
The Epigenetic Link between Prenatal Adverse Environments and Neurodevelopmental Disorders

Abstract
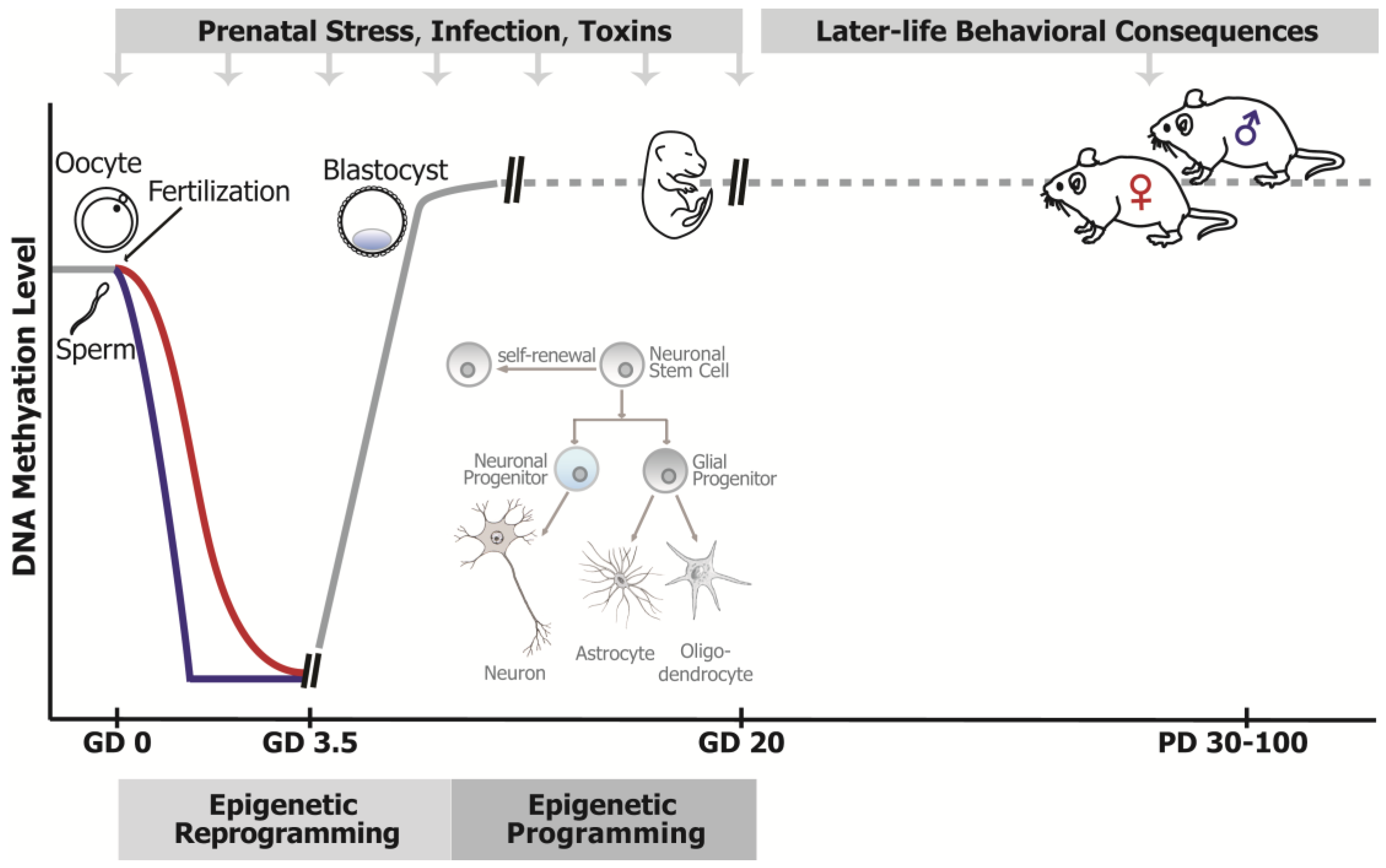
:1. Prenatal Adversity and Neurodevelopmental Disorders
2. Epigenetic Mechanisms and Prenatal Programming of Brain Function
3. Experimental Evidence from Animal Studies
3.1. Maternal Stress
3.2. Maternal Toxicological Exposures
3.3. Maternal Immune Activation
3.4. Maternal Exposure to Drugs
3.4.1. Cannabis
3.4.2. Cocaine
3.4.3. Ethanol
4. Evidence from Human Studies
4.1. Dutch Famine Study
4.2. Prenatal Stress and Maternal Depression
4.3. Toxicological Exposures
5. Future Directions and Challenges
5.1. Dose and Developmental Window of Exposure
5.2. Sex-Specific Effects
5.3. Peripheral Biomarkers
5.4. Technological Advancements and Integrative Approaches
Acknowledgments
Conflicts of Interest
References
- Bale, T.L.; Baram, T.Z.; Brown, A.S.; Goldstein, J.M.; Insel, T.R.; McCarthy, M.M.; Nemeroff, C.B.; Reyes, T.M.; Simerly, R.B.; Susser, E.S. Early life programming and neurodevelopmental disorders. Biol. Psychiatry 2010, 68, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P.; Landrigan, P.J. Neurobehavioural effects of developmental toxicity. Lancet Neurol. 2014, 13, 330–338. [Google Scholar] [CrossRef]
- Meyer, U. Prenatal poly (i: C) exposure and other developmental immune activation models in rodent systems. Biol. Psychiatry 2014, 75, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.J.; Graham, D.L.; Money, K.M.; Stanwood, G.D. Developmental consequences of fetal exposure to drugs: What we know and what we still must learn. Neuropsychopharmacology 2015, 40, 61–87. [Google Scholar] [CrossRef] [PubMed]
- Scheinost, D.; Sinha, R.; Cross, S.N.; Kwon, S.H.; Sze, G.; Constable, R.T.; Ment, L.R. Does prenatal stress alter the developing connectome? Pediatr. Res. 2016, 81, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.R.; Bale, T.L.; Epperson, C.N. Prenatal programming of mental illness: Current understanding of relationship and mechanisms. Curr. Psychiatry Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J.; O’Donovan, M.C.; Thapar, A.; Craddock, N. Neurodevelopmental hypothesis of schizophrenia. Br. J. Psychiatry 2011, 198, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Folsom, T.D. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr. Bull. 2009, 35, 528–548. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.; Weinberger, D. Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Mol. Psychiatry 2004, 10, 40–68. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.H.; Van Tol, H.H. Schizophrenia: From phenomenology to neurobiology. Neurosci. Biobehav. Rev. 2003, 27, 269–306. [Google Scholar] [CrossRef]
- Khashan, A.S.; Abel, K.M.; McNamee, R.; Pedersen, M.G.; Webb, R.T.; Baker, P.N.; Kenny, L.C.; Mortensen, P.B. Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events. Arch. Gen. Psychiatry 2008, 65, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.; Petronis, A. Environmental studies of schizophrenia through the prism of epigenetics. Schizophr. Bull. 2008, 34, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Susser, E.S.; Lin, S.P. Schizophrenia after prenatal exposure to the dutch hunger winter of 1944–1945. Arch. Gen. Psychiatry 1992, 49, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Susser, E.S.; Lin, S.P.; Neugebauer, R.; Gorman, J.M. Increased risk of affective disorders in males after second trimester prenatal exposure to the dutch hunger winter of 1944–45. Br. J. Psychiatry 1995, 166, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; van Os, J.; Driessens, C.; Hoek, H.W.; Susser, E.S. Further evidence of relation between prenatal famine and major affective disorder. Am. J. Psychiatry 2000, 157, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Mill, J.; Petronis, A. Pre- and peri-natal environmental risks for attention-deficit hyperactivity disorder (ADHD): The potential role of epigenetic processes in mediating susceptibility. J. Child Psychol. Psychiatry 2008, 49, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Grayson, D.R.; Guidotti, A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology 2013, 38, 138–166. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J.; Pena, C.J.; Kundakovic, M.; Mitchell, A.; Akbarian, S. Epigenetic basis of mental illness. Neuroscientist 2016, 22, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.E.; Gao, Y.; Deep-Soboslay, A.; Tao, R.; Hyde, T.M.; Weinberger, D.R.; Kleinman, J.E. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat. Neurosci. 2016, 19, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Pidsley, R.; Viana, J.; Hannon, E.; Spiers, H.; Troakes, C.; Al-Saraj, S.; Mechawar, N.; Turecki, G.; Schalkwyk, L.C.; Bray, N.J.; et al. Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kirkbride, J.B.; Susser, E.; Kundakovic, M.; Kresovich, J.K.; Davey Smith, G.; Relton, C.L. Prenatal nutrition, epigenetics and schizophrenia risk: Can we test causal effects? Epigenomics 2012, 4, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, B. A steep learning curve: Decoding epigenetic influence on behavior and mental health. Environ. Health Perspect. 2012, 120, a396–a401. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, A.; Atkinson, S.P.; Lako, M.; Armstrong, L. Epigenetic landscaping during hesc differentiation to neural cells. Stem Cells 2009, 27, 1298–1308. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Casaccia, P. Epigenetic regulation of oligodendrocyte identity. Trends Neurosci. 2010, 33, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.D.; Gauthier, A.S. Timing is everything: Making neurons versus glia in the developing cortex. Neuron 2007, 54, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Scholer, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Coskun, V.; Tao, J.; Xie, W.; Ge, W.; Yoshikawa, K.; Li, E.; Zhang, Y.; Sun, Y.E. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 2010, 329, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Maor, G.L.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Lay, F.D.; Liu, Y.; Kelly, T.K.; Witt, H.; Farnham, P.J.; Jones, P.A.; Berman, B.P. The role of DNA methylation in directing the functional organization of the cancer epigenome. Genome Res. 2015, 25, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Karlić, R.; Chung, H.-R.; Lasserre, J.; Vlahoviček, K.; Vingron, M. Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2926–2931. [Google Scholar] [CrossRef] [PubMed]
- Burgers, W.A.; Fuks, F.; Kouzarides, T. DNA methyltransferases get connected to chromatin. Trends Genet. 2002, 18, 275–277. [Google Scholar] [CrossRef]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, T.; Nakashima, K.; Namihira, M.; Ochiai, W.; Uemura, A.; Yanagisawa, M.; Fujita, N.; Nakao, M.; Taga, T. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev. Cell 2001, 1, 749–758. [Google Scholar] [CrossRef]
- Fan, G.; Martinowich, K.; Chin, M.H.; He, F.; Fouse, S.D.; Hutnick, L.; Hattori, D.; Ge, W.; Shen, Y.; Wu, H. DNA methylation controls the timing of astrogliogenesis through regulation of jak-stat signaling. Development 2005, 132, 3345–3356. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, J.D. The emerging field of neuroepigenetics. Neuron 2013, 80, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Kundakovic, M.; Gudsnuk, K.; Herbstman, J.B.; Tang, D.; Perera, F.P.; Champagne, F.A. DNA methylation of BDNFas a biomarker of early-life adversity. Proc. Natl. Acad. Sci. USA 2015, 112, 6807–6813. [Google Scholar] [CrossRef] [PubMed]
- Basil, P.; Li, Q.; Dempster, E.L.; Mill, J.; Sham, P.C.; Wong, C.C.; McAlonan, G.M. Prenatal maternal immune activation causes epigenetic differences in adolescent mouse brain. Transl. Psychiatry 2014. [Google Scholar] [CrossRef]
- DiNieri, J.A.; Wang, X.; Szutorisz, H.; Spano, S.M.; Kaur, J.; Casaccia, P.; Dow-Edwards, D.; Hurd, Y.L. Maternal cannabis use alters ventral striatal dopamine D2 gene regulation in the offspring. Biol. Psychiatry 2011, 70, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Dong, E.; Dzitoyeva, S.G.; Matrisciano, F.; Tueting, P.; Grayson, D.R.; Guidotti, A. Brain-derived neurotrophic factor epigenetic modifications associated with schizophrenia-like phenotype induced by prenatal stress in mice. Biol. Psychiatry 2015, 77, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Kaminen-Ahola, N.; Ahola, A.; Maga, M.; Mallitt, K.A.; Fahey, P.; Cox, T.C.; Whitelaw, E.; Chong, S. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet. 2010, 6, e1000811. [Google Scholar] [CrossRef] [PubMed]
- Kundakovic, M.; Gudsnuk, K.; Franks, B.; Madrid, J.; Miller, R.L.; Perera, F.P.; Champagne, F.A. Sex-specific epigenetic disruption and behavioral changes following low-dose in utero bisphenol A exposure. Proc. Natl. Acad. Sci. USA 2013, 110, 9956–9961. [Google Scholar] [CrossRef] [PubMed]
- Labouesse, M.A.; Dong, E.; Grayson, D.R.; Guidotti, A.; Meyer, U. Maternal immune activation induces GAD1 and GAD2 promoter remodeling in the offspring prefrontal cortex. Epigenetics 2015, 10, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.R.; Bale, T.L. Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 2008, 28, 9055–9065. [Google Scholar] [CrossRef] [PubMed]
- Matrisciano, F.; Tueting, P.; Dalal, I.; Kadriu, B.; Grayson, D.R.; Davis, J.M.; Nicoletti, F.; Guidotti, A. Epigenetic modifications of gabaergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology 2013, 68, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Novikova, S.I.; He, F.; Bai, J.; Cutrufello, N.J.; Lidow, M.S.; Undieh, A.S. Maternal cocaine administration in mice alters DNA methylation and gene expression in hippocampal neurons of neonatal and prepubertal offspring. PLoS ONE 2008, 3, e1919. [Google Scholar] [CrossRef] [PubMed]
- Onishchenko, N.; Karpova, N.; Sabri, F.; Castren, E.; Ceccatelli, S. Long-lasting depression-like behavior and epigenetic changes of BDNF gene expression induced by perinatal exposure to methylmercury. J. Neurochem. 2008, 106, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Richetto, J.; Massart, R.; Weber-Stadlbauer, U.; Szyf, M.; Riva, M.A.; Meyer, U. Genome-wide DNA Methylation Changes in a Mouse Model of Infection-Mediated Neurodevelopmental Disorders. Biol. Psychiatry 2017, 81, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Schraut, K.G.; Jakob, S.B.; Weidner, M.T.; Schmitt, A.G.; Scholz, C.J.; Strekalova, T.; El Hajj, N.; Eijssen, L.M.; Domschke, K.; Reif, A.; et al. Prenatal stress-induced programming of genome-wide promoter DNA methylation in 5-HTT-deficient mice. Transl. Psychiatry 2014. [Google Scholar] [CrossRef] [PubMed]
- St-Cyr, S.; McGowan, P.O. Programming of stress-related behavior and epigenetic neural gene regulation in mice offspring through maternal exposure to predator odor. Front. Behav. Neurosci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Fan, W.; Zhang, X.; Dong, E. Gestational stress induces depressive-like and anxiety-like phenotypes through epigenetic regulation of BDNFexpression in offspring hippocampus. Epigenetics 2016, 11, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Boulle, F.; van den Hove, D.L.; Jakob, S.B.; Rutten, B.P.; Hamon, M.; van Os, J.; Lesch, K.P.; Lanfumey, L.; Steinbusch, H.W.; Kenis, G. Epigenetic regulation of the BDNFgene: Implications for psychiatric disorders. Mol. Psychiatry 2012, 17, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Weickert, C.S.; Hyde, T.M.; Lipska, B.K.; Herman, M.M.; Weinberger, D.R.; Kleinman, J.E. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol. Psychiatry 2003, 8, 592–610. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, Y.; Rizavi, H.S.; Conley, R.R.; Roberts, R.C.; Tamminga, C.A.; Pandey, G.N. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch. Gen. Psychiatry 2003, 60, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Sarchiapone, M.; Zarrilli, F.; Videtic, A.; Ferraro, A.; Carli, V.; Sacchetti, S.; Lembo, F.; Angiolillo, A.; Jovanovic, N.; et al. Increased BDNF promoter methylation in the wernicke area of suicide subjects. Arch. Gen. Psychiatry 2010, 67, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Rapoport, S.I.; Rao, J.S. Altered expression of apoptotic factors and synaptic markers in postmortem brain from bipolar disorder patients. Neurobiol. Dis. 2010, 37, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Nickl-Jockschat, T.; Michel, T.M. The role of neurotrophic factors in autism. Mol. Psychiatry 2011, 16, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Herbstman, J.B.; Sjödin, A.; Kurzon, M.; Lederman, S.A.; Jones, R.S.; Rauh, V.; Needham, L.L.; Tang, D.; Niedzwiecki, M.; Wang, R.Y.; et al. Prenatal exposure to PBDEs and neurodevelopment. Environ. Health Perspect. 2010, 118, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Horton, M.K.; Rundle, A.; Camann, D.E.; Boyd Barr, D.; Rauh, V.A.; Whyatt, R.M. Impact of prenatal exposure to piperonyl butoxide and permethrin on 36-month neurodevelopment. Pediatrics 2011, 127, e699–e706. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.; Vishnevetsky, J.; Herbstman, J.B.; Calafat, A.M.; Xiong, W.; Rauh, V.; Wang, S. Prenatal bisphenol A exposure and child behavior in an inner city cohort. Environ. Health Perspect. 2012, 120, 1190–1194. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Zhang, X.; Wang, D.; Baccarelli, A. Environmental chemical exposures and human epigenetics. Int. J. Epidemiol. 2012, 41, 79–105. [Google Scholar] [CrossRef] [PubMed]
- Chapin, R.E.; Adams, J.; Boekelheide, K.; Gray, L.E., Jr.; Hayward, S.W.; Lees, P.S.; McIntyre, B.S.; Portier, K.M.; Schnorr, T.M.; Selevan, S.G.; et al. NTP-CERHR expert panel report on the reproductive and developmental toxicity of bisphenol A. Birth Defects Res. B Dev. Reprod. Toxicol. 2008, 83, 157–395. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M.; Kalkbrenner, A.E.; Calafat, A.M.; Yolton, K.; Ye, X.; Dietrich, K.N.; Lanphear, B.P. Impact of early-life bisphenol A exposure on behavior and executive function in children. Pediatrics 2011, 128, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M.; Yolton, K.; Dietrich, K.N.; Hornung, R.; Ye, X.; Calafat, A.M.; Lanphear, B.P. Prenatal bisphenol A exposure and early childhood behavior. Environ. Health Perspect. 2009, 117, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Kundakovic, M.; Champagne, F.A. Epigenetic perspective on the developmental effects of bisphenol A. Brain Behav. Immun. 2011, 25, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; Arnold, A.P. Reframing sexual differentiation of the brain. Nat. Neurosci. 2011, 14, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Patisaul, H.B.; Fortino, A.E.; Polston, E.K. Neonatal genistein or bisphenol-A exposure alters sexual differentiation of the Avpv. Neurotoxicol. Teratol. 2006, 28, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Tetel, M.J.; Pfaff, D.W. Contributions of estrogen receptor-α and estrogen receptor-β to the regulation of behavior. Biochim. Biophys. Acta 2010, 1800, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Derkits, E.J. Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am. J. Psychiatry 2010, 167, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, A.L.; Ponder, K.L.; Padbury, J.F.; Lester, B.M. Fetal effects of psychoactive drugs. Clin. Perinatol. 2009, 36, 595–619. [Google Scholar] [CrossRef] [PubMed]
- Robison, A.J.; Nestler, E.J. Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 2011, 12, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Huizink, A.C.; Mulder, E.J. Maternal smoking, drinking or cannabis use during pregnancy and neurobehavioral and cognitive functioning in human offspring. Neurosci. Biobehav. Rev. 2006, 30, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Porath, A.J.; Fried, P.A. Effects of prenatal cigarette and marijuana exposure on drug use among offspring. Neurotoxicol. TEratol. 2005, 27, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Fowler, J.S.; Wang, G.J.; Swanson, J.M. Dopamine in drug abuse and addiction: Results from imaging studies and treatment implications. Mol. Psychiatry 2004, 9, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Le Foll, B.; Gallo, A.; Le Strat, Y.; Lu, L.; Gorwood, P. Genetics of dopamine receptors and drug addiction: A comprehensive review. Behav. Pharmacol. 2009, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.N.; Volta, M.; Pidsley, R.; Lunnon, K.; Dixit, A.; Lovestone, S.; Coarfa, C.; Harris, R.A.; Milosavljevic, A.; Troakes, C.; et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kundakovic, M. Postnatal risk environments, epigenetics, and psychosis: Putting the pieces together. Soc.Psychiatry Psychiatr. Epidemiol. 2014, 49, 1535–1536. [Google Scholar] [CrossRef] [PubMed]
- Mill, J.; Heijmans, B.T. From promises to practical strategies in epigenetic epidemiology. Nat. Rev. Genet. 2013, 14, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Stein, Z.; Susser, M.; Saenger, G.; Marolla, F. Nutrition and mental performance. Science 1972, 178, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Susser, E.S. Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophr. Bull. 2008, 34, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.; Magnus, P.; Schjolberg, S.; Stoltenberg, C.; Suren, P.; McKeague, I.W.; Davey Smith, G.; Reichborn-Kjennerud, T.; Susser, E. Folic acid supplements in pregnancy and severe language delay in children. JAMA 2011, 306, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Suren, P.; Roth, C.; Bresnahan, M.; Haugen, M.; Hornig, M.; Hirtz, D.; Lie, K.K.; Lipkin, W.I.; Magnus, P.; Reichborn-Kjennerud, T.; et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA 2013, 309, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef] [PubMed]
- Grayson, D.R.; Chen, Y.; Dong, E.; Kundakovic, M.; Guidotti, A. From trans-methylation to cytosine methylation: Evolution of the methylation hypothesis of schizophrenia. Epigenetics 2009, 4, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Aberg, K.A.; McClay, J.L.; Nerella, S.; Clark, S.; Kumar, G.; Chen, W.; Khachane, A.N.; Xie, L.; Hudson, A.; Gao, G.; et al. Methylome-wide association study of schizophrenia: Identifying blood biomarker signatures of environmental insults. JAMA Psychiatry 2014, 71, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Oberlander, T.F.; Weinberg, J.; Papsdorf, M.; Grunau, R.; Misri, S.; Devlin, A.M. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (nr3c1) and infant cortisol stress responses. Epigenetics 2008, 3, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, E.C.; Kundakovic, M.; Ramchandani, P.G.; Murphy, S.E.; Champagne, F.A. Maternal prenatal depressive symptoms predict infant NR3C1 1F and BDNFiv DNA methylation. Epigenetics 2015, 10, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.M.; Ruf, M.; Gunter, H.M.; Dohrmann, K.; Schauer, M.; Meyer, A.; Elbert, T. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Transl. Psychiatry 2011. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Rodriguez, M.; Lotfipour, S.; Leonard, G.; Perron, M.; Richer, L.; Veillette, S.; Pausova, Z.; Paus, T. Maternal smoking during pregnancy is associated with epigenetic modifications of the brain-derived neurotrophic factor-6 exon in adolescent offspring. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Herbstman, J.B.; Tang, D.; Zhu, D.; Qu, L.; Sjodin, A.; Li, Z.; Camann, D.; Perera, F.P. Prenatal exposure to polycyclic aromatic hydrocarbons, benzo[a]pyrene-DNA adducts, and genomic DNA methylation in cord blood. Environ. Health Perspect. 2012, 120, 733–738. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; Auger, A.P.; Bale, T.L.; De Vries, G.J.; Dunn, G.A.; Forger, N.G.; Murray, E.K.; Nugent, B.M.; Schwarz, J.M.; Wilson, M.E. The epigenetics of sex differences in the brain. J. Neurosci. 2009, 29, 12815–12823. [Google Scholar] [CrossRef] [PubMed]
- Kundakovic, M.; Jiang, Y.; Kavanagh, D.H.; Dincer, A.; Brown, L.; Pothula, V.; Zharovsky, E.; Park, R.; Jacobov, R.; Magro, I.; et al. Practical guidelines for high-resolution epigenomic profiling of nucleosomal histones in postmortem human brain tissue. Biol. Psychiatry 2017, 81, 162–170. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study | Species (Strain) | Stress Paradigm | Offspring Sex and Age | Brain Area | Analysis | Gene (Epigenetic Changes) | Epigenetic Regulators | Neurobehavioral Outcome | Sex Specificity |
|---|---|---|---|---|---|---|---|---|---|
| Basil et al., 2014 [49] | Mouse (C57BL/6N) | Maternal immune activation (GD17) | Males and females, PD42 | Hy | Sequenom EpiTYPER assay | Mecp2, LINE1 | - | - | Males and females; more profound changes in females |
| DiNieri et al., 2011 [50] | Rat (Long Evans) | THC rat model (GD5-PD2) | Males, PD62 | NAc | ChIP (H3K9me2, H3K4me3, RNA polymerase II) | Drd2 | - | Increased sensitivity to opiate reward; Drd2 transcriptional changes | - |
| Dong et al., 2015 [51] | Mouse (Swiss albino ND4) | Restrain stress (GD7-GD21) | Males, PD75 | FC, Hy | MeDIP, hMeDIP, ChIP | Bdnf | Dnmt1, Tet1 | Hyperactivity; Impaired social interaction; Bdnf transcriptional changes | - |
| Kaminen-Ahola et al., 2010 [52] | Mouse (C57BL/6J Agoutivy) | Ethanol exposure (GD0.5-GD8.5) | Males and females, PD28–30 | FB | Bisulfite Sequencing, Gene Expression Arrays | IAPs | - | Fetal alcohol syndrome-like features | - |
| Kundakovic et al., 2013 [53] | Mouse (BALB/c) | BPA exposure (GD0-GD19) | Males and females, PD30–70 | PFC, Hi, Hy | Bisulfite Pyrosequencing | Esr1 | Dnmt1, Dnmt3a | Disrupted exploratory, social and anxiety-like behavior; transcriptional changes in Esr1, Esr2 and Esrrg | Sex specific changes |
| Kundakovic et al., 2015 [48] | Mouse (BALB/c) | BPA exposure (GD0-GD19) | Males and females, PD28 and PD60 | Hi | Bisulfite Pyrosequencing | Bdnf IV, Bdnf IX, Grin2b | Dnmt1, Gadd45b | Disrupted exploration of a novel object; Bdnf and Grin2b transcriptional changes | Changes were observed in males |
| Labouesse et al., 2015 [54] | Mouse (C57BL/6N) | Maternal immune activation (GD17) | PD80–100 | mPFC | MeDIP, hMeDIP, ChIP (MeCP2) | Gad1, Gad2 | MeCP2 | Impaired working memory and social interaction deficits; Gad1 and Gad2 transcriptional changes | - |
| Mueller and Bale, 2008 [55] | Mouse (C57BL/6N:129) | Chronic, variable stress during early, mid and late gestation | Males and females, 6–16 weeks | Hi, Am | Bisulfite Pyrosequencing | Crf, Nr3c1 | - | Maladaptive behavioral stress-responsivity, anhedonia, and an increased sensitivity to SSRI treatment in males | Sex specific changes |
| Matrisciano et al., 2013 [56] | Mouse (Swiss albino ND4) | Restrain stress (GD7-GD21) | Males, PD60 | PFC, Hi | MeDIP, hMeDIP, ChIP (Dnmt1, MeCP2) | Reelin, Gad1 | Dnmt 1, Dnmt 3a | Hyperactivity; Impaired social interaction, prepulse inhibition, and fear conditioning; Reelin and Gad1 transcriptional changes | |
| Novikova et al., 2008 [57] | Mouse (CD1) | Cocaine exposure (GD8-GD19) | Males, PD3 and PD30 | Hi | Bisulfite sequencing, MeDIP/CGI array | Genome-wide | Dnmt1, Dnmt 3a | Transcriptional changes in Gpr73, Plk2, Prpn5, Mapk1, Impa1, Pyrk3, Gata4, Mtap6, Gtf3c1, Coq7 | - |
| Onishchenko et al., 2008 [58] | Mouse (C57BL/6/Bkl) | MeHg exposure (GD7-PD7) | Males, 9 weeks | DG | ChIP (H3ac, H3K27me3), Ms-SNuPE | Bdnf | Depresion like behavior; transcriptional changes in Bdnf | - | |
| Richetto et al., 2017 [59] | Mouse (C57BL6/N) | Maternal immune activation (GD9 and GD17) | Males, PD100 | mPFC | SureSelectXT capture sequencing assay and EpiTYPER | Genome wide (Dlx1, Lhx5, Lhx8, Wnt3, Wnt8a, Wnt7b, Efnb3, Mid1, Nlgn1, Nrxn2, Nf2, etc.) | - | Impaired sensorimotor gating, social interaction and spatial memory; transcriptional changes in Dlx1, Wnt3, Mid1, Nlgn1, Nf2 | - |
| Schraut et al., 2014 [60] | Mouse (C57BL6/J, 5-Htt +/+ and 5-Htt +/−) | Restrain stress (GD13-GD17) | Females, PD95 | Hi | MeDIP-on-Chip | Genome wide | - | Anxiety-related behavior; Mdb transcriptional changes | - |
| St-Cyr S and McGowan, 2015 [61] | Mouse (C57BL/6N) | Exposure to predator odor (GD11-GD18) | Females and males, PD90 | Hi, Am | Bisulfite Pyrosequencing | Bdnf | - | Increased avoidance and decreased predator-odor associated activity; Bdnf and Crhr1 transcriptional changes | Males and females, but females showed a greater increase in CORT level |
| Zheng et al., 2016 [62] | Mouse (Kunming) | Restrain stress (GD5-delivery) | Males, PD40 | Hi | MeDIP,ChiP (H3K14ac) | Bdnf | Dnmt1, Hdac1, Hdac2, | Depressive-like and anxiety-like behaviors, Bdnf transcriptional changes | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kundakovic, M.; Jaric, I. The Epigenetic Link between Prenatal Adverse Environments and Neurodevelopmental Disorders. Genes 2017, 8, 104. https://doi.org/10.3390/genes8030104
Kundakovic M, Jaric I. The Epigenetic Link between Prenatal Adverse Environments and Neurodevelopmental Disorders. Genes. 2017; 8(3):104. https://doi.org/10.3390/genes8030104
Chicago/Turabian StyleKundakovic, Marija, and Ivana Jaric. 2017. "The Epigenetic Link between Prenatal Adverse Environments and Neurodevelopmental Disorders" Genes 8, no. 3: 104. https://doi.org/10.3390/genes8030104
APA StyleKundakovic, M., & Jaric, I. (2017). The Epigenetic Link between Prenatal Adverse Environments and Neurodevelopmental Disorders. Genes, 8(3), 104. https://doi.org/10.3390/genes8030104