
The Genetic Architecture of Type 1 Diabetes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structure of Disease Processes
2.1. Heritability Is Variable
2.2. Age-Related Effects on Type 1 Diabetes Onset
2.3. Age-Related Effect on Age at Diagnosis
2.4. Evolution of Genetic Risk
2.5. Non-Genetic Factors Causing Type 1 Diabetes
2.6. Threshold Hypothesis
2.7. Rare Variants
2.8. Seasonal Impact of Gene Effect
2.9. Nature of Genetic Risk
- Cytokine production/signalling.
- Decreased T cell signalling/activation.
- Increased T1 interferon and antiviral response.
- Antigen presentation and T cell repertoire formation.
3. Specific Genetic Factors and Type 1 Diabetes
3.1. Human Leukocyte Antigen (HLA)
3.2. Nature of HLA Risk in Type 1 Diabetes
3.3. HLA Protection Associated with Type 1 Diabetes
3.4. Structure of HLA-Mediated Risk
3.5. HLA Loci Heterozygosity and Disease Risk
3.6. HLA Hyperexpression and Type 1 Diabetes
4. Immune Synapse Genes
4.1. Insulin Gene Plus Insulin as Antigen
4.2. Interferon (IFN) Genes and Virus Infections
4.3. Gut and Genetic Susceptibility to Type 1 Diabetes
5. Genetic Effect on the Variable Clinical Presentation
5.1. Non-HLA Genes Impacting Disease Progression
5.2. Low Risk HLA Genes and Single Autoantibodies
5.3. Immunogenotype and Adult-Onset Diabetes
5.4. Immune-Mediated Destruction and Diabetes
6. Unknown: Features that Could Be Genetically Determined
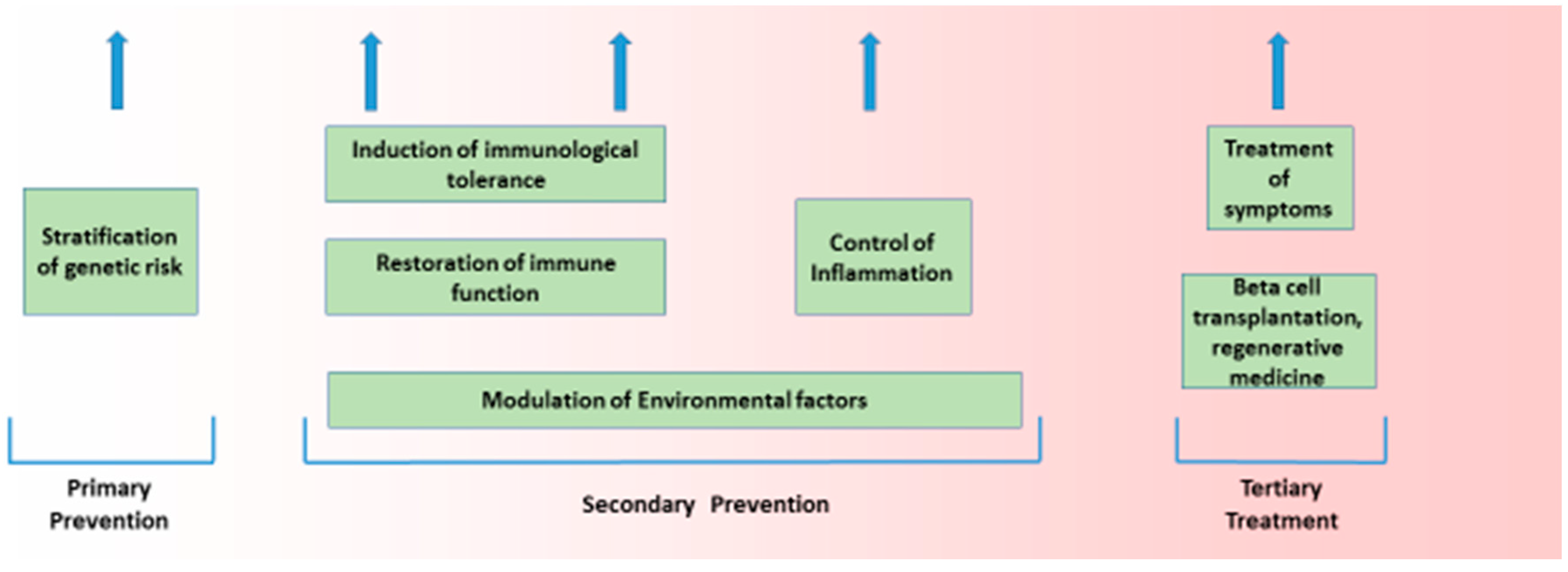
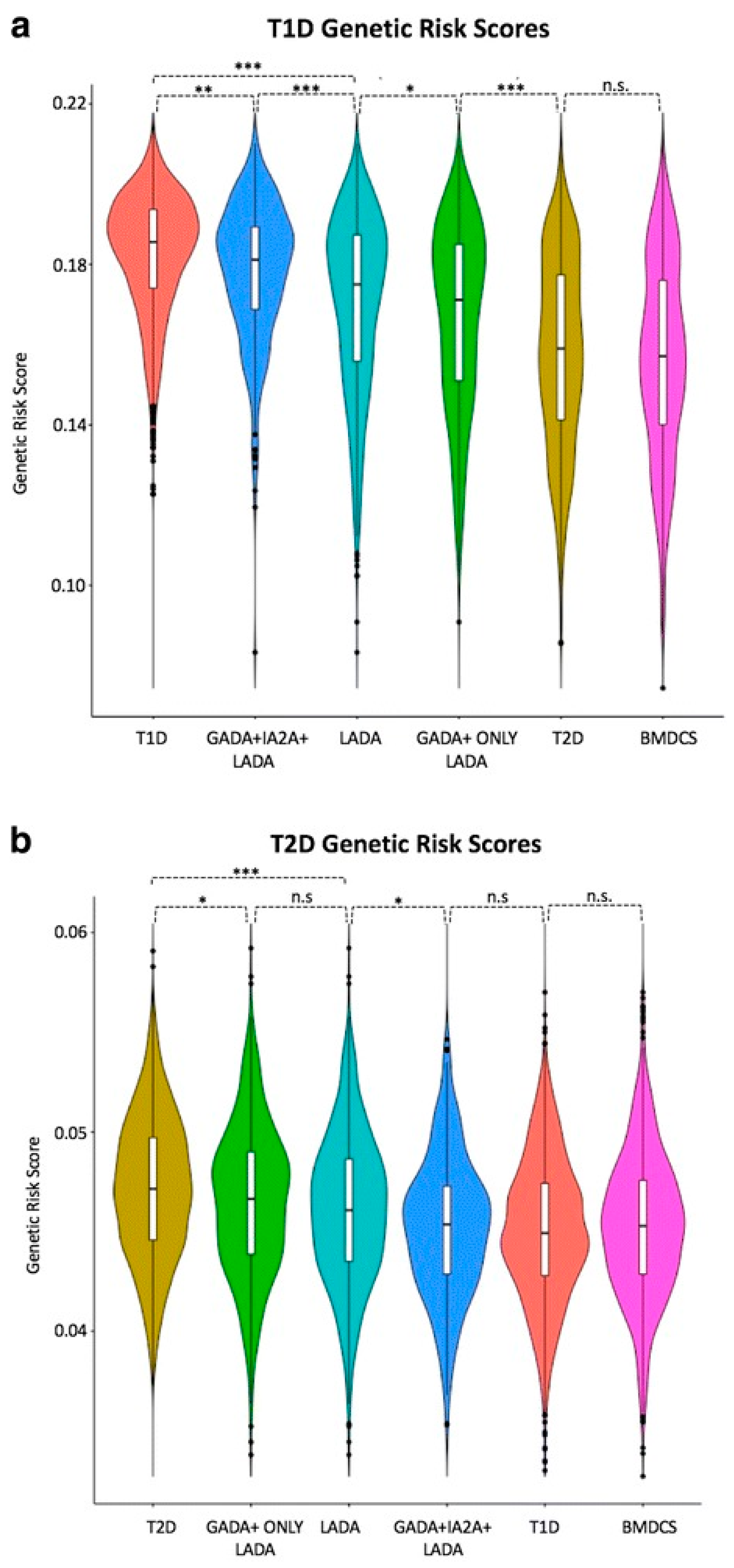
6.1. Stratification of Genetic Risk and Gene Risk Scores (GRS)
6.1.1. Potential Genetic and Non-Genetic Interactions
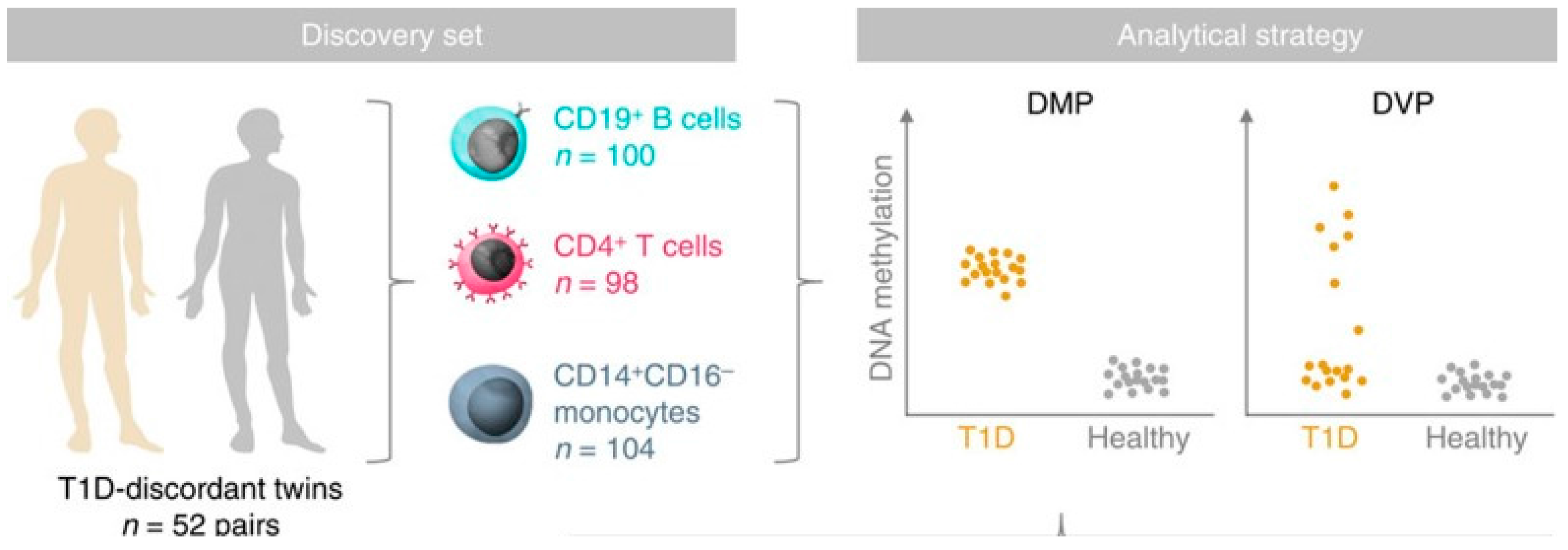
6.1.2. Epigenetic Effects and T1D Risk
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Redondo, M.J.; Yu, L.; Hawa, M.; Mackenzie, T.; Pyke, D.A.; Eisenbarth, G.S.; Leslie, R.D.G. Heterogeneity of type I diabetes: Analysis of monozygotic twins in Great Britain and the United States. Diabetologia 2001, 44, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Salvetti, M.; Ristori, G.; Bomprezzi, R.; Pozzilli, P.; Leslie, R.D.G. Twins: Mirrors of the immune system. Immunol. Today 2000, 21, 342–347. [Google Scholar] [CrossRef]
- Redondo, M.J.; Jeffrey, J.; Fain, P.R.; Eisenbarth, G.S.; Orban, T. Concordance for islet autoimmunity among monozygotic twins. N. Engl. J. Med. 2008, 359, 2849–2850. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.B.; Groop, L. Genetics of type 2 diabetes-pitfalls and possibilities. Genes 2015, 6, 87–123. [Google Scholar] [CrossRef] [PubMed]
- Leslie, R.D.; Castelli, M.D. Age-dependent influences on the origins of autoimmune diabetes: Evidence and implications. Diabetes 2004, 53, 3033–3040. [Google Scholar] [CrossRef] [PubMed]
- Regnell, S.E.; Lernmark, A. Early prediction of autoimmune (type 1) diabetes. Diabetologia 2017, 60, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Krischer, J.P.; Lynch, K.F.; Lernmark, Å.; Hagopian, W.A.; Rewers, M.J.; She, J.X.; Toppari, J.; Ziegler, A.G.; Akolkar, B.; TEDDY Study Group. Genetic and Environmental Interactions Modify the Risk of Diabetes-Related Autoimmunity by 6 Years of Age: The TEDDY Study. In Diabetes Care; American Diabetes Association: Arlington, VA, USA, 2017. [Google Scholar]
- Ziegler, A.G.; Bonifacio, E.; Powers, A.C.; Todd, J.A.; Harrison, L.C.; Atkinson, M.A. Type 1 Diabetes Prevention: A Goal Dependent on Accepting a Diagnosis of an Asymptomatic Disease. Diabetes 2016, 65, 3233–3239. [Google Scholar] [CrossRef] [PubMed]
- Fava, D.; Gardner, S.; Pyke, D.; Leslie, R.D.G. Evidence that the age at diagnosis of IDDM is genetically determined. Diabetes Care 1998, 21, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.F. The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 2002, 347, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Wasserfall, C.; Nead, K.; Mathews, C.; Atkinson, M.A. The threshold hypothesis: Solving the equation of nurture vs nature in type 1 diabetes. Diabetologia 2011, 54, 2232–2236. [Google Scholar] [CrossRef] [PubMed]
- Long, A.E.; Gillespie, K.M.; Rokni, S.; Bingley, P.J.; Williams, A.J. Rising incidence of type 1 diabetes is associated with altered immunophenotype at diagnosis. Diabetes 2012, 61, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.G.; Rewers, M.; Simell, O.; Simell, T.; Lempainen, J.; Steck, A.; Winkler, C.; Ilonen, J.; Veijola, R.; Knip, M.; et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013, 309, 2473–2479. [Google Scholar] [CrossRef] [PubMed]
- Beyerlein, A.; Donnachie, E.; Jergens, S.; Ziegler, A.G. Infections in Early Life and Development of Type 1 Diabetes. JAMA 2016, 315, 1899–1901. [Google Scholar] [CrossRef] [PubMed]
- Tennessen, J.A.; Bigham, A.W.; O’Connor, T.D.; Fu, W.; Kenny, E.E.; Gravel, S.; McGee, S.; Do, R.; Liu, X.; Jun, G.; et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012, 337, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Nejentsev, S.; Walker, N.; Riches, D.; Egholm, M.; Todd, J.A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 2009, 324, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.A.; Mistry, V.; Bockett, N.A.; Ahmad, T.; Ban, M.; Barker, J.N.; Barrett, J.C.; Blackburn, H.; Brand, O.; Burren, O.; et al. Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature 2013, 498, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Dopico, X.C.; Evangelou, M.; Ferreira, R.C.; Guo, H.; Pekalski, M.L.; Smyth, D.J.; Cooper, N.; Burren, O.S.; Fulford, A.J.; Hennig, B.J.; et al. Widespread seasonal gene expression reveals annual differences in human immunity and physiology. Nat. Commun. 2015, 6, 7000. [Google Scholar] [CrossRef] [PubMed]
- Onengut-Gumuscu, S.; Chen, W.M.; Burren, O.; Cooper, N.J.; Quinlan, A.R.; Mychaleckyj, J.C.; Farber, E.; Bonnie, J.K.; Szpak, M.; Schofield, E.; et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet. 2015, 47, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Ram, R.; Morahan, G. Effects of Type 1 Diabetes Risk Alleles on Immune Cell Gene Expression. Genes 2017, 8, 167. [Google Scholar] [CrossRef]
- Storling, J.; Pociot, F. Type 1 diabetes candidate genes linked to pancreatic islet cell inflammation and beta-cell apoptosis. Genes 2017, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Körmendy, D.; Hoff, H.; Hoff, P.; Bröker, B.M.; Burmester, G.R.; Brunner-Weinzierl, M.C. Impact of the CTLA-4/CD28 axis on the processes of joint inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2013, 65, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; Li, J.; Zhao, S.D.; Bradfield, J.P.; Mentch, F.D.; Maggadottir, S.M.; Hou, C.; Abrams, D.J.; Chang, D.; Gao, F.; et al. Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat. Med. 2015, 21, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Bergholdt, R.; Brorsson, C.; Palleja, A.; Berchtold, L.A.; Fløyel, T.; Bang-Berthelsen, C.H.; Frederiksen, K.S.; Jensen, L.J.; Størling, J.; Pociot, F. Identification of novel type 1 diabetes candidate genes by integrating genome-wide association data, protein-protein interactions, and human pancreatic islet gene expression. Diabetes 2012, 61, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Feldman, M. Heterogeneity of autoimmune diseases: Pathophysiologic insights from genetics and implications for new therapies. Nat. Med. 2015, 21, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Arif, S.; Leete, P.; Nguyen, V.; Marks, K.; Nor, N.M.; Estorninho, M.; Kronenberg-Versteeg, D.; Bingley, P.J.; Todd, J.A.; Guy, C.; et al. Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes 2014, 63, 3835–3845. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.K.; Behrens, T.W. Genetics of autoimmune diseases--disorders of immune homeostasis. Nat. Rev. Genet. 2006, 7, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Akolkar, B.; Concannon, P.; Erlich, H.A.; Julier, C.; Morahan, G.; Nierras, C.R.; Todd, J.A.; Rich, S.S.; Nerup, J. Genetics of type 1 diabetes: What’s next? Diabetes 2010, 59, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xiang, Y.; Ji, L.; Jia, W.; Ning, G.; Huang, G.; Yang, L.; Lin, J.; Liu, Z.; Hagopian, W.A.; et al. Frequency, immunogenetics, and clinical characteristics of latent autoimmune diabetes in China (LADA China study): A nationwide, multicenter, clinic-based cross-sectional study. Diabetes 2013, 62, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Noble, J.A.; Valdes, A.M.; Cook, M.; Klitz, W.; Thomson, G.; Erlich, H.A. The role of HLA class II genes in insulin-dependent diabetes mellitus: Molecular analysis of 180 Caucasian, multiplex families. Am. J. Hum. Genet. 1996, 59, 1134–1148. [Google Scholar] [PubMed]
- Zipris, D. Epidemiology of type 1 diabetes and what animal models teach us about the role of viruses in disease mechanisms. Clin. Immunol. 2009, 131, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, H.; Fujisawa, T.; Kawabata, Y.; Noso, S.; Ogihara, T. Genetics of type 1 diabetes: Similarities and differences between Asian and Caucasian populations. Ann. N. Y. Acad. Sci. 2006, 1079, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, Y.; Ikegami, H.; Kawaguchi, Y.; Fujisawa, T.; Shintani, M.; Ono, M.; Nishino, M.; Uchigata, Y.; Lee, I.; Ogihara, T. Asian-specific HLA haplotypes reveal heterogeneity of the contribution of HLA-DR and -DQ haplotypes to susceptibility to type 1 diabetes. Diabetes 2002, 51, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, A.; Boulware, D.; Yu, L.; Babu, S.; Steck, A.K.; Becker, D.; Rodriguez, H.; DiMeglio, L.; Evans-Molina, C.; Harrison, L.C.; et al. HLA-DRB1*15:01-DQA1*01:02-DQB1*06:02 Haplotype Protects Autoantibody-Positive Relatives From Type 1 Diabetes Throughout the Stages of Disease Progression. Diabetes 2016, 65, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.A.; Bell, J.I.; McDevitt, H.O. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature 1987, 329, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Sharon, E.; Sibener, L.V.; Battle, A.; Fraser, H.B.; Garcia, K.C.; Pritchard, J.K. Genetic variation in MHC proteins is associated with T cell receptor expression biases. Nat. Genet. 2016, 48, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Ooi, J.D.; Petersen, J.; Tan, Y.H.; Huynh, M.; Willett, Z.J.; Ramarathinam, S.H.; Eggenhuizen, P.J.; Loh, K.L.; Watson, K.A.; Gan, P.Y.; et al. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature 2017, 545, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Baranzini, S.E. The genetics of autoimmune diseases: A networked perspective. Curr. Opin. Immunol. 2009, 21, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Lettre, G.; Rioux, J.D. Autoimmune diseases: Insights from genome-wide association studies. Hum. Mol. Genet. 2008, 17, R116–R121. [Google Scholar] [CrossRef] [PubMed]
- Zenewicz, L.A.; Abraham, C.; Flavell, R.A.; Cho, J.H. Unraveling the genetics of autoimmunity. Cell 2010, 140, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Rioux, J.D.; Abbas, A.K. Paths to understanding the genetic basis of autoimmune disease. Nature 2005, 435, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Fortune, M.D.; Guo, H.; Burren, O.; Schofield, E.; Walker, N.M.; Ban, M.; Sawcer, S.J.; Bowes, J.; Worthington, J.; Barton, A.; et al. Statistical colocalization of genetic risk variants for related autoimmune diseases in the context of common controls. Nat. Genet. 2015, 47, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Lenz, T.L.; Deutsch, A.J.; Han, B.; Hu, X.; Okada, Y.; Eyre, S.; Knapp, M.; Zhernakova, A.; Huizinga, T.W.; Abecasis, G.; et al. Widespread non-additive and interaction effects within HLA loci modulate the risk of autoimmune diseases. Nat. Genet. 2015, 47, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Bottazzo, G.F. Banting Lecture. On the honey disease: A dialogue with Socrates. Diabetes 1993, 42, 778–800. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.J.; Rodriguez-Calvo, T.; Gerling, I.C.; Mathews, C.E.; Kaddis, J.S.; Russell, M.A.; Zeissler, M.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K.; et al. Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 2016, 59, 2448–2458. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.; Cutler, A.J.; Pontikos, N.; Pekalski, M.L.; Burren, O.S.; Cooper, J.D.; Garcia, A.R.; Ferreira, R.C.; Guo, H.; Walker, N.M.; et al. Dissection of a Complex Disease Susceptibility Region Using a Bayesian Stochastic Search Approach to Fine Mapping. PLoS Genet. 2015, 11, e1005272. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.T.; Todd, J.A. Human type 1 diabetes and the insulin gene: Principles of mapping polygenes. Annu. Rev. Genet. 1996, 30, 343–370. [Google Scholar] [CrossRef] [PubMed]
- Lucassen, A.M.; Julier, C.; Beressi, J.P.; Boitard, C.; Froguel, P.; Lathrop, M.; Bell, J.I. Susceptibility to insulin dependent diabetes mellitus maps to a 4.1 kb segment of DNA spanning the insulin gene and associated VNTR. Nat. Genet. 1993, 4, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.T.; Lucassen, A.M.; Gough, S.C.L.; Powell, E.E.; Undlien, D.E.; Pritchard, L.E.; Merriman, M.E.; Kawaguchi, Y.; Dronsfield, M.J.; Pociot, F.; et al. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat. Genet. 1995, 9, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Julier, C.; Hyer, R.N.; Davies, J.; Merlin, F.; Soularue, P.; Briant, L.; Cathelineau, G.; Deschamps, I.; Rotter, J.I.; Froguel, P.; et al. Insulin-IGF2 region on chromosome 11p encodes a gene implicated in HLA-DR4-dependent diabetes susceptibility. Nature 1991, 354, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, K.A.; Hitman, G.A.; Rowe, R.E.; Hawa, M.; Huang, X.; Stewart, T.; Leslie, R.D.G. Concordance for type 1 diabetes in identical twins is affected by insulin genotype. Diabetes Care 2001, 24, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Chesi, A.; Cousminer, D.L.; Hawa, M.I.; Bradfield, J.P.; Hodge, K.M.; Guy, V.C.; Hakonarson, H.; Mauricio, D.; Schloot, N.C.; et al. Relative contribution of type 1 and type 2 diabetes loci to the genetic etiology of adult-onset, non-insulin-requiring autoimmune diabetes. BMC Med. 2017, 15, 88. [Google Scholar] [CrossRef] [PubMed]
- Foulis, A.K.; Farquharson, M.A.; Meager, A. Immunoreactive alpha-interferon in insulin-secreting beta cells in type 1 diabetes mellitus. Lancet 1987, 2, 1423–1427. [Google Scholar] [CrossRef]
- Heinig, M.; Petretto, E.; Wallace, C.; Bottolo, L.; Rotival, M.; Lu, H.; Li, Y.; Sarwar, R.; Langley, S.R.; Bauerfeind, A.; et al. A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature 2010, 467, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.; Lauber, C.; Adler, K.; Grallert, H.; Illig, T.; Ziegler, A.G.; Bonifacio, E. An interferon-induced helicase (IFIH1) gene polymorphism associates with different rates of progression from autoimmunity to type 1 diabetes. Diabetes 2011, 60, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Bonifacio, E.; Warncke, K.; Winkler, C.; Wallner, M.; Ziegler, A.G. Cesarean section and interferon-induced helicase gene polymorphisms combine to increase childhood type 1 diabetes risk. Diabetes 2011, 60, 3300–3306. [Google Scholar] [CrossRef] [PubMed]
- Beyan, H.; Wen, L.; Leslie, R.D. Guts, germs, and meals: The origin of type 1 diabetes. Curr. Diabetes Rep. 2012, 12, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Alkanani, A.K.; Hara, N.; Gottlieb, P.A.; Ir, D.; Robertson, C.E.; Wagner, B.D.; Frank, D.N.; Zipris, D. Alterations in Intestinal Microbiota Correlate With Susceptibility to Type 1 Diabetes. Diabetes 2015, 64, 3510–3520. [Google Scholar] [CrossRef] [PubMed]
- Vatanen, T.; Kostic, A.D.; d’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.M.; et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 2016, 165, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyötyläinen, T.; Hämäläinen, A.M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I.; et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Uusitalo, U.; Liu, X.; Yang, J.; Aronsson, C.A.; Hummel, S.; Butterworth, M.; Lernmark, Å.; Rewers, M.; Hagopian, W.; She, J.X.; et al. Association of Early Exposure of Probiotics and Islet Autoimmunity in the TEDDY Study. JAMA Pediatr. 2016, 170, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Achenbach, P.; Hummel, M.; Thümer, L.; Boerschmann, H.; Höfelmann, D.; Ziegler, A.G. Characteristics of rapid vs slow progression to type 1 diabetes in multiple islet autoantibody-positive children. Diabetologia 2013, 56, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Lempainen, J.; Hermann, R.; Veijola, R.; Simell, O.; Knip, M.; Ilonen, J. Effect of the PTPN22 and INS risk genotypes on the progression to clinical type 1 diabetes after the initiation of beta-cell autoimmunity. Diabetes 2012, 61, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Howson, J.M.; Cooper, J.D.; Smyth, D.J.; Walker, N.M.; Stevens, H.; She, J.X.; Eisenbarth, G.S.; Rewers, M.; Todd, J.A.; Akolkar, B.; et al. Evidence of gene-gene interaction and age-at-diagnosis effects in type 1 diabetes. Diabetes 2012, 61, 3012–3017. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fei, M.; Fu, D.; Zhang, L.; Ma, Y.; Wang, Y.; Zhang, F.; Xia, Q.; Wang, X. Association between cytotoxic T lymphocyte antigen-4 polymorphism and type 1 diabetes: A meta-analysis. Gene 2013, 516, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, I.; Weets, I.; Costa, O.; Asanghanwa, M.; Verhaeghen, K.; Decochez, K.; Ruige, J.; Casteels, K.; Wenzlau, J.; Hutton, J.C.; et al. An important minority of prediabetic first-degree relatives of type 1 diabetic patients derives from seroconversion to persistent autoantibody positivity after 10 years of age. Diabetologia 2012, 55, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Howson, J.M.; Rosinger, S.; Smyth, D.J.; Boehm, B.O.; Todd, J.A.; ADBW-END study group. Genetic analysis of adult-onset autoimmune diabetes. Diabetes 2011, 60, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.K.; Sterner, M.; Forsén, T.; Käräjämäki, A.; Rolandsson, O.; Forsblom, C.; Groop, P.H.; Lahti, K.; Nilsson, P.M.; Groop, L.; et al. Type 2 diabetes susceptibility gene variants predispose to adult-onset autoimmune diabetes. Diabetologia 2014, 57, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Cervin, C.; Lyssenko, V.; Bakhtadze, E.; Lindholm, E.; Nilsson, P.; Tuomi, T.; Cilio, C.M.; Groop, L. Genetic similarities between latent autoimmune diabetes in adults, type 1 diabetes, and type 2 diabetes. Diabetes 2008, 57, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.C.; Dahlquist, G.; Soltesz, G.; Green, A.; Eurodiab ACE Study Group. Is childhood-onset type I diabetes a wealth-related disease? An ecological analysis of European incidence rates. Diabetologia 2001, 44 (Suppl. 3), B9–B16. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Thompson, M.; Wasserfall, C.; Montgomery, E.L.; Atkinson, M.A.; Kaddis, J.S. Pancreas organ weight in individuals with disease-associated autoantibodies at risk for type 1 diabetes. JAMA 2012, 308, 2337–2339. [Google Scholar] [CrossRef] [PubMed]
- Gregg, B.E.; Moore, P.C.; Demozay, D.; Hall, B.A.; Li, M.; Husain, A.; Wright, A.J.; Atkinson, M.A.; Rhodes, C.J. Formation of a human beta-cell population within pancreatic islets is set early in life. J. Clin. Endocrinol. Metab. 2012, 97, 3197–3206. [Google Scholar] [CrossRef] [PubMed]
- Oram, R.A.; Patel, K.; Hill, A.; Shields, B.; McDonald, T.J.; Jones, A.; Hattersley, A.T.; Weedon, M.N. A Type 1 Diabetes Genetic Risk Score Can Aid Discrimination Between Type 1 and Type 2 Diabetes in Young Adults. Diabetes Care 2016, 39, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.A.; Oram, R.A.; Flanagan, S.E.; De Franco, E.; Colclough, K.; Ellard, S.; Weedon, M.N.; Hattersley, A.T. Type 1 Diabetes Genetic Risk Score: A Novel Tool to Discriminate Monogenic and Type 1 Diabetes. Diabetes 2016, 65, 2094–2099. [Google Scholar] [CrossRef] [PubMed]
- Brorsson, C.A.; Pociot, F. Shared Genetic Basis for Type 1 Diabetes, Islet Autoantibodies, and Autoantibodies Associated With Other Immune-Mediated Diseases in Families With Type 1 Diabetes. Diabetes Care 2015, 38 (Suppl. 2), S8–S13. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.H.; Volkov, P.; Bacos, K.; Dayeh, T.; Hall, E.; Nilsson, E.A.; Ladenvall, C.; Rönn, T.; Ling, C. Genome-wide associations between genetic and epigenetic variation influence mRNA expression and insulin secretion in human pancreatic islets. PLoS Genet. 2014, 10, e1004735. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.S.; Teschendorff, A.E.; Dang, M.A.; Lowe, R.; Hawa, M.I.; Ecker, S.; Beyan, H.; Cunningham, S.; Fouts, A.R.; Ramelius, A.; et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat. Commun. 2016, 7, 13555. [Google Scholar] [CrossRef] [PubMed]
- Ecker, S.; Chen, L.; Pancaldi, V.; Bagger, B.O.; Fernández, J.M.; de Santa Pau, E.C.; Juan, D.; Mann, A.L.; Watt, S.; Casale, F.P.; et al. Genome-wide analysis of differential transcriptional and epigenetic variability across human immune cell types. Genome Biol. 2017, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- Snowhite, I.V.; Allende, G.; Sosenko, J.; Pastori, R.L.; Messinger, C.S.; Pugliese, A. Association of serum microRNAs with islet autoimmunity, disease progression and metabolic impairment in relatives at risk of type 1 diabetes. Diabetologia 2017, 60, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Dorajoo, R.; Liu, J.; Boehm, B.O. Genetics of Type 2 Diabetes and Clinical Utility. Genes 2015, 6, 372–384. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jerram, S.T.; Leslie, R.D. The Genetic Architecture of Type 1 Diabetes. Genes 2017, 8, 209. https://doi.org/10.3390/genes8080209
Jerram ST, Leslie RD. The Genetic Architecture of Type 1 Diabetes. Genes. 2017; 8(8):209. https://doi.org/10.3390/genes8080209
Chicago/Turabian StyleJerram, Samuel T, and Richard David Leslie. 2017. "The Genetic Architecture of Type 1 Diabetes" Genes 8, no. 8: 209. https://doi.org/10.3390/genes8080209
APA StyleJerram, S. T., & Leslie, R. D. (2017). The Genetic Architecture of Type 1 Diabetes. Genes, 8(8), 209. https://doi.org/10.3390/genes8080209