
Chromatin Switches during Neural Cell Differentiation and Their Dysregulation by Prenatal Alcohol Exposure


Abstract
:1. Introduction
2. Chromatin Structure and Gene Expression
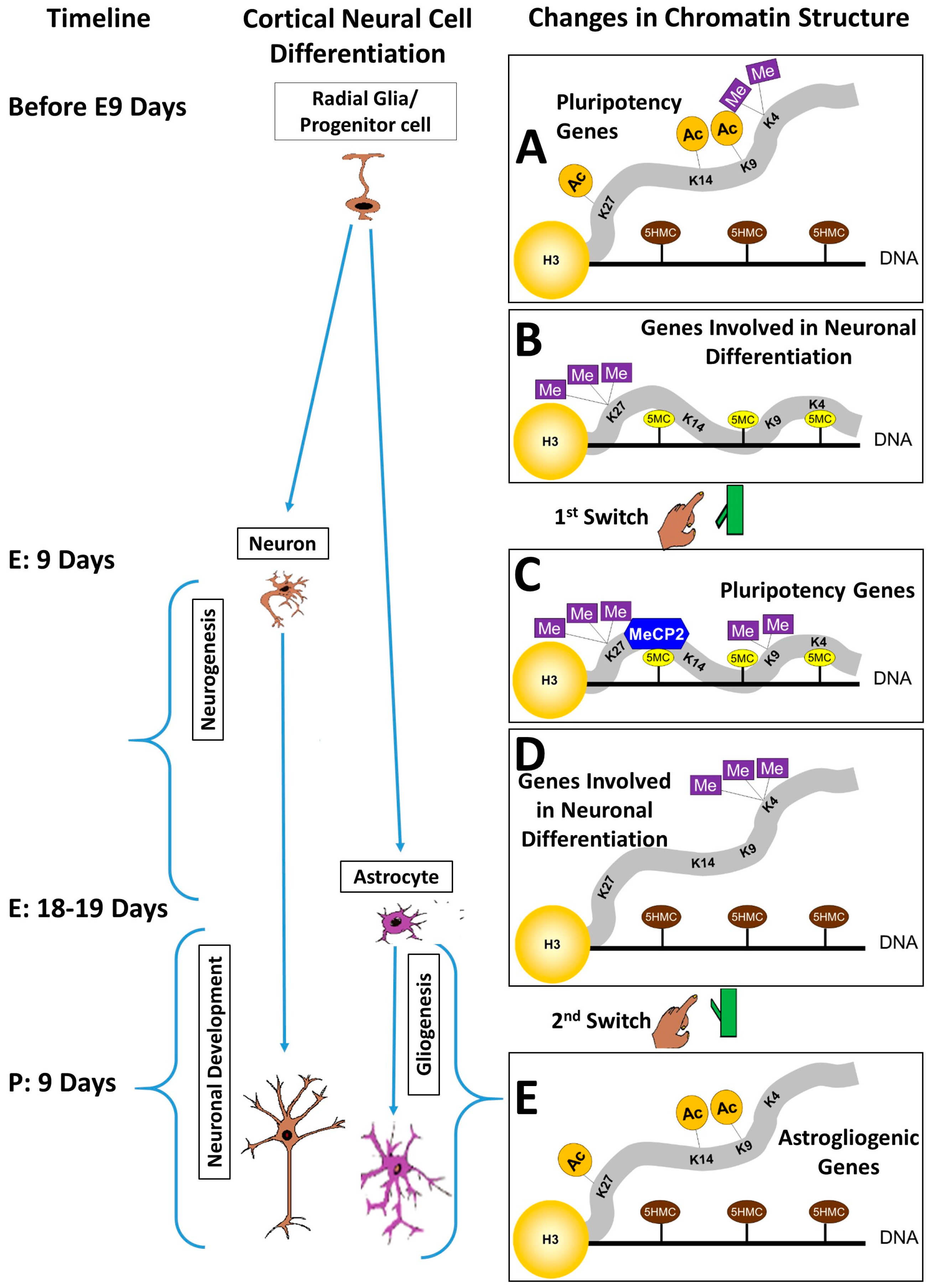
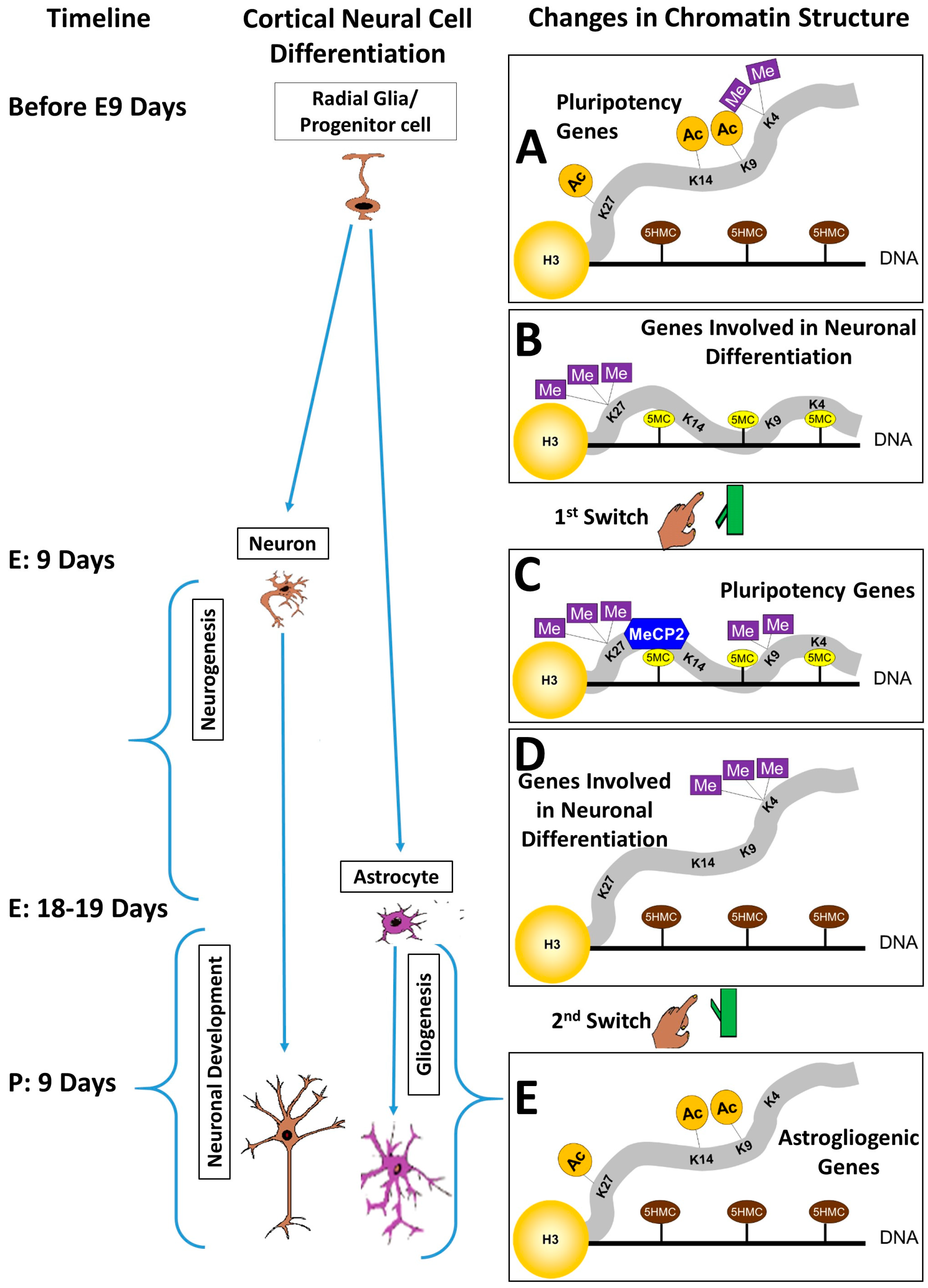
3. The Pluripotency/Neurogenesis Switch
4. The Neurogenesis/Gliogenesis Switch
5. Abnormal Differentiation in FASD
6. FASD, Cell Fate, and Chromatin Structure
7. FASD and DNA Methylation
8. FASD and Histone Modifications
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lunde, E.R.; Washburn, S.E.; Golding, M.C.; Bake, S.; Miranda, R.C.; Ramadoss, J. Alcohol-induced developmental origins of adult-onset diseases. Alcohol. Clin. Exp. Res. 2016, 40, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Chakrapani, B.P.; Schwegler, H.; Hofmann, H.D.; Kirsch, M. Neurogenesis in the dentate gyrus depends on ciliary neurotrophic factor and signal transducer and activator of transcription 3 signaling. Stem Cells 2009, 27, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Grumet, M. BMP and LIF signaling coordinately regulate lineage restriction of radial glia in the developing forebrain. Glia 2007, 55, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Nardelli, J. Cellular and molecular introduction to brain development. Neurobiol. Dis. 2016, 92, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Song, M.R.; Ghosh, A. FGF2-induced chromatin remodeling regulates CNTF-mediated gene expression and astrocyte differentiation. Nat. Neurosci. 2004, 7, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Dhara, S.K.; Swetenburg, R.; Mithani, M.; Cao, K.; Medrzycki, M.; Fan, Y.; Stice, S.L. Inhibition of DNA methyltransferases and histone deacetylases induces astrocytic differentiation of neural progenitors. Stem Cell Res. 2013, 11, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Urayama, S.; Semi, K.; Sanosaka, T.; Hori, Y.; Namihira, M.; Kohyama, J.; Takizawa, T.; Nakashima, K. Chromatin accessibility at a STAT3 target site is altered prior to astrocyte differentiation. Cell Struct. Funct. 2013, 38, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Language: Disputed definitions. Nature 2008, 455, 1023–1028. [Google Scholar] [PubMed]
- Laird, P.W. Cancer epigenetics. Hum. Mol. Genet. 2005, 14, R65–R76. [Google Scholar] [CrossRef] [PubMed]
- Levenson, J.M.; Sweatt, J.D. Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 2005, 6, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Barth, T.K.; Imhof, A. Fast signals and slow marks: The dynamics of histone modifications. Trends Biochem. Sci. 2010, 35, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Dent, S.Y. Chromatin modifiers and remodellers: Regulators of cellular differentiation. Nat. Rev. Genet. 2014, 15, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell. Biol. 2009, 10, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Chong, S.; Champ, M.E.; Cuthbert, P.C.; Morgan, H.D.; Luu, K.V.; Whitelaw, E. Transgenerational inheritance of epigenetic states at the murine axin(fu) allele occurs after maternal and paternal transmission. Proc. Natl. Acad. Sci. USA 2003, 100, 2538–2543. [Google Scholar] [CrossRef] [PubMed]
- Almouzni, G.; Cedar, H. Maintenance of epigenetic information. Cold Spring Harb. Perspect. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Commerford, S.L.; Carsten, A.L.; Cronkite, E.P. Histone turnover within nonproliferating cells. Proc. Natl. Acad. Sci. USA 1982, 79, 1163–1165. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, K.P.; Gendreizig, S.; White, D.A.; Bradbury, C.; Hollfelder, F.; Turner, B.M. Cross-talk between histone modifications in response to histone deacetylase inhibitors: MLL4 links histone H3 acetylation and histone H3K4 methylation. J. Biol. Chem. 2007, 282, 4408–4416. [Google Scholar] [CrossRef] [PubMed]
- Balaraman, S.; Tingling, J.D.; Tsai, P.C.; Miranda, R.C. Dysregulation of microRNA expression and function contributes to the etiology of fetal alcohol spectrum disorders. Alcohol Res. 2013, 35, 18–24. [Google Scholar] [PubMed]
- Fierz, B. Dynamic chromatin regulation from a single molecule perspective. ACS Chem. Biol. 2016, 11, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.M. Cellular memory and the histone code. Cell 2002, 111, 285–291. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.P.; Gavin, D.P.; Chase, K.A. Heterochromatin as an incubator for pathology and treatment non-response: Implication for neuropsychiatric illness. Pharmacogenom. J. 2012, 12, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Trošelj, K.G.; Kujundzic, R.N.; Ugarkovic, D. Polycomb repressive complex’s evolutionary conserved function: The role of EZH2 status and cellular background. Clin. Epigenet. 2016, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- De Napoles, M.; Mermoud, J.E.; Wakao, R.; Tang, Y.A.; Endoh, M.; Appanah, R.; Nesterova, T.B.; Silva, J.; Otte, A.P.; Vidal, M.; et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 2004, 7, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Kriukiene, E.; Liutkeviciute, Z.; Klimasauskas, S. 5-hydroxymethylcytosine—The elusive epigenetic mark in mammalian DNA. Chem. Soc. Rev. 2012, 41, 6916–6930. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Ait-Si-Ali, S.; Yokochi, T.; Wade, P.A.; Jones, P.L.; Wolffe, A.P. DNMT1 forms a complex with rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet. 2000, 25, 338–342. [Google Scholar] [PubMed]
- Wen, L.; Li, X.; Yan, L.; Tan, Y.; Li, R.; Zhao, Y.; Wang, Y.; Xie, J.; Zhang, Y.; Song, C.; et al. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 2014, 15, R49. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Coskun, V.; Tao, J.; Xie, W.; Ge, W.; Yoshikawa, K.; Li, E.; Zhang, Y.; Sun, Y.E. DNMT3A-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 2010, 329, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Damayanti, N.P.; Irudayaraj, J.; Dunn, K.; Zhou, F.C. Diversity of two forms of DNA methylation in the brain. Front. Genet. 2014, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Gavin, D.P.; Chase, K.A.; Sharma, R.P. Active DNA demethylation in post-mitotic neurons: A reason for optimism. Neuropharmacology 2013, 75, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Mellen, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Song, C.X.; Szulwach, K.E.; Fu, Y.; Dai, Q.; Yi, C.; Li, X.; Li, Y.; Chen, C.H.; Zhang, W.; Jian, X.; et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011, 29, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.A.; Qiu, R.; Wu, X.; Li, A.X.; Zhang, H.; Wang, J.; Jui, J.; Jin, S.G.; Jiang, Y.; Pfeifer, G.P.; et al. Dynamics of 5-hydroxymethylcytosine and chromatin marks in mammalian neurogenesis. Cell Rep. 2013, 3, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.G.; Wu, X.; Li, A.X.; Pfeifer, G.P. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011, 39, 5015–5024. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed]
- Khare, T.; Pai, S.; Koncevicius, K.; Pal, M.; Kriukiene, E.; Liutkeviciute, Z.; Irimia, M.; Jia, P.; Ptak, C.; Xia, M.; et al. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat. Struct. Mol. Biol. 2012, 19, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Chambers, I. The molecular basis of pluripotency in mouse embryonic stem cells. Cloning Stem Cells 2004, 6, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H. How is pluripotency determined and maintained? Development 2007, 134, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, R.; Ohtsuka, T.; Hatakeyama, J.; Ohsawa, R. Roles of bHLH genes in neural stem cell differentiation. Exp. Cell Res. 2005, 306, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Petell, C.J.; Alabdi, L.; He, M.; San Miguel, P.; Rose, R.; Gowher, H. An epigenetic switch regulates de novo DNA methylation at a subset of pluripotency gene enhancers during embryonic stem cell differentiation. Nucleic Acids Res. 2016, 44, 7605–7617. [Google Scholar] [CrossRef] [PubMed]
- Bhanu, N.V.; Sidoli, S.; Garcia, B.A. Histone modification profiling reveals differential signatures associated with human embryonic stem cell self-renewal and differentiation. Proteomics 2016, 16, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Liang, J.; Xiong, Y.; Shi, F.; Zhang, Y.; Lu, W.; He, Q.; Yang, D.; Chen, R.; Liu, D.; et al. The trithorax group protein Ash2l is essential for pluripotency and maintaining open chromatin in embryonic stem cells. J. Biol. Chem. 2013, 288, 5039–5048. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Tada, M.; Nakatsuji, N.; Tada, T. Histone code modifications on pluripotential nuclei of reprogrammed somatic cells. Mol. Cell. Biol. 2004, 24, 5710–5720. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Nishino, K.; Ko, Y.G.; Hattori, N.; Ohgane, J.; Tanaka, S.; Shiota, K. Epigenetic control of mouse oct-4 gene expression in embryonic stem cells and trophoblast stem cells. J. Biol. Chem. 2004, 279, 17063–17069. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.P.; VerMilyea, M.D.; Turner, B.M. Epigenetic characterization of the early embryo with a chromatin immunoprecipitation protocol applicable to small cell populations. Nat. Genet. 2006, 38, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Bruce, A.W.; Jedrusik, A.; Ellis, P.D.; Andrews, R.M.; Langford, C.F.; Glover, D.M. CARM1 is required in embryonic stem cells to maintain pluripotency and resist differentiation. Stem Cells 2009, 27, 2637–2645. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.H.; Wu, Q.; Chew, J.L.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J.; et al. The Oct4 and nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006, 38, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Andres, M.E.; Burger, C.; Peral-Rubio, M.J.; Battaglioli, E.; Anderson, M.E.; Grimes, J.; Dallman, J.; Ballas, N.; Mandel, G. CoREST: A functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. USA 1999, 96, 9873–9878. [Google Scholar] [CrossRef] [PubMed]
- Grimes, J.A.; Nielsen, S.J.; Battaglioli, E.; Miska, E.A.; Speh, J.C.; Berry, D.L.; Atouf, F.; Holdener, B.C.; Mandel, G.; Kouzarides, T. The co-repressor mSin3A is a functional component of the REST-CoREST repressor complex. J. Biol. Chem. 2000, 275, 9461–9467. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, M.A.; Bochar, D.A.; Chenoweth, J.; Lane, W.S.; Mandel, G.; Shiekhattar, R. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc. Natl. Acad. Sci. USA 2002, 99, 7420–7425. [Google Scholar] [CrossRef] [PubMed]
- Roopra, A.; Qazi, R.; Schoenike, B.; Daley, T.J.; Morrison, J.F. Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Mol. Cell 2004, 14, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, P.; Westbrook, T.F.; Ottinger, M.; Pavlova, N.; Chang, B.; Macia, E.; Shi, Y.J.; Barretina, J.; Liu, J.; Howley, P.M.; et al. CDYL bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Mol. Cell 2008, 32, 718–726. [Google Scholar] [CrossRef] [PubMed]
- McGann, J.C.; Oyer, J.A.; Garg, S.; Yao, H.; Liu, J.; Feng, X.; Liao, L.; Yates, J.R., 3rd; Mandel, G. Polycomb- and REST-associated histone deacetylases are independent pathways toward a mature neuronal phenotype. eLife 2014, 3, e04235. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of developmental regulators by polycomb in human embryonic stem cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Endoh, M.; Endo, T.A.; Endoh, T.; Endoh, T.; Fujimura, Y.; Ohara, O.; Toyoda, T.; Otte, A.P.; Okano, M.; Brockdorff, N.; et al. Polycomb group proteins Ring1A/B are functionally linked to the core transcriptional regulatory circuitry to maintain ES cell identity. Development 2008, 135, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Jepsen, K.; Solum, D.; Zhou, T.; McEvilly, R.J.; Kim, H.J.; Glass, C.K.; Hermanson, O.; Rosenfeld, M.G. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 2007, 450, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Park, D.H.; Hong, S.J.; Salinas, R.D.; Liu, S.J.; Sun, S.W.; Sgualdino, J.; Testa, G.; Matzuk, M.M.; Iwamori, N.; Lim, D.A. Activation of neuronal gene expression by the JMJD3 demethylase is required for postnatal and adult brain neurogenesis. Cell Rep. 2014, 8, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.D.; Sansom, S.N.; Smith, J.; Dobenecker, M.W.; Tarakhovsky, A.; Livesey, F.J. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 15957–15962. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.A.; Huang, Y.C.; Swigut, T.; Mirick, A.L.; Garcia-Verdugo, J.M.; Wysocka, J.; Ernst, P.; Alvarez-Buylla, A. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 2009, 458, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Guizzetti, M.; Zhang, X.; Goeke, C.; Gavin, D.P. Glia and neurodevelopment: Focus on fetal alcohol spectrum disorders. Front. Pediatr. 2014, 2, 123. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Krencik, R.; Ullian, E.M.; Tsai, H.H.; Deneen, B.; Richardson, W.D.; Barres, B.A.; Rowitch, D.H. Astrocytes and disease: A neurodevelopmental perspective. Genes Dev. 2012, 26, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Heneka, M.T.; Montana, V.; Oliet, S.H.; Schousboe, A.; Haydon, P.G.; Stout, R.F., Jr; Spray, D.C.; Reichenbach, A.; Pannicke, T.; et al. Glial cells in (patho) physiology. J. Neurochem. 2012, 121, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Haydon, P.G. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc. Natl. Acad. Sci. USA 2000, 97, 8629–8634. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, T.; Nakashima, K.; Namihira, M.; Ochiai, W.; Uemura, A.; Yanagisawa, M.; Fujita, N.; Nakao, M.; Taga, T. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev. Cell 2001, 1, 749–758. [Google Scholar] [CrossRef]
- Fan, G.; Martinowich, K.; Chin, M.H.; He, F.; Fouse, S.D.; Hutnick, L.; Hattori, D.; Ge, W.; Shen, Y.; Wu, H.; et al. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development 2005, 132, 3345–3356. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Nadal-Vicens, M.; Misono, S.; Lin, M.Z.; Zubiaga, A.; Hua, X.; Fan, G.; Greenberg, M.E. Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell 2001, 104, 365–376. [Google Scholar] [CrossRef]
- Lee, H.J.; Dreyfus, C.; DiCicco-Bloom, E. Valproic acid stimulates proliferation of glial precursors during cortical gliogenesis in developing rat. Dev. Neurobiol. 2015, 76, 780–798. [Google Scholar] [CrossRef] [PubMed]
- Shaked, M.; Weissmuller, K.; Svoboda, H.; Hortschansky, P.; Nishino, N.; Wolfl, S.; Tucker, K.L. Histone deacetylases control neurogenesis in embryonic brain by inhibition of BMP2/4 signaling. PLoS ONE 2008, 3, e2668. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.; Nakashima, K.; Kuwabara, T.; Mejia, E.; Gage, F.H. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 16659–16664. [Google Scholar] [CrossRef] [PubMed]
- Laeng, P.; Pitts, R.L.; Lemire, A.L.; Drabik, C.E.; Weiner, A.; Tang, H.; Thyagarajan, R.; Mallon, B.S.; Altar, C.A. The mood stabilizer valproic acid stimulates GABA neurogenesis from rat forebrain stem cells. J. Neurochem. 2004, 91, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Greenway, D.J.; Street, M.; Jeffries, A.; Buckley, N.J. RE1 silencing transcription factor maintains a repressive chromatin environment in embryonic hippocampal neural stem cells. Stem Cells 2007, 25, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Liang, M.H.; Marinova, Z.; Yahyavi, A.; Chuang, D.M. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol. Psychiatry 2009, 14, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef] [PubMed]
- Kanski, R.; Sneeboer, M.A.; van Bodegraven, E.J.; Sluijs, J.A.; Kropff, W.; Vermunt, M.W.; Creyghton, M.P.; De Filippis, L.; Vescovi, A.; Aronica, E.; et al. Histone acetylation in astrocytes suppresses GFAP and stimulates a reorganization of the intermediate filament network. J. Cell Sci. 2014, 127, 4368–4380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yoon, H.G.; Wong, J. JMJD2A is a novel N-CoR-interacting protein and is involved in repression of the human transcription factor achaete scute-like homologue 2 (ASCL2/Hash2). Mol. Cell. Biol. 2005, 25, 6404–6414. [Google Scholar] [CrossRef] [PubMed]
- Cascante, A.; Klum, S.; Biswas, M.; Antolin-Fontes, B.; Barnabe-Heider, F.; Hermanson, O. Gene-specific methylation control of H3K9 and H3K36 on neurotrophic BDNF versus astroglial GFAP genes by KDM4A/C regulates neural stem cell differentiation. J. Mol. Biol. 2014, 426, 3467–3477. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, Y.; Suzki, N.; Tsuboi, M.; Endo, T.A.; Toyoda, T.; Shinga, J.; Koseki, H.; Vidal, M.; Gotoh, Y. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 2009, 63, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Levers, T.E.; Edgar, J.M.; Price, D.J. The fates of cells generated at the end of neurogenesis in developing mouse cortex. J. Neurobiol. 2001, 48, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Sparmann, A.; Xie, Y.; Verhoeven, E.; Vermeulen, M.; Lancini, C.; Gargiulo, G.; Hulsman, D.; Mann, M.; Knoblich, J.A.; van Lohuizen, M. The chromodomain helicase Chd4 is required for polycomb-mediated inhibition of astroglial differentiation. EMBO J. 2013, 32, 1598–1612. [Google Scholar] [CrossRef] [PubMed]
- Amarger, V.; Lecouillard, A.; Ancellet, L.; Grit, I.; Castellano, B.; Hulin, P.; Parnet, P. Protein content and methyl donors in maternal diet interact to influence the proliferation rate and cell fate of neural stem cells in rat hippocampus. Nutrients 2014, 6, 4200–4217. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Huang, K.; Yu, J.; Le, T.; Namihira, M.; Liu, Y.; Zhang, J.; Xue, Z.; Cheng, L.; Fan, G. Dnmt3a regulates both proliferation and differentiation of mouse neural stem cells. J. Neurosci. Res. 2012, 90, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Andoh-Noda, T.; Akamatsu, W.; Miyake, K.; Matsumoto, T.; Yamaguchi, R.; Sanosaka, T.; Okada, Y.; Kobayashi, T.; Ohyama, M.; Nakashima, K.; et al. Differentiation of multipotent neural stem cells derived from rett syndrome patients is biased toward the astrocytic lineage. Mol. Brain 2015, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Namihira, M.; Kohyama, J.; Semi, K.; Sanosaka, T.; Deneen, B.; Taga, T.; Nakashima, K. Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev. Cell 2009, 16, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yanagisawa, M.; Arakawa, H.; Kimura, N.; Hisatsune, T.; Kawabata, M.; Miyazono, K.; Taga, T. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science 1999, 284, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Ballas, N.; Lioy, D.T.; Grunseich, C.; Mandel, G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 2009, 12, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Kishi, N.; Macklis, J.D. MeCP2 functions largely cell-autonomously, but also non-cell-autonomously, in neuronal maturation and dendritic arborization of cortical pyramidal neurons. Exp. Neurol. 2010, 222, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Lioy, D.T.; Garg, S.K.; Monaghan, C.E.; Raber, J.; Foust, K.D.; Kaspar, B.K.; Hirrlinger, P.G.; Kirchhoff, F.; Bissonnette, J.M.; Ballas, N.; et al. A role for glia in the progression of rett’s syndrome. Nature 2011, 475, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, R.; Skrzypiec, A.; Sulkowski, S.; Buczko, W. Ethanol-induced neurotoxicity is counterbalanced by increased cell proliferation in mouse dentate gyrus. Neurosci. Lett. 2002, 327, 83–86. [Google Scholar] [CrossRef]
- Nash, R.J.; Heimburg-Molinaro, J.; Nash, R.J. Heparin binding epidermal growth factor-like growth factor reduces ethanol-induced apoptosis and differentiation in human embryonic stem cells. Growth Factors 2009, 27, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Hicks, S.D.; Middleton, F.A.; Miller, M.W. Ethanol-induced methylation of cell cycle genes in neural stem cells. J. Neurochem. 2010, 114, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, M.; Gerwe, B.A.; Scharer, C.D.; Heimburg-Molinaro, J.; Gregory, F.; Nash, R.J.; Arumugham, J.; Stewart, B.; Stice, S.L.; Nash, R.J. Low ethanol concentration alters CHRNA5 RNA levels during early human development. Reprod. Toxicol. 2010, 30, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, M.; Gerwe, B.A.; Scharer, C.D.; Heimburg-Molinaro, J.; Gregory, F.; Nash, R.J.; Arumugham, J.; Usta, S.N.; Eilertson, C.D.; Stice, S.L.; et al. GABRB3 gene expression increases upon ethanol exposure in human embryonic stem cells. J. Recept. Signal. Transduct. Res. 2011, 31, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Go, H.S.; Bak, H.R.; Choi, C.S.; Choi, I.; Kim, P.; Han, S.H.; Han, S.M.; Shin, C.Y.; Ko, K.H. Prenatal exposure of ethanol induces increased glutamatergic neuronal differentiation of neural progenitor cells. J. Biomed. Sci. 2010, 17, 85. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, M.; Gerwe, B.A.; Scharer, C.D.; Sahasranaman, V.; Eilertson, C.D.; Nash, R.J.; Usta, S.N.; Kelly, S.; Rose, M.; Peraza, R.; et al. Ethanol alters proliferation and differentiation of normal and chromosomally abnormal human embryonic stem cell-derived neurospheres. Birth Defects Res. B Dev. Reprod. Toxicol. 2013, 98, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Walker, D.W. Prenatal ethanol exposure permanently reduces the number of pyramidal neurons in rat hippocampus. Brain Res. 1981, 227, 333–340. [Google Scholar] [CrossRef]
- Miller, M.W. Generation of neurons in the rat dentate gyrus and hippocampus: Effects of prenatal and postnatal treatment with ethanol. Alcohol. Clin. Exp. Res. 1995, 19, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Wigal, T.; Amsel, A. Behavioral and neuroanatomical effects of prenatal, postnatal, or combined exposure to ethanol in weanling rats. Behav. Neurosci. 1990, 104, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Bayer, S.A.; Yackel, J.W.; Puri, P.S. Neurons in the rat dentate gyrus granular layer substantially increase during juvenile and adult life. Science 1982, 216, 890–892. [Google Scholar] [CrossRef] [PubMed]
- Rami, A.; Patel, A.J.; Rabie, A. Thyroid hormone and development of the rat hippocampus: Morphological alterations in granule and pyramidal cells. Neuroscience 1986, 19, 1217–1226. [Google Scholar] [CrossRef]
- Bonthius, D.J.; West, J.R. Alcohol-induced neuronal loss in developing rats: Increased brain damage with binge exposure. Alcohol. Clin. Exp. Res. 1990, 14, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Bonthius, D.J.; West, J.R. Permanent neuronal deficits in rats exposed to alcohol during the brain growth spurt. Teratology 1991, 44, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Bonthius, D.J.; Woodhouse, J.; Bonthius, N.E.; Taggard, D.A.; Lothman, E.W. Reduced seizure threshold and hippocampal cell loss in rats exposed to alcohol during the brain growth spurt. Alcohol. Clin. Exp. Res. 2001, 25, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Greene, P.L.; Diaz-Granados, J.L.; Amsel, A. Blood ethanol concentration from early postnatal exposure: Effects on memory-based learning and hippocampal neuroanatomy in infant and adult rats. Behav. Neurosci. 1992, 106, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.D.; Kelly, S.J. Critical periods for ethanol-induced cell loss in the hippocampal formation. Neurotoxicol. Teratol. 2003, 25, 519–528. [Google Scholar] [CrossRef]
- Borges, S.; Lewis, P.D. A study of alcohol effects on the brain during gestation and lactation. Teratology 1982, 25, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Borges, S.; Lewis, P.D. The effect of ethanol on the cellular composition of the cerebellum. Neuropathol. Appl. Neurobiol. 1983, 9, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.C.; Cragg, B.G. A change in susceptibility of rat cerebellar purkinje cells to damage by acetaldehyde during fetal, neonatal and adult life. Neuropathol. Appl. Neurobiol. 1982, 8, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Volk, B. Cerebellar histogenesis and synaptic maturation following pre- and postnatal alcohol administration: An electron-microscopic investigation of the rat cerebellar cortex. Acta Neuropathol. 1984, 63, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W. Effects of alcohol on the generation and migration of cerebral cortical neurons. Science 1986, 233, 1308–1311. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W. Effect of prenatal exposure to alcohol on the distribution and time of origin of corticospinal neurons in the rat. J. Comp. Neurol. 1987, 257, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Cragg, B.; Phillips, S. Natural loss of purkinje cells during development and increased loss with alcohol. Brain Res. 1985, 325, 151–160. [Google Scholar] [CrossRef]
- Marcussen, B.L.; Goodlett, C.R.; Mahoney, J.C.; West, J.R. Developing rat purkinje cells are more vulnerable to alcohol-induced depletion during differentiation than during neurogenesis. Alcohol 1994, 11, 147–156. [Google Scholar] [CrossRef]
- Hamre, K.M.; West, J.R. The effects of the timing of ethanol exposure during the brain growth spurt on the number of cerebellar purkinje and granule cell nuclear profiles. Alcohol. Clin. Exp. Res. 1993, 17, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Kotkoskie, L.A.; Norton, S. Cerebral cortical morphology and behavior in rats following acute prenatal ethanol exposure. Alcohol. Clin. Exp. Res. 1989, 13, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Pennington, S.N.; Boyd, J.W.; Kalmus, G.W.; Wilson, R.W. The molecular mechanism of fetal alcohol syndrome (FAS). I. ethanol-induced growth suppression. Neurobehav. Toxicol. Teratol. 1983, 5, 259–262. [Google Scholar] [PubMed]
- Ledig, M.; Ciesielski, L.; Simler, S.; Lorentz, J.G.; Mandel, P. Effect of pre- and postnatal alcohol consumption on GABA levels of various brain regions in the rat offspring. Alcohol Alcohol. 1988, 23, 63–67. [Google Scholar] [PubMed]
- Miller, M.W. Effect of prenatal exposure to ethanol on glutamate and GABA immunoreactivity in macaque somatosensory and motor cortices: Critical timing of exposure. Neuroscience 2006, 138, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.D.; Brien, J.F.; Reynolds, J.N. Chronic prenatal ethanol exposure alters the proportion of GABAergic neurons in layers II/III of the adult guinea pig somatosensory cortex. Neurotoxicol. Teratol. 2004, 26, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, S.H.; Parrish, A.R.; Nahm, S.S.; Abbott, L.C.; McCool, B.A.; Frye, G.D. Effects of early postnatal ethanol intubation on GABAergic synaptic proteins. Dev. Brain Res. 2002, 138, 177–185. [Google Scholar] [CrossRef]
- Hsiao, S.H.; DuBois, D.W.; Miranda, R.C.; Frye, G.D. Critically timed ethanol exposure reduces GABAAR function on septal neurons developing in vivo but not in vitro. Brain Res. 2004, 1008, 69–80. [Google Scholar] [CrossRef] [PubMed]
- DuBois, D.W.; Parrish, A.R.; Trzeciakowski, J.P.; Frye, G.D. Binge ethanol exposure delays development of GABAergic miniature postsynaptic currents in septal neurons. Dev. Brain Res. 2004, 152, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Park, J.H.; Choi, C.S.; Choi, I.; Joo, S.H.; Kim, M.K.; Kim, S.Y.; Kim, K.C.; Park, S.H.; Kwon, K.J.; et al. Effects of ethanol exposure during early pregnancy in hyperactive, inattentive and impulsive behaviors and MeCP2 expression in rodent offspring. Neurochem. Res. 2013, 38, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W. Effect of prenatal exposure to ethanol on the development of cerebral cortex: I. Neuronal generation. Alcohol. Clin. Exp. Res. 1988, 12, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W. Effects of prenatal exposure to ethanol on callosal projection neurons in rat somatosensory cortex. Brain Res. 1997, 766, 121–128. [Google Scholar] [CrossRef]
- Al-Rabiai, S.; Miller, M.W. Effect of prenatal exposure to ethanol on the ultrastructure of layer V of mature rat somatosensory cortex. J. Neurocytol. 1989, 18, 711–729. [Google Scholar] [CrossRef] [PubMed]
- Benuskova, L.; Rema, V.; Armstrong-James, M.; Ebner, F.F. Theory for normal and impaired experience-dependent plasticity in neocortex of adult rats. Proc. Natl. Acad. Sci. USA 2001, 98, 2797–2802. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.; Zachariah, R.M.; Davie, J.R.; Rastegar, M. Ethanol deregulates Mecp2/MeCP2 in differentiating neural stem cells via interplay between 5-methylcytosine and 5-hydroxymethylcytosine at the Mecp2 regulatory elements. Exp. Neurol. 2015, 265, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Guizzetti, M.; Costa, L.G. Inhibition of muscarinic receptor-stimulated glial cell proliferation by ethanol. J. Neurochem. 1996, 67, 2236–2245. [Google Scholar] [CrossRef] [PubMed]
- Guerri, C.; Pascual, M.; Renau-Piqueras, J. Glia and fetal alcohol syndrome. Neurotoxicology 2001, 22, 593–599. [Google Scholar] [CrossRef]
- Valles, S.; Sancho-Tello, M.; Minana, R.; Climent, E.; Renau-Piqueras, J.; Guerri, C. Glial fibrillary acidic protein expression in rat brain and in radial glia culture is delayed by prenatal ethanol exposure. J. Neurochem. 1996, 67, 2425–2433. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, T.L.; Shain, W. Ethanol-induced changes in astrocyte gene expression during rat central nervous system development. Alcohol. Clin. Exp. Res. 1993, 17, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Goodlett, C.R.; Leo, J.T.; O’Callaghan, J.P.; Mahoney, J.C.; West, J.R. Transient cortical astrogliosis induced by alcohol exposure during the neonatal brain growth spurt in rats. Dev. Brain Res. 1993, 72, 85–97. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Peterson, S.D.; Lundahl, K.R.; Pearlman, A.D. Binge-like alcohol exposure of neonatal rats via intragastric intubation induces both purkinje cell loss and cortical astrogliosis. Alcohol. Clin. Exp. Res. 1997, 21, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Topper, L.A.; Baculis, B.C.; Valenzuela, C.F. Exposure of neonatal rats to alcohol has differential effects on neuroinflammation and neuronal survival in the cerebellum and hippocampus. J. Neuroinflamm. 2015, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Veazey, K.J.; Parnell, S.E.; Miranda, R.C.; Golding, M.C. Dose-dependent alcohol-induced alterations in chromatin structure persist beyond the window of exposure and correlate with fetal alcohol syndrome birth defects. Epigenet. Chromatin 2015, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Numata, S.; Ye, T.; Hyde, T.M.; Guitart-Navarro, X.; Tao, R.; Wininger, M.; Colantuoni, C.; Weinberger, D.R.; Kleinman, J.E.; Lipska, B.K. DNA methylation signatures in development and aging of the human prefrontal cortex. Am. J. Hum. Genet. 2012, 90, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.R.; Ho, M.F.; Vega, M.C.; Burne, T.H.; Chong, S. Prenatal ethanol exposure alters adult hippocampal VGLUT2 expression with concomitant changes in promoter DNA methylation, H3K4 trimethylation and miR-467b-5p levels. Epigenet. Chromatin 2015, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Bundo, M.; Ueda, J.; Oldham, M.C.; Ukai, W.; Hashimoto, E.; Saito, T.; Geschwind, D.H.; Kato, T. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 2011, 21, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 2015, 86, 1369–1384. [Google Scholar] [CrossRef] [PubMed]
- Kozlenkov, A.; Roussos, P.; Timashpolsky, A.; Barbu, M.; Rudchenko, S.; Bibikova, M.; Klotzle, B.; Byne, W.; Lyddon, R.; Di Narzo, A.F.; et al. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 2014, 42, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Kaminen-Ahola, N.; Ahola, A.; Maga, M.; Mallitt, K.A.; Fahey, P.; Cox, T.C.; Whitelaw, E.; Chong, S. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet. 2010, 6, e1000811. [Google Scholar] [CrossRef] [PubMed]
- Downing, C.; Johnson, T.E.; Larson, C.; Leakey, T.I.; Siegfried, R.N.; Rafferty, T.M.; Cooney, C.A. Subtle decreases in DNA methylation and gene expression at the mouse Igf2 locus following prenatal alcohol exposure: Effects of a methyl-supplemented diet. Alcohol 2011, 45, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Varela-Rey, M.; Woodhoo, A.; Martinez-Chantar, M.L.; Mato, J.M.; Lu, S.C. Alcohol, DNA methylation, and cancer. Alcohol Res. 2013, 35, 25–35. [Google Scholar] [PubMed]
- Mandaviya, P.R.; Stolk, L.; Heil, S.G. Homocysteine and DNA methylation: A review of animal and human literature. Mol. Genet. Metab. 2014, 113, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, S.H.; Nicolaou, A.; Gibbons, W.A. The effect of ethanol and its metabolites upon methionine synthase activity in vitro. Alcohol 1998, 15, 305–309. [Google Scholar] [CrossRef]
- Hutson, J.R.; Stade, B.; Lehotay, D.C.; Collier, C.P.; Kapur, B.M. Folic acid transport to the human fetus is decreased in pregnancies with chronic alcohol exposure. PLoS ONE 2012, 7, e38057. [Google Scholar] [CrossRef] [PubMed]
- Barak, A.J.; Beckenhauer, H.C.; Tuma, D.J.; Badakhsh, S. Effects of prolonged ethanol feeding on methionine metabolism in rat liver. Biochem. Cell Biol. 1987, 65, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Barak, A.J.; Tuma, D.J.; Beckenhauer, H.C. Ethanol, the choline requirement, methylation and liver injury. Life Sci. 1985, 37, 789–791. [Google Scholar] [CrossRef]
- Wang, L.L.; Zhang, Z.; Li, Q.; Yang, R.; Pei, X.; Xu, Y.; Wang, J.; Zhou, S.F.; Li, Y. Ethanol exposure induces differential microRNA and target gene expression and teratogenic effects which can be suppressed by folic acid supplementation. Hum. Reprod. 2009, 24, 562–579. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Idrus, N.M.; Monk, B.R.; Dominguez, H.D. Prenatal choline supplementation mitigates behavioral alterations associated with prenatal alcohol exposure in rats. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kusumo, H.; Sakharkar, A.J.; Pandey, S.C.; Guizzetti, M. Regulation of DNA methylation by ethanol induces tissue plasminogen activator expression in astrocytes. J. Neurochem. 2014, 128, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rezzoug, F.; Kaikaus, J.; Greene, R.M.; Pisano, M.M. Alcohol modulates expression of DNA methyltranferases and methyl CpG-/CpG domain-binding proteins in murine embryonic fibroblasts. Reprod. Toxicol. 2013, 37, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chen, Y.; Carreon, S.; Qiang, M. Chronic intermittent ethanol exposure and its removal induce a different miRNA expression pattern in primary cortical neuronal cultures. Alcohol. Clin. Exp. Res. 2012, 36, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Garro, A.J.; McBeth, D.L.; Lima, V.; Lieber, C.S. Ethanol consumption inhibits fetal DNA methylation in mice: Implications for the fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1991, 15, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Nagre, N.N.; Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Basavarajappa, B.S. CB1-receptor knockout neonatal mice are protected against ethanol-induced impairments of DNMT1, DNMT3A, and DNA methylation. J. Neurochem. 2015, 132, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.C.; Chen, Y.; Love, A. Cellular DNA methylation program during neurulation and its alteration by alcohol exposure. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ozturk, N.C.; Zhou, F.C. DNA methylation program in developing hippocampus and its alteration by alcohol. PLoS ONE 2013, 8, e60503. [Google Scholar] [CrossRef] [PubMed]
- Otero, N.K.; Thomas, J.D.; Saski, C.A.; Xia, X.; Kelly, S.J. Choline supplementation and DNA methylation in the hippocampus and prefrontal cortex of rats exposed to alcohol during development. Alcohol. Clin. Exp. Res. 2012, 36, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Chater-Diehl, E.J.; Laufer, B.I.; Castellani, C.A.; Alberry, B.L.; Singh, S.M. Alteration of gene expression, DNA methylation, and histone methylation in free radical scavenging networks in adult mouse hippocampus following fetal alcohol exposure. PLoS ONE 2016, 11, e0154836. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Lehmann, C.; Lawrence, R.C.; Kelly, S.J. Alcohol exposure during development: Impact on the epigenome. Int. J. Dev. Neurosci. 2013, 31, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in beta-endorphin-producing POMC neurons of the hypothalamus. Alcohol. Clin. Exp. Res. 2013, 37, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Govorko, D.; Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biol. Psychiatry 2012, 72, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Nagre, N.N.; Shivakumar, M.; Umapathy, N.S.; Psychoyos, D.; Basavarajappa, B.S. Ethanol induced acetylation of histone at G9a exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice. Neuroscience 2014, 258, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Tunc-Ozcan, E.; Ullmann, T.M.; Shukla, P.K.; Redei, E.E. Low-dose thyroxine attenuates autism-associated adverse effects of fetal alcohol in male offspring’s social behavior and hippocampal gene expression. Alcohol. Clin. Exp. Res. 2013, 37, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Gangisetty, O.; Bekdash, R.; Maglakelidze, G.; Sarkar, D.K. Fetal alcohol exposure alters proopiomelanocortin gene expression and hypothalamic-pituitary-adrenal axis function via increasing MeCP2 expression in the hypothalamus. PLoS ONE 2014, 9, e113228. [Google Scholar] [CrossRef] [PubMed]
- Portales-Casamar, E.; Lussier, A.A.; Jones, M.J.; MacIsaac, J.L.; Edgar, R.D.; Mah, S.M.; Barhdadi, A.; Provost, S.; Lemieux-Perreault, L.P.; Cynader, M.S.; et al. DNA methylation signature of human fetal alcohol spectrum disorder. Epigenet. Chromatin 2016, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Laufer, B.I.; Kapalanga, J.; Castellani, C.A.; Diehl, E.J.; Yan, L.; Singh, S.M. Associative DNA methylation changes in children with prenatal alcohol exposure. Epigenomics 2015, 7, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.C.; Balaraman, Y.; Teng, M.; Liu, Y.; Singh, R.P.; Nephew, K.P. Alcohol alters DNA methylation patterns and inhibits neural stem cell differentiation. Alcohol. Clin. Exp. Res. 2011, 35, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Laufer, B.I.; Mantha, K.; Kleiber, M.L.; Diehl, E.J.; Addison, S.M.; Singh, S.M. Long-lasting alterations to DNA methylation and ncRNAs could underlie the effects of fetal alcohol exposure in mice. Dis. Models Mech. 2013, 6, 977–992. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Balaraman, Y.; Wang, G.; Nephew, K.P.; Zhou, F.C. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 2009, 4, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.; Pitarch, J.; Renau-Piqueras, J.; Guerri, C. Ethanol exposure affects glial fibrillary acidic protein gene expression and transcription during rat brain development. J. Neurochem. 1997, 69, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Ouko, L.A.; Shantikumar, K.; Knezovich, J.; Haycock, P.; Schnugh, D.J.; Ramsay, M. Effect of alcohol consumption on CpG methylation in the differentially methylated regions of H19 and IG-DMR in male gametes: Implications for fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2009, 33, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Masemola, M.L.; van der Merwe, L.; Lombard, Z.; Viljoen, D.; Ramsay, M. Reduced DNA methylation at the PEG3 DMR and KvDMR1 loci in children exposed to alcohol in utero: A south african fetal alcohol syndrome cohort study. Front. Genet. 2015, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Ngai, Y.F.; Sulistyoningrum, D.C.; O’Neill, R.; Innis, S.M.; Weinberg, J.; Devlin, A.M. Prenatal alcohol exposure alters methyl metabolism and programs serotonin transporter and glucocorticoid receptor expression in brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R613–R622. [Google Scholar] [CrossRef] [PubMed]
- Marjonen, H.; Sierra, A.; Nyman, A.; Rogojin, V.; Grohn, O.; Linden, A.M.; Hautaniemi, S.; Kaminen-Ahola, N. Early maternal alcohol consumption alters hippocampal DNA methylation, gene expression and volume in a mouse model. PLoS ONE 2015, 10, e0124931. [Google Scholar] [CrossRef] [PubMed]
- Veazey, K.J.; Carnahan, M.N.; Muller, D.; Miranda, R.C.; Golding, M.C. Alcohol-induced epigenetic alterations to developmentally crucial genes regulating neural stemness and differentiation. Alcohol. Clin. Exp. Res. 2013, 37, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Bielawski, D.M.; Zaher, F.M.; Svinarich, D.M.; Abel, E.L. Paternal alcohol exposure affects sperm cytosine methyltransferase messenger RNA levels. Alcohol. Clin. Exp. Res. 2002, 26, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Crossey, E.L.; Zhang, L.; Zucca, S.; George, O.L.; Valenzuela, C.F.; Zhao, X. Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS ONE 2011, 6, e19351. [Google Scholar] [CrossRef] [PubMed]
- Kleiber, M.L.; Mantha, K.; Stringer, R.L.; Singh, S.M. Neurodevelopmental alcohol exposure elicits long-term changes to gene expression that alter distinct molecular pathways dependent on timing of exposure. J. Neurodev. Disord. 2013, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Shivakumar, M.; Umapathy, N.S.; Saito, M.; Mohan, P.S.; Kumar, A.; Nixon, R.A.; Verin, A.D.; Psychoyos, D.; Basavarajappa, B.S. G9a-mediated histone methylation regulates ethanol-induced neurodegeneration in the neonatal mouse brain. Neurobiol. Dis. 2013, 54, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Nagre, N.N.; Umapathy, N.S.; Pace, B.S.; Basavarajappa, B.S. Ethanol exposure induces neonatal neurodegeneration by enhancing CB1R Exon1 histone H4K8 acetylation and up-regulating CB1R function causing neurobehavioral abnormalities in adult mice. Int J. Neuropsychopharmacol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Varadinova, M.; Boyadjieva, N. Epigenetic mechanisms: A possible link between autism spectrum disorders and fetal alcohol spectrum disorders. Pharmacol. Res. 2015, 102, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Tyler, C.R.; Allan, A.M. Prenatal alcohol exposure alters expression of neurogenesis-related genes in an ex vivo cell culture model. Alcohol 2014, 48, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Gressens, P.; Lammens, M.; Picard, J.J.; Evrard, P. Ethanol-induced disturbances of gliogenesis and neuronogenesis in the developing murine brain: An in vitro and in vivo immunohistochemical and ultrastructural study. Alcohol Alcohol. 1992, 27, 219–226. [Google Scholar] [PubMed]
- Miller, M.W.; Robertson, S. Prenatal exposure to ethanol alters the postnatal development and transformation of radial glia to astrocytes in the cortex. J. Comp. Neurol. 1993, 337, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Rubert, G.; Minana, R.; Pascual, M.; Guerri, C. Ethanol exposure during embryogenesis decreases the radial glial progenitorpool and affects the generation of neurons and astrocytes. J. Neurosci. Res. 2006, 84, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.H.; Williams, J.K.; Thomas, J.D. Choline supplementation attenuates learning deficits associated with neonatal alcohol exposure in the rat: Effects of varying the timing of choline administration. Brain Res. 2008, 1237, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Biane, J.S.; O’Bryan, K.A.; O’Neill, T.M.; Dominguez, H.D. Choline supplementation following third-trimester-equivalent alcohol exposure attenuates behavioral alterations in rats. Behav. Neurosci. 2007, 121, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Rastegar, M.; Hotta, A.; Pasceri, P.; Makarem, M.; Cheung, A.Y.; Elliott, S.; Park, K.J.; Adachi, M.; Jones, F.S.; Clarke, I.D.; et al. MECP2 isoform-specific vectors with regulated expression for rett syndrome gene therapy. PLoS ONE 2009, 4, e6810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bhattacharyya, S.; Kusumo, H.; Goodlett, C.R.; Tobacman, J.K.; Guizzetti, M. Arylsulfatase B modulates neurite outgrowth via astrocyte chondroitin-4-sulfate: Dysregulation by ethanol. Glia 2014, 62, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Guerri, C. The peptide NAP promotes neuronal growth and differentiation through extracellular signal-regulated protein kinase and akt pathways, and protects neurons co-cultured with astrocytes damaged by ethanol. J. Neurochem. 2007, 103, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Kleiber, M.L.; Laufer, B.I.; Stringer, R.L.; Singh, S.M. Third trimester-equivalent ethanol exposure is characterized by an acute cellular stress response and an ontogenetic disruption of genes critical for synaptic establishment and function in mice. Dev. Neurosci. 2014, 36, 499–519. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Basavarajappa, B.S. Pre-administration of G9a/GLP inhibitor during synaptogenesis prevents postnatal ethanol-induced LTP deficits and neurobehavioral abnormalities in adult mice. Exp. Neurol. 2014, 261, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Rogic, S.; Wong, A.; Pavlidis, P. Meta-analysis of gene expression patterns in animal models of prenatal alcohol exposure suggests role for protein synthesis inhibition and chromatin remodeling. Alcohol. Clin. Exp. Res. 2016, 40, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Shilatifard, A. Histone modification: Cause or cog? Trends Genet. 2011, 27, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Badeaux, A.I.; Shi, Y. Emerging roles for chromatin as a signal integration and storage platform. Nat. Rev. Mol. Cell Biol. 2013, 14, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Coles, C.D.; Kable, J.A.; Keen, C.L.; Jones, K.L.; Wertelecki, W.; Granovska, I.V.; Pashtepa, A.O.; Chambers, C.D.; CIFASD. Dose and timing of prenatal alcohol exposure and maternal nutritional supplements: Developmental effects on 6-month-old infants. Matern. Child Health J. 2015, 19, 2605–2614. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Risbud, R.D.; Mattson, S.N.; Chambers, C.D.; Thomas, J.D. Randomized, double-blind, placebo-controlled clinical trial of choline supplementation in school-aged children with fetal alcohol spectrum disorders. Am. J. Clin. Nutr. 2016, 104, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.D.; Thomas, J.D. Adolescent choline supplementation attenuates working memory deficits in rats exposed to alcohol during the third trimester equivalent. Alcohol. Clin. Exp. Res. 2016, 40, 897–905. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| 5-methylcytosine (5mC) | |||||||
| Global or Gene Specific | Increase or Decrease | Time Alcohol Administered | Time Studied | Brain Region/Cell Culture | Species | Sex | Reference |
| Global | ↓ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| ↓ | E8.25 | E10 | Neural tube | mice | U | [175] | |
| ↓ | E9–11 | E11 | whole embryo | mice | B | [161] | |
| NC | E5–16 | E17 | ammonic neuroepithelium | mice | B | [164] | |
| ↑ | E5–16 | E17 | intermediate zone | mice | B | [164] | |
| ↑ | E5–16 | E17 | hippocampus CA1 | mice | B | [164] | |
| ↓ | P7 | P8 | hippocampus | mice | B | [162] | |
| ↓ | P7 | P8 | neocortex | mice | B | [162] | |
| ↑ | P2–10 | P21 | Prefrontal cortex | rats | B | [165] | |
| ↑ | P2–10 | P21 | hippocampus | rats | B | [165] | |
| NC | E7–21 | P60–80 | hypothalamus | rats | B | [169] | |
| Genome-wide | mostly ↓ | 48 h | NSC | B | [163] | ||
| ↑↓ | E8–10 * | E10 | whole embryo | mice | B | [177] | |
| mostly ↑ | P4 & P7 | P70 | Hippocampus | mice | M | [166] | |
| ↑↓ | 5–18 yo | Buccal Epithelial Cells | human | B | [173] | ||
| Cell cycle genes | ↑ | 48 h | NSC | F | [97] | ||
| MeCP2 promoter | ↑ | D0–2 | D2 | NSC | mice | B | [134] |
| ↓ | D0–8 | D8 | NSC | mice | B | [134] | |
| Igf2 | ↓ | E9 | E9 | whole embryo | mice | B | [149] |
| Gfap promoter | ↑ | E1–21 | E21 | whole brain | rats | B | [178] |
| Plat promoter | ↓ | 24 h | primary cortical astrocytes | rats | B | [158] | |
| Imprinted genes (H19 and IG-DMR) | ↓ | correlated with alcohol drinking | sperm | human | M | [179] | |
| KCNQ1OT1 | ↓ | 1–26 yo | blood | human | B | [180] | |
| PEG3 promoter | ↓ | 1–26 yo | blood | human | B | [180] | |
| Slc6a4 promoter | ↑ | E1–21 | P55 | hypothalamus | rats | F | [181] |
| Pomc promoter | ↑ | E7–21 | P60–80 | hypothalamus | rats | B | [169] |
| Gm9268 promoter | ↑ | E0.5–8.5 | P28 | hippocampus | mice | M | [182] |
| Vpreb2 promoter | ↑ | E0.5–8.5 | P28 | hippocampus | mice | M | [182] |
| Olfr601 promoter | ↓ | E0.5–8.5 | P28 | hippocampus | mice | M | [182] |
| Slc17a6 promoter | ↓ | E0.5–8.5 | P120 | hippocampus | mice | M | [144] |
| 5-hydroxymethylcytosine (5hmC) | |||||||
| Global or Gene Specific | Increase or Decrease | Time Alcohol Administered | Time Studied | Brain Region/Cell Culture | Species | Sex | Reference |
| Global | ↓ | E5–16 | E17 | ammonic neuroepithelium | mice | B | [164] |
| ↓ | E5–16 | E17 | intermediate zone | mice | B | [164] | |
| DNMT Expression/Activity | |||||||
| DNMT Isoform mRNA, Protein, Activity | Increase or Decrease | Time Alcohol Administered | Time Studied | Brain Region/Cell Culture | Species | Sex | Reference |
| Dnmt1 mRNA | ↓ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| ↑ | 5D | neurospheres | mice | B | [183] | ||
| ↑ | D1–3 | D7 | neurospheres | mice | B | [142] | |
| NC | 24 h | primary cortical astrocytes | rats | B | [158] | ||
| ↓ | P90–155 | P155 | sperm | rats | M | [184] | |
| ↓ | P7 | P8 | hippocampus | mice | B | [162] | |
| ↑ | P7 | P8 | neocortex | mice | B | [162] | |
| ↑ | E7–21 | P60–65 | hypothalamus | rats | M | [168] | |
| DNMT1 protein | ↓ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| ↑ | 48 h | NSC | F | [97] | |||
| NC | 24 h | primary cortical astrocytes | rats | B | [158] | ||
| NC | E6–15 | P35 | striatum | mice | U | [129] | |
| ↓ | P7 | P8 | hippocampus | mice | B | [162] | |
| NC | E6–15 | P35 | cortex | mice | U | [129] | |
| ↓ | P7 | P8 | neocortex | mice | B | [162] | |
| ↑ | E7–21 | P60–65 | hypothalamus | rats | M | [168] | |
| Dnmt3a mRNA | ↑ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| NC | 24 h | primary cortical astrocytes | rats | B | [158] | ||
| ↓ | P7 (high dose) | P8 | hippocampus | mice | B | [162] | |
| ↑ | P7 (low dose) | P8 | hippocampus | mice | B | [170] | |
| ↓ | P7 (high dose) | P8 | neocortex | mice | B | [162] | |
| ↑ | P7 (low dose) | P8 | neocortex | mice | B | [170] | |
| DNMT3A protein | ↓ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| ↓ | 24 h | primary cortical astrocytes | rats | B | [158] | ||
| ↓ | P7 (high dose) | P8 | hippocampus | mice | B | [162] | |
| ↑ | P7 (low dose) | P8 | hippocampus | mice | B | [170] | |
| ↓ | P7 (high dose) | P8 | neocortex | mice | B | [162] | |
| ↑ | P7 (low dose) | P8 | neocortex | mice | B | [170] | |
| ↑ | E7–21 | P60–65 | hypothalamus | rats | M | [168] | |
| Dnmt3b mRNA | ↑ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| DNMT3B protein | ↓ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| DNMT activity | ↑ | 48 h | NSC | F | [97] | ||
| ↓ | 24 or 48 h | primary cortical astrocytes | rats | B | [158] | ||
| ↓ | E9–11 | E11 | whole embryo | mice | B | [161] | |
| ↑ | E1-P10 | P21 | hippocampus | rats | B | [167] | |
| MBD Expression | |||||||
| MBD Isoform mRNA/Protein | Increase or Decrease | Time Alcohol Administered | Time Studied | Brain Region/Cell Culture | Species | Sex | Reference |
| MeCP2 mRNA | ↑ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| ↑ | D0–2 | D2 | NSC | mice | B | [134] | |
| ↑ | D0–8 | D8 | NSC | mice | B | [134] | |
| ↓ | D0–2 | D8 | NSC | mice | B | [134] | |
| ↓ | D3–13 | D13 | primary cortical neurons | mice | B | [160] | |
| ↓ | D3–8 | D13 | primary cortical neurons | mice | B | [160] | |
| ↑ | E7–21 | P60–65 | hypothalamus | rats | M | [168] | |
| NC | P7 (low dose) | P8 | hippocampus | mice | B | [170] | |
| NC | P7 (low dose) | P8 | neocortex | mice | B | [170] | |
| MeCP2 protein | ↓ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| ↑ | D0–2 | D2 | NSC | mice | B | [134] | |
| ↑ | D0–8 | D8 | NSC | mice | B | [134] | |
| ↓ | D0–2 | D8 | NSC | mice | B | [134] | |
| ↓ | D3–13 | D13 | primary cortical neurons | mice | B | [160] | |
| ↓ | D3–8 | D13 | primary cortical neurons | mice | B | [160] | |
| ↓ | E6–15 | P35 | striatum | mice | U | [129] | |
| ↓ | E5–16 | E17 | hippocampus | mice | B | [164] | |
| ↑ | P7 (low dose) | P8 | hippocampus | mice | B | [170] | |
| ↑ | E8–21 | Adult | hippocampus | rats | M | [171] | |
| ↓ | E6–15 | P35 | cortex | mice | U | [129] | |
| ↑ | P7 (low dose) | P8 | neocortex | mice | B | [170] | |
| ↑ | E7–21 | P60–65 | hypothalamus | rats | M | [168] | |
| Mbd2 mRNA | ↓ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| MBD2 protein | ↓ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| Mbd3 mRNA | ↓ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| MBD3 protein | ↓ | 48 h | embryonic fibroblasts | mice | M | [159] | |
| Histone Acetylation | |||||||
| Global or Gene Specific | Increase or Decrease | Time Alcohol Administered | Time Studied | Brain Region/Cell Culture | Species | Sex | Reference |
| Global | ↓ | E7–21 | P60–80 | hypothalamus | rats | M | [168] |
| ↓ | P2–10 | P2–10 | cerebellum | rats | B | [185] | |
| NC | P2–12 | P12 | cerebellum | rats | B | [185] | |
| 22 Growth factor genes | ↑ | D1–3 (low dose) | D3 | NSC | mice | B | [142] |
| ↓ | D1–3 (high dose) | D3 | NSC | mice | B | [142] | |
| ↑ | D1–3 (low dose) | D7 | NSC | mice | B | [142] | |
| ↓ | D1–3 (high dose) | D7 | NSC | mice | B | [142] | |
| Ehmt2 promoter | ↑ | P7 (low dose) | P8 | neocortex | mice | B | [170] |
| Cnr1 promoter | ↑ | P7 | P8 | neocortex, hippocamus | mice | B | [177] |
| Dlx2 Promoter | ↑ | E7 | E17 | neocortex | mice | B | [142] |
| HAT/HDAC Expression | |||||||
| HAT/HDAC isoform mRNA/protein | Increase or Decrease | Time Alcohol Administered | Time Studied | Brain Region/Cell Culture | Species | Sex | Reference |
| Crebbp mRNA | ↓ | E7–21 | P60–80 | hypothalamus | rats | B | [169] |
| CREBBP protein | ↓ | P2–10 | P2–10 | cerebellum | rats | B | [185] |
| CREBBP protein | NC | P2–12 | P12 | cerebellum | rats | B | [185] |
| Hdac1 mRNA | ↓ | P7 | P7 | whole brain | mice | M | [186] |
| Hdac2 mRNA | ↑ | E7–21 | P60–80 | hypothalamus | rats | B | [169] |
| H3K4 Methylation | |||||||
| Global or Gene Specific | Increase or Decrease | Time Alcohol Administered | Time Studied | Brain Region/Cell Culture | Species | Sex | Reference |
| Global | ↓ | E7–21 | P60–65 | hypothalamus | rats | M | [168] |
| 22 Growth factor genes | ↑ | D1–3 (low dose) | D3 | NSC | mice | B | [142] |
| ↓ | D1–3 (high dose) | D3 | NSC | mice | B | [142] | |
| ↑↓ | D1–3 (low dose) | D7 | NSC | mice | B | [142] | |
| ↑↓ | D1–3 (high dose) | D7 | NSC | mice | B | [142] | |
| Genome-wide | ↑↓ | P4 & P7 | P70 | Hippocampus | mice | M | [166] |
| Slc17a6 promoter | ↑ | E0.5–8.5 | P120 | hippocampus | mice | M | [144] |
| Sox2 Promoter | ↓ | D1–5 | D5 | neurospheres | mice | B | [183] |
| Dlx2 Promoter | ↓ | D1–5 | D5 | neurospheres | mice | B | [183] |
| Pax6 Promoter | ↓ | D1–5 | D5 | neurospheres | mice | B | [183] |
| H3K4 Methyltransferase/Demethylase Expression | |||||||
| HMT/Demethylase isoform mRNA/protein | Increase or Decrease | Time Alcohol Administered | Time Studied | Brain Region/Cell Culture | Species | Sex | Reference |
| Ash2l1 mRNA | ↓ | D1–5 | D5 | neurospheres | mice | B | [183] |
| Setd7 mRNA | ↓ | E7–21 | P60–80 | hypothalamus | rats | M | [168] |
| Kdm1b mRNA | ↓ | D1–5 | D5 | neurospheres | mice | B | [183] |
| H3K9 Methylation | |||||||
| Global or Gene Specific | Increase or Decrease | Time Alcohol Administered | Time Studied | Brain Region/Cell Culture | Species | Sex | Reference |
| Global | ↑ | E7–21 | P60–65 | hypothalamus | rats | M | [168] |
| 22 Growth factor genes | ↑ | P7 (high dose) | P8 | hippocampus | mice | B | [187] |
| ↑ | P7 (high dose) | P8 | neocortex | mice | B | [187] | |
| ↓ | D1–3 (low dose) | D3 | NSC | mice | B | [142] | |
| ↓ | D1–3 (high dose) | D3 | NSC | mice | B | [142] | |
| ↑ | D1–3 (low dose) | D7 | NSC | mice | B | [142] | |
| ↑ | D1–3 (high dose) | D7 | NSC | mice | B | [142] | |
| Cnr1 promoter | ↓ | P7 | P8 | neocortex, hippocampus | mice | B | [188] |
| Dlx2 Promoter | ↑ | E7 | E17 | neocortex | mice | B | [142] |
| Dlx3 Promoter | ↑ | E7 | E17 | neocortex | mice | B | [142] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavin, D.P.; Grayson, D.R.; Varghese, S.P.; Guizzetti, M. Chromatin Switches during Neural Cell Differentiation and Their Dysregulation by Prenatal Alcohol Exposure. Genes 2017, 8, 137. https://doi.org/10.3390/genes8050137
Gavin DP, Grayson DR, Varghese SP, Guizzetti M. Chromatin Switches during Neural Cell Differentiation and Their Dysregulation by Prenatal Alcohol Exposure. Genes. 2017; 8(5):137. https://doi.org/10.3390/genes8050137
Chicago/Turabian StyleGavin, David P., Dennis R. Grayson, Sajoy P. Varghese, and Marina Guizzetti. 2017. "Chromatin Switches during Neural Cell Differentiation and Their Dysregulation by Prenatal Alcohol Exposure" Genes 8, no. 5: 137. https://doi.org/10.3390/genes8050137
APA StyleGavin, D. P., Grayson, D. R., Varghese, S. P., & Guizzetti, M. (2017). Chromatin Switches during Neural Cell Differentiation and Their Dysregulation by Prenatal Alcohol Exposure. Genes, 8(5), 137. https://doi.org/10.3390/genes8050137