on 9 March 2018).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, Z.; Fu, L.; Niu, B.; Li, W. WebMGA: A customizable web server for fast metagenomic sequence analysis. BMC Genom. 2011, 12, 444. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.S.; Lin, C.J.; Hwang, J.K. Predicting subcellular localization of proteins for gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004, 13, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, J.D.; Kiemer, L.; Fausbøll, A.; Brunak, S. Non-classical protein secretion in bacteria. BMC Microbiol. 2005, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Juncker, A.S.; Willenbrock, H.; Von Heijne, G.; Brunak, S.; Nielsen, H.; Krogh, A. Prediction of lipoprotein signal peptides in gram-negative bacteria. Protein Sci. 2003, 12, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H., Jr.; Reddy, V.S.; Tsu, B.V.; Ahmed, M.S.; Li, C.; Moreno-Hagelsieb, G. The transporter classification database (TCDB): Recent advances. Nucleic Acids Res. 2015, 44, D372–D379. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Smith, J.; Lam, M.; Zemla, A.; Dyer, M.D.; Slezak, T. MvirDB—A microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res. 2006, 35, D391–D394. [Google Scholar] [CrossRef] [PubMed]
- Eichinger, V.; Nussbaumer, T.; Platzer, A.; Jehl, M.-A.; Arnold, R.; Rattei, T. EffectiveDB—Updates and novel features for a better annotation of bacterial secreted proteins and type III, IV, VI secretion systems. Nucleic Acids Res. 2015, 44, D669–D674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ou, H.Y.; Zhang, C.T. Deg: A database of essential genes. Nucleic Acids Res. 2004, 32, D271–D272. [Google Scholar] [CrossRef] [PubMed]
- Barakat, M.; Ortet, P.; Whitworth, D.E. P2RP: A web-based framework for the identification and analysis of regulatory proteins in prokaryotic genomes. BMC Genom. 2013, 14, 269. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Míguez, A.; Gutiérrez-Jácome, A.; Fdez-Riverola, F.; Lourenço, A.; Sánchez, B. MAHMI database: A comprehensive metaHit-based resource for the study of the mechanism of action of the human microbiota. Database 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, J.; Schleker, S.; Klein-Seetharaman, J.; Oliva, B. BIPS: BIANA interolog prediction server. A tool for protein-protein interaction inference. Nucleic Acids Res. 2012, 40, W147–W151. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2014, 43, D204–D212. [Google Scholar] [CrossRef] [PubMed]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.; Brinkman, F.S.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond—Recent updates and continuing curation. Nucleic Acids Res. 2012, 41, D1228–D1233. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.R.; Otasek, D.; Ali, M.; McGuffin, M.J.; Xie, W.; Devani, B.; Toch, I.L.v.; Jurisica, I. NAViGaTOR: Network analysis, visualization and graphing toronto. Bioinformatics 2009, 25, 3327–3329. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein—Protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45. [Google Scholar] [CrossRef] [PubMed]
- Carlos Guimaraes, L.; Benevides de Jesus, L.; Vinicius Canario Viana, M.; Silva, A.; Thiago Juca Ramos, R.; de Castro Soares, S.; Azevedo, V. Inside the pan-genome-methods and software overview. Curr. Genom. 2015, 16, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.M.; Ji, G.E.; Cho, S.H.; Park, M.S.; Lee, H.J. Characterization of a Bifidobacterium longum BORI dipeptidase belonging to the U34 family. Appl. Environ. Microbiol. 2007, 73, 5598–5606. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]
- Öner, Ö.; Aslim, B.; Aydaş, S.B. Mechanisms of cholesterol-lowering effects of lactobacilli and bifidobacteria strains as potential probiotics with their bsh gene analysis. J. Mol. Microbiol. Biotechnol. 2014, 24, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Pokusaeva, K.; Fitzgerald, G.F.; Sinderen, D. Carbohydrate metabolism in bifidobacteria. Genes Nutr. 2011, 6, 285–306. [Google Scholar] [CrossRef] [PubMed]
- El Kaoutari, A.; Armougom, F.; Gordon, J.I.; Raoult, D.; Henrissat, B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013, 11, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, L.; Delgado, S.; Ruas-Madiedo, P.; Sánchez, B.; Margolles, A. Bifidobacteria and their molecular communication with the immune system. Front. Microbiol. 2017, 8, 2345. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Ma, C.; Ye, G.; Shi, Y.; Xu, W.; Zhong, L.; Wang, J.; Yin, Y.; Zhang, X.; Wang, H. DnaJ (hsp40) of Streptococcus pneumoniae is involved in bacterial virulence and elicits a strong natural immune reaction via PI3K/JNK. Mol. Immunol. 2017, 83, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Koliński, T.; Marek-Trzonkowska, N.; Trzonkowski, P.; Siebert, J. Heat shock proteins (HSPs) in the homeostasis of regulatory T cells (Tregs). Cent. Eur. J. Immunol. 2016, 41, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, C.A.; Hynes, R.O. Distribution and evolution of von willebrand/integrin a domains: Widely dispersed domains with roles in cell adhesion and elsewhere. Mol. Biol. Cell 2002, 13, 3369–3387. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Kenny, J.G.; Zhang, Z.; Fitzgerald, G.F.; van Sinderen, D. The clpB gene of Bifidobacterium breve UCC 2003: Transcriptional analysis and first insights into stress induction. Microbiology 2005, 151, 2861–2872. [Google Scholar] [CrossRef] [PubMed]
- Moussatova, A.; Kandt, C.; O'Mara, M.L.; Tieleman, D.P. ATP-binding cassette transporters in Escherichia coli. Biochim. Biophys. Acta -Biomembranes 2008, 1778, 1757–1771. [Google Scholar] [CrossRef] [PubMed]
- Krastel, K.; Senadheera, D.B.; Mair, R.; Downey, J.S.; Goodman, S.D.; Cvitkovitch, D.G. Characterization of a glutamate transporter operon, glnQHMP, in Streptococcus mutans and its role in acid tolerance. J. Bacteriol. 2010, 192, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, M.G.; Wanner, B.L.; Crépin, S.; Harel, J. The phosphate regulon and bacterial virulence: A regulatory network connecting phosphate homeostasis and pathogenesis. FEMS Microbiol. Rev. 2008, 32, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Green, E.R.; Mecsas, J. Bacterial secretion systems—An overview. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Driessen, A.J.; Nouwen, N. Protein translocation across the bacterial cytoplasmic membrane. Annu. Rev. Biochem. 2008, 77, 643–667. [Google Scholar] [CrossRef] [PubMed]
- Breidenstein, E.B.; Janot, L.; Strehmel, J.; Fernandez, L.; Taylor, P.K.; Kukavica-Ibrulj, I.; Gellatly, S.L.; Levesque, R.C.; Overhage, J.; Hancock, R.E. The Lon protease is essential for full virulence in Pseudomonas aeruginosa. PLoS ONE 2012, 7, e49123. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; He, X.; Brancaccio, V.F.; Yuan, J.; Riedel, C.U. Bifidobacteria exhibit LuxS-dependent autoinducer 2 activity and biofilm formation. PLoS ONE 2014, 9, e88260. [Google Scholar] [CrossRef] [PubMed]
- Aseev, L.; Boni, I. Extraribosomal functions of bacterial ribosomal proteins. Mol. Biol. 2011, 45, 739. [Google Scholar] [CrossRef]
- Melnikov, S.; Manakongtreecheep, K.; Söll, D. Revising the structural diversity of ribosomal proteins across the three domains of life. Mol. Biol. Evol. 2018, 35, 1588–1598. [Google Scholar] [CrossRef] [PubMed]
- Kenley, E.C.; Kirk, L.; Cho, Y.-R. Differentiating party and date hubs in protein interaction networks using semantic similarity measures. In Proceedings of the 2nd ACM Conference on Bioinformatics, Computational Biology and Biomedicine, Chicago, IL, USA, 1–3 August 2011; pp. 641–645. [Google Scholar]
- Rouli, L.; Merhej, V.; Fournier, P.-E.; Raoult, D. The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 2015, 7, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Mobeen, F.; Prakash, T. Comparative genomics of herpesviridae family to look for potential signatures of human infecting strains. Int. J. Genom. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Duranti, S.; Bottacini, F.; Guglielmetti, S.; Van Sinderen, D.; Ventura, M. Bifidobacterium bifidum as an example of a specialized human gut commensal. Front. Microbiol. 2014, 5, 437. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, K.S.; Gierasch, L.M. How bacteria survive an acid trip. Proc. Natl. Acad. Sci. USA 2013, 110, 5279–5280. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.S.; Chen, W.; Jin, J.; Tai, P.C.; Wang, B. SecA: A potential antimicrobial target. Future Med. Chem. 2015, 7, 989–1007. [Google Scholar] [CrossRef] [PubMed]
- Bensing, B.A.; Seepersaud, R.; Yen, Y.T.; Sullam, P.M. Selective transport by SecA2: An expanding family of customized motor proteins. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1674–1686. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, S.K.; Chowdhury, I.; Singh, R. Understanding the mechanism of bacterial biofilms resistance to antimicrobial agents. Open Microbiol. J. 2017, 11, 53. [Google Scholar] [CrossRef] [PubMed]
- Licandro-Seraut, H.; Scornec, H.; Pédron, T.; Cavin, J.-F.; Sansonetti, P.J. Functional genomics of Lactobacillus casei establishment in the gut. Proc. Natl. Acad. Sci. USA 2014, 111, 3101–3109. [Google Scholar] [CrossRef] [PubMed]
- Abby, S.S.; Tannier, E.; Gouy, M.; Daubin, V. Lateral gene transfer as a support for the tree of life. Proc. Natl. Acad. Sci. USA 2012, 109, 4962–4967. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Hang, X.; Zhang, M.; Liu, X.; Yang, H. Analysis of newly detected tetracycline resistance genes and their flanking sequences in human intestinal bifidobacteria. Sci. Rep. 2017, 7, 6267. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Hill, C.; Gahan, C.G. Bile salt hydrolase activity in probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Braga, R.M.; Dourado, M.N.; Araújo, W.L. Microbial interactions: Ecology in a molecular perspective. Braz. J. Microbiol. 2016, 47, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Rottner, K.; Stradal, T.E.; Wehland, J. Bacteria-host-cell interactions at the plasma membrane: Stories on actin cytoskeleton subversion. Dev. Cell 2005, 9, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.L.; Chan, J. Immunology of tuberculosis. Annu. Rev. Immunol. 2001, 19, 93–129. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The Self-Assembly and Dynamic Structure of Cytoskeletal Filaments, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Kast, D.J.; Dominguez, R. The cytoskeleton–autophagy connection. Curr. Biol. 2017, 27, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Chorell, E.; Andersson, E.; Evans, M.L.; Jain, N.; Götheson, A.; Åden, J.; Chapman, M.R.; Almqvist, F.; Wittung-Stafshede, P. Bacterial chaperones CsgE and CsgC differentially modulate human α-synuclein amyloid formation via transient contacts. PLoS ONE 2015, 10, e0140194. [Google Scholar] [CrossRef] [PubMed]
- Soufi, B.; Soares, N.C.; Ravikumar, V.; Macek, B. Proteomics reveals evidence of cross-talk between protein modifications in bacteria: Focus on acetylation and phosphorylation. Curr. Opin. Microbiol. 2012, 15, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Modell, A.E.; Blosser, S.L.; Arora, P.S. Systematic targeting of protein-protein interactions. Trends Pharmacol. Sci. 2016, 37, 702–713. [Google Scholar] [CrossRef]
- Bojadzic, D.; Chen, J.; Alcazar, O.; Buchwald, P. Design, synthesis, and evaluation of novel immunomodulatory small molecules targeting the CD40–CD154 costimulatory protein-protein interaction. Molecules 2018, 23, 1153. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.; Medzhitov, R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 2011, 140, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Doria, M.; Klein, N.; Lucito, R.; Schneider, R. The hepatitis B virus HBx protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of transcription factors. EMBO J. 1995, 14, 4747–4757. [Google Scholar] [CrossRef] [PubMed]
- Meusel, T.R.; Imani, F. Viral induction of inflammatory cytokines in human epithelial cells follows a p38 mitogen-activated protein kinase-dependent but NF-κB-independent pathway. J. Immunol. 2003, 171, 3768–3774. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.-T.; Liu, H.J. PI3K-Akt signaling and viral infection. Recent Pat. Biotechnol. 2008, 2, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fernandez, M.E.; Rueda, C.M.; Velilla, P.A.; Rugeles, M.T.; Chougnet, C.A. cAMP during HIV infection: Friend or foe? AIDS Res. Hum. Retrovirus. 2012, 28, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Kamranvar, S.A.; Masucci, M.G. Regulation of telomere homeostasis during Epstein-Barr virus infection and immortalization. Viruses 2017, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Tzahar, E.; Moyer, J.D.; Waterman, H.; Barbacci, E.G.; Bao, J.; Levkowitz, G.; Shelly, M.; Strano, S.; Pinkas-Kramarski, R.; Pierce, J.H. Pathogenic poxviruses reveal viral strategies to exploit the ErbB signaling network. EMBO J. 1998, 17, 5948–5963. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.B. Viral inhibition of the IFN-induced JAK/STAT signalling pathway: Development of live attenuated vaccines by mutation of viral-encoded IFN-antagonists. Vaccines 2016, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Schaulies, J.; Schneider-Schaulies, S. Sphingolipids in viral infection. J. Biol. Chem. 2015, 396, 585–595. [Google Scholar]
- Willeaume, V.; Kruys, V.; Mijatovic, T.; Huez, G. Tumor necrosis factor-alpha production induced by viruses and by lipopolysaccharides in macrophages: Similarities and differences. J. Inflamm. 1995, 46, 1–12. [Google Scholar] [PubMed]
- Cui, M.; Huang, Y.; Zhao, Y.; Zheng, J. New insights for FOXO and cell-fate decision in HIV infection and HIV associated neurocognitive disorder. Adv. Exp. Med. Biol. 2009, 665, 143–159. [Google Scholar] [PubMed]
- Zuylen, W.J.; Rawlinson, W.D.; Ford, C.E. The Wnt pathway: A key network in cell signalling dysregulated by viruses. Rev. Med. Virol. 2016, 26, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Kant, M.; AKGÜL, B.; ALDANMAZ, A.N. Endogenous heat shock protein GRoEL of A. actinomycetemcomitans preferentially targets primary human CD8+ T cells. Turk. J. Biol. 2015, 39, 104–110. [Google Scholar] [CrossRef]
- Dobbin, C.A.; Smith, N.C.; Johnson, A.M. Heat shock protein 70 is a potential virulence factor in murine toxoplasma infection via immunomodulation of host NF-κB and nitric oxide. J. Immunol. 2002, 169, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Honda-Ogawa, M.; Sumitomo, T.; Mori, Y.; Hamd, D.T.; Ogawa, T.; Yamaguchi, M.; Nakata, M.; Kawabata, S. Streptococcus pyogenes endopeptidase O contributes to evasion from complement-mediated bacteriolysis via binding to human complement factor C1q. J. Biol. Chem. 2017, 292, 4244–4254. [Google Scholar] [CrossRef] [PubMed]
- Querol-García, J.; Fernández, F.J.; Marin, A.V.; Gómez, S.; Fullà, D.; Melchor-Tafur, C.; Franco-Hidalgo, V.; Albertí, S.; Juanhuix, J.; Rodríguez de Córdoba, S. Crystal structure of glyceraldehyde-3-phosphate dehydrogenase from the gram-positive bacterial pathogen A. vaginae, an immunoevasive factor that interacts with the human C5a anaphylatoxin. Front. Microbiol. 2017, 8, 541. [Google Scholar] [CrossRef] [PubMed]
- Blom, A.M.; Bergmann, S.; Fulde, M.; Riesbeck, K.; Agarwal, V. Streptococcus pneumoniae phosphoglycerate kinase is a novel complement inhibitor affecting the membrane attack complex formation. J. Biol. Chem. 2014, 289, 32499–32511. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 3160–3165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, X.; He, Q.; Dong, W.; Zhang, X.; Fan, F.; Peng, D.; Huang, W.; Yin, H. Gene turnover contributes to the evolutionary adaptation of Acidithiobacillus caldus: Insights from comparative genomics. Front. Microbiol. 2016, 7, 1960. [Google Scholar] [CrossRef] [PubMed]
- Davids, W.; Zhang, Z. The impact of horizontal gene transfer in shaping operons and protein interaction networks—Direct evidence of preferential attachment. BMC Evol. Biol. 2008, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Kanhere, A.; Vingron, M. Horizontal gene transfers in prokaryotes show differential preferences for metabolic and translational genes. BMC Evol. Biol. 2009, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Ngugi, D.K.; Blom, J.; Stepanauskas, R.; Stingl, U. Diversification and niche adaptations of Nitrospina-like bacteria in the polyextreme interfaces of Red Sea brines. ISME J. 2016, 10, 1383. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, P.; Bottacini, F.; Mahony, J.; Kilcawley, K.N.; van Sinderen, D. Comparative and functional genomics of the Lactococcus lactis taxon; insights into evolution and niche adaptation. BMC Genom. 2017, 18, 267. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.; Duranti, S.; Milani, C.; Mancabelli, L.; Lugli, G.A.; Turroni, F.; Mangifesta, M.; Viappiani, A.; Ossiprandi, M.C.; van Sinderen, D. Exploring amino acid auxotrophy in Bifidobacterium bifidum PRL2010. Front. Microbiol. 2015, 6, 1331. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Gómez, B.; Fernàndez-Guerra, A.; Casamayor, E.O.; González, J.M.; Pedrós-Alió, C.; Acinas, S.G. Patterns and architecture of genomic islands in marine bacteria. BMC Genom. 2012, 13, 347. [Google Scholar] [CrossRef] [PubMed]
- Touchon, M.; Rocha, E.P. Causes of insertion sequences abundance in prokaryotic genomes. Mol. Biol. Evol. 2007, 24, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Touchon, M.; Bernheim, A.; Rocha, E.P. Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J. 2016, 10, 2744. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly | Organism/Name | Strain | Isolation Source | Probiotic Potential [Reference] |
|---|---|---|---|---|
| GCA_000695895.1 | B. animalis RH | RH | Feces * | Yes [20] |
| GCA_000260715.1 | B. animalis subsp. animalis ATCC 25527 | ATCC 25527 | Sewage | Yes [21] |
| GCA_000021425.1 | B. animalis subsp. lactis AD011 | AD011 | Infant fecal sample * | Yes [3] |
| GCA_000022965.1 | B. animalis subsp. lactis DSM 10140 | DSM 10140 | Commercially available probiotic strain | Yes [3] |
| GCA_000224965.2 | B. animalis subsp. lactis BLC1 | BLC1 | Commercially available probiotic strain | Yes [3] |
| GCA_000277325.1 | B. animalis subsp. lactis B420 | B420 | Commercially available probiotic strain | Yes [22] |
| GCA_000414215.1 | B. animalis subsp. lactis Bl12 | Bl12 | Colonoscopic sample * | No |
| GCA_000818055.1 | B. animalis subsp. lactis BF052 | BF052 | Feces of breast-fed infant * | Yes [23] |
| GCA_000022705.1 | B. animalis subsp. lactis Bl-04 | Bl-04; ATCC SD5219 | Fecal sample from a healthy adult * | Yes [3] |
| GCA_000277345.1 | B. animalis subsp. lactis Bi-07 | Bi-07 | Commercially available probiotic strain | Yes [22] |
| GCA_000025245.1 | B. animalis subsp. lactis BB-12 | BB-12 | Commercially available probiotic strain | Yes [3] |
| GCA_000220885.1 | B. animalis subsp. lactis CNCM I-2494 | CNCM I-2494 | Commercially available probiotic strain | Yes [24] |
| GCA_000092765.1 | B. animalis subsp. lactis V9 | V9 | Feces of healthy Mongolian infants * | Yes [3] |
| GCA_000816205.1 | B. animalis subsp. lactis KLDS2.0603 | KLDS2.0603 | Adult feces * | Yes [25] |
| GCA_000817045.1 | B. animalis A6 | A6 | Feces * | Yes [26] |
| GCA_001025155.1 | B. angulatum DSM 20098 = JCM 7096 | JCM 7096 | Human feces * | Yes [27] |
| GCA_000966445.2 | B. angulatum GT102 | GT102 | Feces * | No |
| GCA_001025195.1 | B. catenulatum DSM 16992 = JCM 1194 = LMG 11043 | JCM 1194 | Human feces * | Yes [3] |
| GCA_000010425.1 | B. adolescentis ATCC 15703 | ATCC 15703 | Human adult intestine * | Yes [3] |
| GCA_000817995.1 | B. adolescentis BBMN23 | BBMN23 | Human feces * | Yes [28] |
| GCA_000164965.1 | B. bifidum S17 | S17 | Feces of a breast-fed infant * | Yes [3] |
| GCA_000737885.1 | B. adolescentis 22L | 22L | Milk * | Yes [29] |
| GCA_001281345.1 | B. bifidum BF3 | BF3 | Feces * | Yes [30] |
| GCA_001025135.1 | B. bifidum ATCC 29521 = JCM 1255 = DSM 20456 | JCM 1255 | Stool of breast-fed infant * | Yes [31] |
| GCA_000165905.1 | B. bifidum PRL2010 | PRL2010 | Infant stool samples * | Yes [3] |
| GCA_000265095.1 | B. bifidum BGN4 | BGN4 | Human feces * | Yes [32] |
| GCA_000568955.1 | B. breve 12L | 12L | Human milk * | No |
| GCA_000007525.1 | B. longum NCC2705 | NCC2705 | Infant feces * | Yes [3] |
| GCA_001719085.1 | B. longum 35624 | 35624 | Ileal mucosa of an individual free of gastrointestinal disease * | Yes [33] |
| GCA_000166315.1 | B. longum subsp. longum BBMN68 | BBMN68 | Long-lived man’s intestinal tract * | Yes [3] |
| GCA_001025175.1 | B. breve DSM 20213 = JCM 1192 | JCM 1192 | Infant feces * | Yes [3] |
| GCA_000568975.1 | B. breve JCM 7017 | JCM 7017 | Infant feces * | No |
| GCA_000829295.1 | B. longum 105-A | 105-A | Human feces * | Yes [34] |
| GCA_000347695.1 | B. thermophilum RBL67 | RBL67 | Baby feces * | Yes [3] |
| GCA_000569075.1 | B. breve S27 | S27 | Infant feces * | No |
| GCA_001025215.1 | B. pseudocatenulatum DSM 20438 = JCM 1200 = LMG 10505 | JCM 1200 | Infant feces * | Yes [3] |
| GCA_000569035.1 | B. breve NCFB 2258 | NCFB 2258 | Infant feces * | Yes [35] |
| GCA_000569055.1 | B. breve 689b | 689b | Infant feces * | No |
| GCA_001042615.1 | B. kashiwanohense JCM 15439 = DSM 21854 | JCM 15439 | Feces of a healthy Japanese infant * | No |
| GCA_000772485.1 | B. longum subsp. longum GT15 | GT15 | The gastrointestinal tract (GIT) of a healthy adult from Central region of Russia * | Yes [36] |
| GCA_001446255.1 | B. longum subsp. longum NCIMB8809 | NCIMB8809 | Stool sample * | Yes [5] |
| GCA_000569015.1 | B. breve JCM 7019 | JCM 7019 | Adult feces * | No |
| GCA_000196555.1 | B. longum subsp. longum JCM 1217 | JCM 1217 | Intestine of adult * | Yes [3] |
| GCA_000008945.1 | B. longum DJO10A | DJO10A | Healthy young adult’s feces * | Yes [3] |
| GCA_000219455.1 | B. longum subsp. longum KACC 91563 | KACC 91563 | Feces of neonates * | Yes [3] |
| GCA_000196575.1 | B. longum subsp. infantis 157F | 157F | Human infant feces * | Yes [3] |
| GCA_001725985.1 | B. longum subsp. longum AH1206 | AH1206 | Stool sample * | Yes [37] |
| GCA_000220135.1 | B. breve UCC2003 | UCC2003 | Infant nursing stool * | Yes [3] |
| GCA_001281425.1 | B. breve BR3 | BR3 | Feces * | Yes [38] |
| GCA_001293145.1 | B. longum BG7 | BG7 | Feces * | Yes [39] |
| GCA_001446275.1 | B. longum subsp. longum CCUG30698 | CCUG30698 | Human adult intestine * | No |
| GCA_000092325.1 | B. longum subsp. longum JDM301 | JDM301 | Human infant feces * | Yes [3] |
| GCA_000730205.1 | B. longum BXY01 | BXY01 | Gut * | No |
| GCA_001281305.1 | B. longum subsp. Infantis BT1 | BT1 | Feces * | No |
| GCA_000269965.1 | B. longum subsp. infantis ATCC 15697 = JCM 1222 = DSM 20088 | JCM 1222 | Intestine of infant * | Yes [40] |
| GCA_000020425.1 | B. longum subsp. infantis ATCC 15697 = JCM 1222 = DSM 20088 | ATCC 15697 | Human infant feces * | Yes [40] |
| Feature | Feature Count * | Sub-Feature | Sub Feature Count # |
|---|---|---|---|
| Protein Encoding Genes (PEGs) | 613 (100%) | ||
| PEGs predicted with the COGs functions | 442 (72.1%) | Cellular Processes and Signaling | 57 (9.3%) |
| Information Storage and Processing | 135 (22.02%) | ||
| Metabolism | 179 (29.2%) | ||
| Multiple Classes | 31 (5.06%) | ||
| Poorly Characterized | 40 (6.53%) | ||
| PEGs mapped to the KEGG functions | 488 (79.61%) | ||
| PEGs assigned to the Transporter Proteins | 60 (9.79%) | ||
| PEGs assigned to the Virulence Factors | 118 (19.25%) | ||
| Subcellular Localization of PEGs | 613 (100%) | Cell Wall | 1 (0.16%) |
| Cytoplasmic | 497 (81.08%) | ||
| Extracellular | 29 (4.73%) | ||
| Membrane | 86 (14.03%) | ||
| PEGs predicted with the Transmembrane Helices | 78 (12.72%) | ||
| PEGs predicted with the Signal Peptide Cleavage Sites | 9 (1.47%) | ||
| PEGs predicted with the Lipoprotein Signal Peptides | 613 (100%) | Cytoplasmic Proteins | 561 (91.52%) |
| SPaseI-cleaved Proteins | 12 (1.96%) | ||
| Lipoproteins (SPaseII-cleaved Proteins) | 1 (0.16%) | ||
| Transmembrane Proteins | 39 (6.36%) | ||
| PEGs predicted with the Non-Classical (Not Signal Peptide Triggered) Secretion | 94 (15.33%) | ||
| PEGs assigned to the Effector Proteins | 69 (11.26%) | Endoplasmic Reticulum as an Effector Target | 34 (5.55%) |
| Mitochondrion as an Effector Target | 7 (1.14%) | ||
| Endoplasmic Reticulum as a Possible Effector Target | 9 (1.47%) | ||
| Mitochondrion as a Possible Effector Target | 19 (3.1%) | ||
| PEGs assigned to the Essential Genes | 496 (80.91%) | ||
| PEGs assigned to the Types of Other DNA-binding Proteins | 1 (0.16%) | ||
| PEGs assigned to the Types of Transcription Factors | 19 (3.1%) | ||
| PEGs assigned to the Types of Two-Component Systems | 7 (1.14%) | ||
| PEGs assigned to the Carbohydrate Active Enzymes | 11 (1.8%) | Carbohydrate-Binding Modules | 2 (0.32%) |
| Glycoside Hydrolases | 6 (0.97%) | ||
| Glycosyl Transferases | 3 (0.48%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, V.; Mobeen, F.; Prakash, T. Exploration of Survival Traits, Probiotic Determinants, Host Interactions, and Functional Evolution of Bifidobacterial Genomes Using Comparative Genomics. Genes 2018, 9, 477. https://doi.org/10.3390/genes9100477
Sharma V, Mobeen F, Prakash T. Exploration of Survival Traits, Probiotic Determinants, Host Interactions, and Functional Evolution of Bifidobacterial Genomes Using Comparative Genomics. Genes. 2018; 9(10):477. https://doi.org/10.3390/genes9100477
Chicago/Turabian StyleSharma, Vikas, Fauzul Mobeen, and Tulika Prakash. 2018. "Exploration of Survival Traits, Probiotic Determinants, Host Interactions, and Functional Evolution of Bifidobacterial Genomes Using Comparative Genomics" Genes 9, no. 10: 477. https://doi.org/10.3390/genes9100477
APA StyleSharma, V., Mobeen, F., & Prakash, T. (2018). Exploration of Survival Traits, Probiotic Determinants, Host Interactions, and Functional Evolution of Bifidobacterial Genomes Using Comparative Genomics. Genes, 9(10), 477. https://doi.org/10.3390/genes9100477