Targeting MYC Dependence by Metabolic Inhibitors in Cancer

Abstract
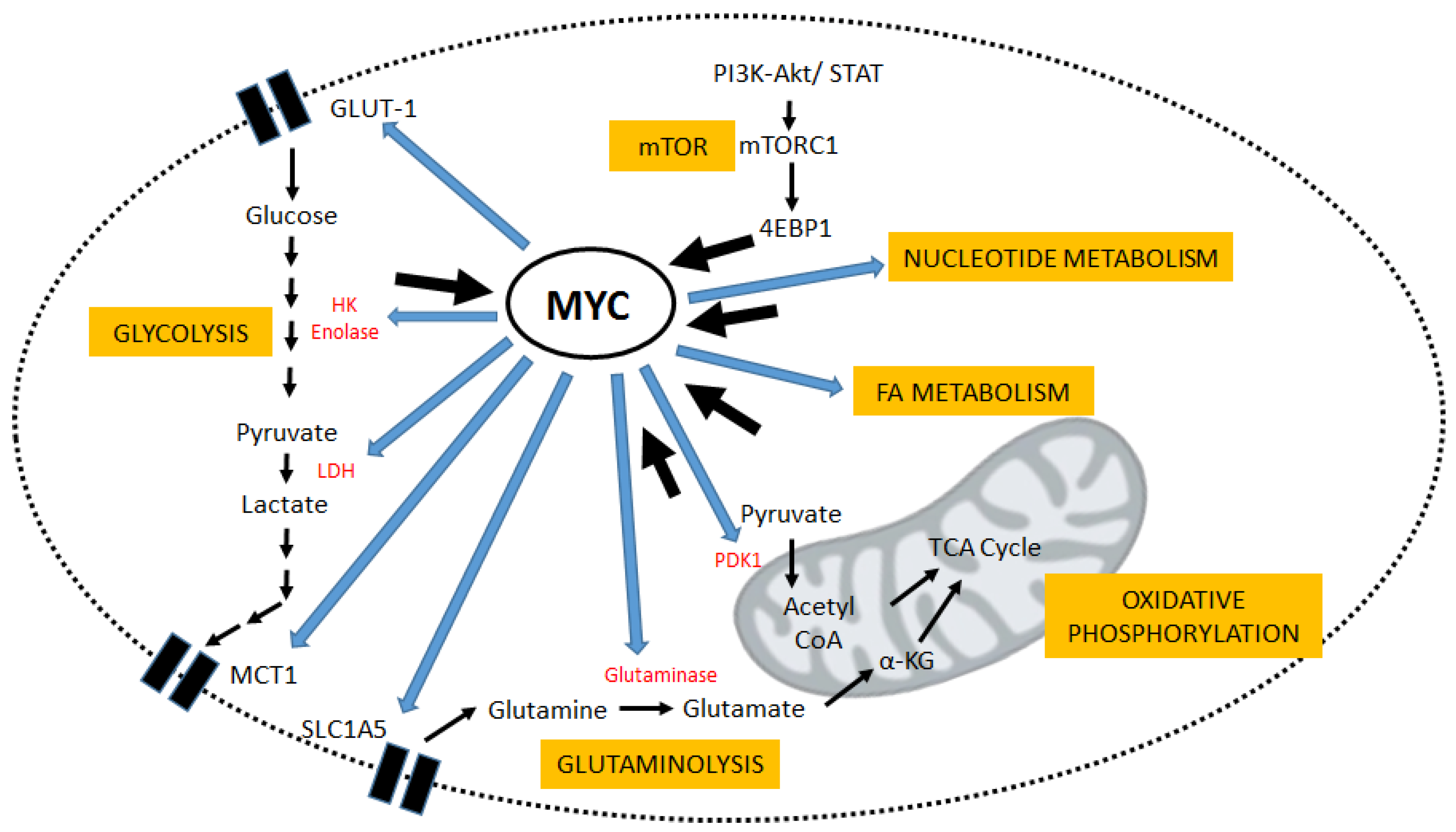
:1. Introduction
2. MYC and Glucose Metabolism
2.1. Transcriptional Control of Glycolytic Genes
2.2. Indirect Transcriptional Control of Glycolytic Genes
3. Targeting MYC Dependence through Glucose Metabolism
3.1. GLUT-1 Inhibitors
3.2. Hexokinase Inhibitors
- a.
- 3-Bromopyruvate3-Bromopyruvate (3BP) was initially described as a metabolic inhibitor in the early 1960s, but has since been studied extensively for its effect on glycolysis and glycolytic enzymes. In a liver cancer model, 3BP was shown to effectively inhibit hexokinase 2, and inhibit glycolysis, facilitating death of hepatoma cells [32]. 3BP is a structural analog of pyruvic acid and is thought to be taken up by cells via MCTs [33]. High levels of MYC expression in tumor cells drives the overexpression of MCTs, enabling efficient uptake of 3BP and enhanced lethality of the tumor cells in response to this drug [23]. In addition to HK2, 3BP effectively inhibits the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) enzyme, another important enzyme in glycolysis, resulting in significant depletion of cellular ATP and cell death [34,35].
- b.
- 2-DeoxyglucoseIn the early 1950s, 2-deoxy-d-glucose (2DG) was shown to inhibit both aerobic and anaerobic glycolysis in rat tumor tissue [36]. It is a synthetic glucose analog, in which the C-2-hydroxyl group is replaced by hydrogen, and for the last several decades it has been used extensively to study tumor cell metabolism. While the effects of 2DG on glycolysis are the focus of most studies, this agent acts in several different ways to kill cancer cells [37]. 2DG enters cells via GLUTs and is phosphorylated by HK to form 2-Deoxy-d-glucose-6-phosphate (2DG-6-P) which then inhibits both HK and phosphoglucose isomerase (PGI) activity, thereby decreasing glycolysis and reducing the ATP/AMP ratio in cells [38,39]. Increased levels of HIF-1 and MYC can induce resistance to 2DG via upregulating the levels of glycolytic enzymes [40]. Drugs like methylprednisolone, cisplatin or ABT-737 which reduce HIF-1 and MYC levels can synergize with 2DG to inhibit cell proliferation and induce apoptosis [41,42,43].
3.3. Metformin
3.4. LDH Inhibitors
- a
- GalloflavinGalloflavin, identified as a LDH inhibitor in 2012, was capable of inhibiting both LDH A and B isoforms of the enzyme [52]. It is capable of blocking glycolysis and inducing cell death. LDH inhibition also results in lower NAD levels and lower activity of SIRT1, thereby decreasing MYC protein levels. In Burkitt lymphoma cells, the down regulation of MYC results in inhibition of lymphoma cell growth [53].
- b
- Other LDH inhibitorsGossypol, Oxamate and FX11 are some of the LDH inhibitors currently being studied for their efficacy in various cancer models [54].
3.5. Pyruvate Dehydrogenase Kinase Inhibitor—Dichloroacetate
3.6. Other Glucose Metabolism Inhibitor Targeting MYC
4. MYC and mTOR Pathway
4.1. Post Translational Regulation of MYC by mTOR
4.2. Targeting MYC Dependence through mTOR Inhibition
4.3. Dual PI3K and mTOR Inhibitors
4.4. Other Agents with mTOR Inhibitory Activity
5. MYC and Nucleotide and Fatty Acid Metabolism
5.1. Targeting MYC Dependence through Nucleotide Metabolism
5.2. Targeting MYC Dependence through Fatty Acid Metabolism
6. MYC and Glutamine Metabolism
7. Combination Therapies Targeting MYC
7.1. Arsenic Trioxide and Dichloroacetate
7.2. 6-BT and Metformin
8. Summary and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sharma, S.V.; Settleman, J. Oncogene addiction: Setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007, 21, 3214–3231. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Sabo, A.; Guccione, E. Targeting MYC in cancer therapy: RNA processing offers new opportunities. BioEssays 2016, 38, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Nagel, R.; Semenova, E.A.; Berns, A. Drugging the addict: Non-oncogene addiction as a target for cancer therapy. EMBO Rep. 2016, 17, 1516–1531. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, D.B.; Zilz, N.D.; Towle, H.C. Sequences within the 5′-flanking region of the S14 gene confer responsiveness to glucose in primary hepatocytes. J. Biol. Chem. 1989, 264, 17623–17626. [Google Scholar] [PubMed]
- Foufelle, F.; Girard, J.; Ferre, P. Regulation of lipogenic enzyme expression by glucose in liver and adipose tissue: A review of the potential cellular and molecular mechanisms. Adv. Enzym. Regul. 1996, 36, 199–226. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose catabolism in cancer cells. Isolation, sequence, and activity of the promoter for type II hexokinase. J. Biol. Chem. 1995, 270, 16918–16925. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.S.; Towle, H.C. Localization of the carbohydrate response element of the rat l-type pyruvate kinase gene. J. Biol. Chem. 1991, 266, 8679–8682. [Google Scholar] [PubMed]
- Dang, C.V.; Lewis, B.C.; Dolde, C.; Dang, G.; Shim, H. Oncogenes in tumor metabolism, tumorigenesis, and apoptosis. J. Bioenerg. Biomembr. 1997, 29, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.C.; Cheung, A.F.; Simkevich, C.P.; Tam, W.; Ren, X.; Mateyak, M.K.; Sedivy, J.M. A large scale genetic analysis of c-Myc-regulated gene expression patterns. J. Biol. Chem. 2003, 278, 12563–12573. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of Myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [PubMed]
- Dewhirst, M.W. Intermittent hypoxia furthers the rationale for hypoxia-inducible factor-1 targeting. Cancer Res. 2007, 67, 854–855. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Kim, J.W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Longatto-Filho, A.; Azevedo-Silva, J.; Casal, M.; Schmitt, F.C.; Baltazar, F. Role of monocarboxylate transporters in human cancers: State of the art. J. Bioenerg. Biomembr. 2012, 44, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Yang, C.; Scott, K.E.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking lactate export by inhibiting the Myc target MCT1 disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Xiu, R.; Ren, P.; Yue, M.; Su, H.; Guo, G.; Xiao, D.; Yu, J.; Jiang, H.; Liu, H.; et al. Metabolic targeting of oncogene myc by selective activation of the proton-coupled monocarboxylate family of transporters. Oncogene 2016, 35, 3037–3048. [Google Scholar] [CrossRef] [PubMed]
- Moriizumi, S.; Gourdon, L.; Lefrancois-Martinez, A.M.; Kahn, A.; Raymondjean, M. Effect of different basic helix-loop-helix leucine zipper factors on the glucose response unit of the L-type pyruvate kinase gene. Gene Expr. 1998, 7, 103–113. [Google Scholar] [PubMed]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.J.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Metukuri, M.R.; Bindom, S.M.; Prochownik, E.V.; O’Doherty, R.M.; Scott, D.K. c-Myc is required for the ChREBP-dependent activation of glucose-responsive genes. Mol. Endocrinol. 2010, 24, 1274–1286. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.J.; Doan, T.T.; Daniels, M.C.; Schurr, J.R.; Kolls, J.K.; Scott, D.K. c-Myc is required for the glucose-mediated induction of metabolic enzyme genes. J. Biol. Chem. 2003, 278, 6588–6595. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Collier, J.J.; Scott, D.K. Camp opposes the glucose-mediated induction of the L-PK gene by preventing the recruitment of a complex containing ChREBP, HNF4alpha, and CBP. FASEB J. 2009, 23, 2855–2865. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.E.; Dalili, S.; Simpson, C.D.; Hurren, R.; Mao, X.; Saiz, F.S.; Gronda, M.; Eberhard, Y.; Minden, M.D.; Bilan, P.J.; et al. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol. Cancer Ther. 2008, 7, 3546–3555. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Pedersen, P.L.; Geschwind, J.F. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: Characterization and targeting hexokinase. Cancer Lett. 2001, 173, 83–91. [Google Scholar] [CrossRef]
- Lis, P.; Dylag, M.; Niedzwiecka, K.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ulaszewski, S. The hk2 dependent “warburg effect” and mitochondrial oxidative phosphorylation in cancer: Targets for effective therapy with 3-bromopyruvate. Molecules 2016, 21, 1730. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Wang, T.; Possemato, R.; Yilmaz, O.H.; Koch, C.E.; Chen, W.W.; Hutchins, A.W.; Gultekin, Y.; Peterson, T.R.; Carette, J.E.; et al. MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat. Genet. 2013, 45, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Kunjithapatham, R.; Geschwind, J.F. Anticancer efficacy of the metabolic blocker 3-bromopyruvate: Specific molecular targeting. Anticancer Res. 2013, 33, 13–20. [Google Scholar] [PubMed]
- Woodward, G.E.; Hudson, M.T. The effect of 2-desoxy-d-glucose on glycolysis and respiration of tumor and normal tissues. Cancer Res. 1954, 14, 599–605. [Google Scholar] [PubMed]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-deoxy-d-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Bachelard, H.S.; Clark, A.G.; Thompson, M.F. Cerebral-cortex hexokinase. Elucidation of reaction mechanisms by substrate and dead-end inhibitor kinetic analysis. Biochem. J. 1971, 123, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.; Kolbe, H.; Lange, K.; Herken, H. Behaviour of the glycolytic system of rat brain and kidney in vivo after in hibition of the glucosephosphate isomerase. II. Substrate concentrations under the influence of ischemia, 6-aminonicotinamide, and 2-deoxyglucose. Hoppe Seylers Z. Physiol. Chem. 1972, 353, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.H.; Chou, M.H.; Tai, M.H.; Lin, T.K.; Liou, C.W.; Chen, T.; Hsu, W.M.; Wang, P.W. 2-Deoxyglucose treatment complements the cisplatin- or BH3-only mimetic-induced suppression of neuroblastoma cell growth. Int. J. Biochem. Cell Biol. 2013, 45, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.Y.; Wang, T.; Chen, F.Y.; Wu, Y.L.; Shao, X.; Xiao, F.; Huang, H.H.; Zhong, H.; Zhong, J.H. Glycolytic inhibitor 2-deoxy-d-glucose suppresses cell proliferation and enhances methylprednisolone sensitivity in non-hodgkin lymphoma cells through down-regulation of HIF-1α and c-Myc. Leuk. Lymphoma 2015, 56, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Casinelli, G.; LaRosa, J.; Sharma, M.; Cherok, E.; Banerjee, S.; Branca, M.; Edmunds, L.; Wang, Y.; Sims-Lucas, S.; Churley, L.; et al. N-Myc overexpression increases cisplatin resistance in neuroblastoma via deregulation of mitochondrial dynamics. Cell Death Discov. 2016, 2, 16082. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, K.C.; Hogarty, M.D. Small-molecule BH3 mimetics to antagonize BCL-2-homolog survival functions in cancer. Curr. Opin. Investig. Drugs 2009, 10, 559–571. [Google Scholar] [PubMed]
- Yu, X.; Mao, W.; Zhai, Y.; Tong, C.; Liu, M.; Ma, L.; Yu, X.; Li, S. Anti-tumor activity of metformin: From metabolic and epigenetic perspectives. Oncotarget 2017, 8, 5619–5628. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Cazzaniga, M.; Bonanni, B. Relationship between metabolic reprogramming and mitochondrial activity in cancer cells. Understanding the anticancer effect of metformin and its clinical implications. Anticancer Res. 2015, 35, 5789–5796. [Google Scholar] [PubMed]
- Akinyeke, T.; Matsumura, S.; Wang, X.; Wu, Y.; Schalfer, E.D.; Saxena, A.; Yan, W.; Logan, S.K.; Li, X. Metformin targets c-MYC oncogene to prevent prostate cancer. Carcinogenesis 2013, 34, 2823–2832. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Valerio, M.; Cioce, M.; Mori, F.; Casadei, L.; Pulito, C.; Sacconi, A.; Biagioni, F.; Cortese, G.; Galanti, S.; et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat. Commun. 2012, 3, 865. [Google Scholar] [CrossRef] [PubMed]
- Javeshghani, S.; Zakikhani, M.; Austin, S.; Bazile, M.; Blouin, M.J.; Topisirovic, I.; St-Pierre, J.; Pollak, M.N. Carbon source and myc expression influence the antiproliferative actions of metformin. Cancer Res. 2012, 72, 6257–6267. [Google Scholar] [CrossRef] [PubMed]
- Augoff, K.; Hryniewicz-Jankowska, A.; Tabola, R. Lactate dehydrogenase 5: An old friend and a new hope in the war on cancer. Cancer Lett. 2015, 358, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Menssen, A.; Hydbring, P.; Kapelle, K.; Vervoorts, J.; Diebold, J.; Luscher, B.; Larsson, L.G.; Hermeking, H. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc. Natl. Acad. Sci. USA 2012, 109, E187–E196. [Google Scholar] [CrossRef] [PubMed]
- Manerba, M.; Vettraino, M.; Fiume, L.; Di Stefano, G.; Sartini, A.; Giacomini, E.; Buonfiglio, R.; Roberti, M.; Recanatini, M. Galloflavin (CAS 568-80-9): A novel inhibitor of lactate dehydrogenase. ChemMedChem 2012, 7, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Vettraino, M.; Manerba, M.; Govoni, M.; Di Stefano, G. Galloflavin suppresses lactate dehydrogenase activity and causes MYC downregulation in burkitt lymphoma cells through NAD/NADH-dependent inhibition of sirtuin-1. Anticancer Drugs 2013, 24, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Paterni, I.; Rani, R.; Minutolo, F. Small-molecule inhibitors of human LDH5. Futur. Med. Chem. 2013, 5, 1967–1991. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Kawano, Y.; Yuki, H.; Okuno, Y.; Nosaka, K.; Mitsuya, H.; Hata, H. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br. J. Cancer 2013, 108, 170–178. [Google Scholar] [CrossRef] [PubMed]
- James, M.O.; Jahn, S.C.; Zhong, G.; Smeltz, M.G.; Hu, Z.; Stacpoole, P.W. Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1. Pharmacol. Ther. 2016, 170, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Kankotia, S.; Stacpoole, P.W. Dichloroacetate and cancer: New home for an orphan drug? Biochim. Biophys. Acta 2014, 1846, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Lang, S.A.; Renner, K.; Bosserhoff, A.; Gronwald, W.; Rehli, M.; Einhell, S.; Gedig, I.; Singer, K.; Seilbeck, A.; et al. New aspects of an old drug—Diclofenac targets MYC and glucose metabolism in tumor cells. PLoS ONE 2013, 8, e66987. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Sabatini, D.M. A central role for mTOR in lipid homeostasis. Cell Metab. 2013, 18, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, L.; Geng, Y.D.; An, F.L.; Xia, Y.Z.; Guo, C.; Luo, J.G.; Zhang, L.Y.; Guo, Q.L.; Kong, L.Y. Icariside II, a natural mTOR inhibitor, disrupts aberrant energy homeostasis via suppressing mTORC1-4E-BP1 axis in sarcoma cells. Oncotarget 2016, 7, 27819–27837. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sharma, L.; Lu, J.; Finch, P.; Fletcher, S.; Prochownik, E.V. Structurally diverse c-Myc inhibitors share a common mechanism of action involving atp depletion. Oncotarget 2015, 6, 15857–15870. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Oudard, S.; Medioni, J.; Aylllon, J.; Barrascourt, E.; Elaidi, R.T.; Balcaceres, J.; Scotte, F. Everolimus (RAD001): An mTOR inhibitor for the treatment of metastatic renal cell carcinoma. Expert Rev. Anticancer Ther. 2009, 9, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Toral-Barza, L.; Discafani, C.; Zhang, W.G.; Skotnicki, J.; Frost, P.; Gibbons, J.J. mTOR, a novel target in breast cancer: The effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocr. Relat. Cancer 2001, 8, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Pourdehnad, M.; Truitt, M.L.; Siddiqi, I.N.; Ducker, G.S.; Shokat, K.M.; Ruggero, D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 11988–11993. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Gery, S.; Koeffler, H.P.; Yokoyama, A. Inhibition of mammalian target of rapamycin signaling potentiates the effects of all-trans retinoic acid to induce growth arrest and differentiation of human acute myelogenous leukemia cells. Int. J. Cancer 2009, 125, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Leu, W.J.; Swain, S.P.; Chan, S.H.; Hsu, J.L.; Liu, S.P.; Chan, M.L.; Yu, C.C.; Hsu, L.C.; Chou, Y.L.; Chang, W.L.; et al. Non-immunosuppressive triazole-based small molecule induces anticancer activity against human hormone-refractory prostate cancers: The role in inhibition of PI3K/AKT/mTOR and c-Myc signaling pathways. Oncotarget 2016, 7, 76995–77009. [Google Scholar] [CrossRef] [PubMed]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Bjornsti, M.A.; Houghton, P.J. The tor pathway: A target for cancer therapy. Nat. Rev. Cancer 2004, 4, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Lee, G.; Yoon, S.O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the EIF4B-dependent control of c-Myc translation. Curr. Biol. 2014, 24, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Lonetti, A.; Teti, G.; Orsini, E.; Bressanin, D.; Cappellini, A.; Ricci, F.; Tazzari, P.L.; Ognibene, A.; Falconi, M.; et al. A combination of temsirolimus, an allosteric mTOR inhibitor, with clofarabine as a new therapeutic option for patients with acute myeloid leukemia. Oncotarget 2012, 3, 1615–1628. [Google Scholar] [CrossRef] [PubMed]
- Frost, P.; Moatamed, F.; Hoang, B.; Shi, Y.; Gera, J.; Yan, H.; Frost, P.; Gibbons, J.; Lichtenstein, A. In Vivo antitumor effects of the mTOR inhibitor CCI-779 against human multiple myeloma cells in a xenograft model. Blood 2004, 104, 4181–4187. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Koeffler, H.P.; Yokoyama, A. Blockade of mTOR signaling potentiates the ability of histone deacetylase inhibitor to induce growth arrest and differentiation of acute myelogenous leukemia cells. Leukemia 2008, 22, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Campone, M.; Noel, B.; Couriaud, C.; Grau, M.; Guillemin, Y.; Gautier, F.; Gouraud, W.; Charbonnel, C.; Campion, L.; Jezequel, P.; et al. c-Myc dependent expression of pro-apoptotic bim renders HER2-overexpressing breast cancer cells dependent on anti-apoptotic Mcl-1. Mol. Cancer 2011, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.X.; Li, Y.; Yue, P.; Owonikoko, T.K.; Ramalingam, S.S.; Khuri, F.R.; Sun, S.Y. The combination of rad001 and nvp-bez235 exerts synergistic anticancer activity against non-small cell lung cancer in vitro and in vivo. PLoS ONE 2011, 6, e20899. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, S.; Meja, K.; Shepherd, C.; Khwaja, A. Synergistic induction of cell death in haematological malignancies by combined phosphoinositide-3-kinase and bet bromodomain inhibition. Br. J. Haematol. 2015, 170, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Kang, H.J.; Bae, E.J.; Oh, S.; Seong, Y.S.; Bae, I. β-TRCP1 degradation is a novel action mechanism of PI3K/mTOR inhibitors in triple-negative breast cancer cells. Exp. Mol. Med. 2015, 47, e143. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.; Yue, P.; Deng, X.; Khuri, F.R.; Sun, S.Y. mTOR complex 2 stabilizes mcl-1 protein by suppressing its glycogen synthase kinase 3-dependent and SCF-FBXW7-mediated degradation. Mol. Cell. Biol. 2015, 35, 2344–2355. [Google Scholar] [CrossRef] [PubMed]
- Kannan, A.; Lin, Z.; Shao, Q.; Zhao, S.; Fang, B.; Moreno, M.A.; Vural, E.; Stack, B.C., Jr.; Suen, J.Y.; Kannan, K.; et al. Dual mTOR inhibitor MLN0128 suppresses Merkel cell carcinoma (MCC) xenograft tumor growth. Oncotarget 2016, 7, 6576–6592. [Google Scholar] [PubMed]
- Yun, S.; Vincelette, N.D.; Knorr, K.L.; Almada, L.L.; Schneider, P.A.; Peterson, K.L.; Flatten, K.S.; Dai, H.; Pratz, K.W.; Hess, A.D.; et al. 4EBP1/c-MYC/PUMA and nf-κb/egr1/bim pathways underlie cytotoxicity of mTOR dual inhibitors in malignant lymphoid cells. Blood 2016, 127, 2711–2722. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.B.; Aiba, I.; Long, Y.; Lin, H.K.; Feun, L.; Savaraj, N.; Kuo, M.T. Activation of RAS/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res. 2012, 72, 2622–2633. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.; Lee, J.; Landon, K.A.; Benavides-Serrato, A.; Bashir, T.; Jung, M.E.; Lichtenstein, A.; Gera, J. Mechanistic target of rapamycin (mTOR) inhibition synergizes with reduced internal ribosome entry site (IRES)-mediated translation of cyclin d1 and c-MYC mRNAs to treat glioblastoma. J. Biol. Chem. 2016, 291, 14146–14159. [Google Scholar] [CrossRef] [PubMed]
- Chan, S. Targeting the mammalian target of rapamycin (mTOR): A new approach to treating cancer. Br. J. Cancer 2004, 91, 1420–1424. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.J.; Chung, J.; Fiorentino, D.F.; Flanagan, W.M.; Blenis, J.; Crabtree, G.R. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 1992, 358, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol. 2008, 1, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Vignot, S.; Faivre, S.; Aguirre, D.; Raymond, E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann. Oncol. 2005, 16, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Willems, L.; Chapuis, N.; Puissant, A.; Maciel, T.T.; Green, A.S.; Jacque, N.; Vignon, C.; Park, S.; Guichard, S.; Herault, O.; et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia 2012, 26, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Chen, Y.; Meng, T.; Ma, L.; Meng, L.; Wang, X.; Yu, T.; Zask, A.; Shen, J.; Yu, K. Molecular regulation of apoptotic machinery and lipid metabolism by mTORC1/mTORC2 dual inhibitors in preclinical models of HER2+/PIK3CAmut breast cancer. Oncotarget 2016, 7, 67071–67086. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Sementino, E.; Pei, J.; Kadariya, Y.; Ito, T.K.; Testa, J.R. Co-targeting of Akt and Myc inhibits viability of lymphoma cells from Lck-Dlx5 mice. Cancer Biol. Ther. 2015, 16, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, N.; Tamburini, J.; Green, A.S.; Vignon, C.; Bardet, V.; Neyret, A.; Pannetier, M.; Willems, L.; Park, S.; Macone, A.; et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin. Cancer Res. 2010, 16, 5424–5435. [Google Scholar] [CrossRef] [PubMed]
- Shortt, J.; Martin, B.P.; Newbold, A.; Hannan, K.M.; Devlin, J.R.; Baker, A.J.; Ralli, R.; Cullinane, C.; Schmitt, C.A.; Reimann, M.; et al. Combined inhibition of PI3K-related DNA damage response kinases and mTORC1 induces apoptosis in MYC-driven B-cell lymphomas. Blood 2013, 121, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Mazzoletti, M.; Bortolin, F.; Brunelli, L.; Pastorelli, R.; Di Giandomenico, S.; Erba, E.; Ubezio, P.; Broggini, M. Combination of PI3K/mTOR inhibitors: Antitumor activity and molecular correlates. Cancer Res. 2011, 71, 4573–4584. [Google Scholar] [CrossRef] [PubMed]
- Boulware, S.B.; Christensen, L.A.; Thames, H.; Coghlan, L.; Vasquez, K.M.; Finch, R.A. Triplex-forming oligonucleotides targeting c-MYC potentiate the anti-tumor activity of gemcitabine in a mouse model of human cancer. Mol. Carcinog. 2014, 53, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Dickman, K.G.; Zong, W.X. Akt and c-Myc differentially activate cellular metabolic programs and prime cells to bioenergetic inhibition. J. Biol. Chem. 2010, 285, 7324–7333. [Google Scholar] [CrossRef] [PubMed]
- Zirath, H.; Frenzel, A.; Oliynyk, G.; Segerstrom, L.; Westermark, U.K.; Larsson, K.; Munksgaard Persson, M.; Hultenby, K.; Lehtio, J.; Einvik, C.; et al. Myc inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10258–10263. [Google Scholar] [CrossRef] [PubMed]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1c promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Wallington-Beddoe, C.T.; Powell, J.A.; Tong, D.; Pitson, S.M.; Bradstock, K.F.; Bendall, L.J. Sphingosine kinase 2 promotes acute lymphoblastic leukemia by enhancing Myc expression. Cancer Res. 2014, 74, 2803–2815. [Google Scholar] [CrossRef] [PubMed]
- Wald, D.N.; Vermaat, H.M.; Zang, S.; Lavik, A.; Kang, Z.; Peleg, G.; Gerson, S.L.; Bunting, K.D.; Agarwal, M.L.; Roth, B.L.; et al. Identification of 6-benzylthioinosine as a myeloid leukemia differentiation-inducing compound. Cancer Res. 2008, 68, 4369–4376. [Google Scholar] [CrossRef] [PubMed]
- Gortz, A.; Franklin, T.J.; Dive, C.; Hickman, J.A. Cell cycle specific induction of HL-60 cell differentiation and apoptosis by mycophenolic acid. Cell Death Differ. 1997, 4, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Sokoloski, J.A.; Blair, O.C.; Sartorelli, A.C. Alterations in glycoprotein synthesis and guanosine triphosphate levels associated with the differentiation of HL-60 leukemia cells produced by inhibitors of inosine 5′-phosphate dehydrogenase. Cancer Res. 1986, 46, 2314–2319. [Google Scholar] [PubMed]
- Lucas, D.L.; Webster, H.K.; Wright, D.G. Purine metabolism in myeloid precursor cells during maturation. Studies with the HL-60 cell line. J. Clin. Investig. 1983, 72, 1889–1900. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Gathy, K.; Santiago, L.; Chen, E.; Huang, M.; Graves, L.M.; Mitchell, B.S. Induction of apoptosis in il-3-dependent hematopoietic cell lines by guanine nucleotide depletion. Blood 2003, 101, 4958–4965. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Santiago, L.; Mitchell, B.S. Synergy between imatinib and mycophenolic acid in inducing apoptosis in cell lines expressing Bcr-Abl. Blood 2005, 105, 3270–3277. [Google Scholar] [CrossRef] [PubMed]
- Al Safarjalani, O.N.; Rais, R.H.; Kim, Y.A.; Chu, C.K.; Naguib, F.N.; El Kouni, M.H. Carbocyclic 6-benzylthioinosine analogues as subversive substrates of toxoplasma gondii adenosine kinase: Biological activities and selective toxicities. Biochem. Pharmacol. 2010, 80, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.; Buolamwini, J.K.; Yadav, V.; Chu, C.K.; Naguib, F.N.; El Kouni, M.H. 6-benzylthioinosine analogues: Promising anti-toxoplasmic agents as inhibitors of the mammalian nucleoside transporter ent1 (es). Biochem. Pharmacol. 2005, 19, 69–73. [Google Scholar] [CrossRef] [PubMed]
- El Kouni, M.H.; Guarcello, V.; Al Safarjalani, O.N.; Naguib, F.N. Metabolism and selective toxicity of 6-nitrobenzylthioinosine in toxoplasma gondii. Antimicrob. Agents Chemother. 1999, 43, 2437–2443. [Google Scholar] [PubMed]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for myc-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.S.; Voelkel-Johnson, C.; Smith, C.D. Suppression of c-Myc and RRM2 expression in pancreatic cancer cells by the sphingosine kinase-2 inhibitor ABC294640. Oncotarget 2016, 7, 60181–60192. [Google Scholar] [CrossRef] [PubMed]
- Venkata, J.K.; An, N.; Stuart, R.; Costa, L.J.; Cai, H.; Coker, W.; Song, J.H.; Gibbs, K.; Matson, T.; Garrett-Mayer, E.; et al. Inhibition of sphingosine kinase 2 downregulates the expression of c-Myc and Mcl-1 and induces apoptosis in multiple myeloma. Blood 2014, 124, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H. Nutrition needs of mammalian cells in tissue culture. Science 1955, 122, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Souba, W.W. Glutamine and cancer. Ann. Surg. 1993, 218, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Knox, W.E.; Horowitz, M.L.; Friedell, G.H. The proportionality of glutaminase content to growth rate and morphology of rat neoplasms. Cancer Res. 1969, 29, 669–680. [Google Scholar] [PubMed]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces Myc-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Fogal, V.; Richardson, A.D.; Karmali, P.P.; Scheffler, I.E.; Smith, J.W.; Ruoslahti, E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell. Biol. 2010, 30, 1303–1318. [Google Scholar] [CrossRef] [PubMed]
- Fogal, V.; Babic, I.; Chao, Y.; Pastorino, S.; Mukthavaram, R.; Jiang, P.; Cho, Y.J.; Pingle, S.C.; Crawford, J.R.; Piccioni, D.E.; et al. Mitochondrial p32 is upregulated in Myc expressing brain cancers and mediates glutamine addiction. Oncotarget 2015, 6, 1157–1170. [Google Scholar] [CrossRef] [PubMed]
- Bott, A.J.; Peng, I.C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D.; et al. Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.M.; Lee, A.; Lee, J.; Haigis, M.C. SIRT4 protein suppresses tumor formation in genetic models of Myc-induced B cell lymphoma. J. Biol. Chem. 2014, 289, 4135–4144. [Google Scholar] [CrossRef] [PubMed]
- Perez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; Van Hee, V.F.; De Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.J.; Brisson, L.; et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 2016, 15, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.K.; Kolhe, R.; Black, S.M.; Keller, J.R.; Mivechi, N.F.; Satyanarayana, A. Inhibitor of differentiation 1 transcription factor promotes metabolic reprogramming in hepatocellular carcinoma cells. FASEB J. 2016, 30, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, J.; Sun, X.; Guo, Y.; Chu, D.; Wei, L.; Li, X.; Yang, G.; Liu, X.; Yao, L.; et al. Tumor suppressor NDRG2 inhibits glycolysis and glutaminolysis in colorectal cancer cells by repressing c-Myc expression. Oncotarget 2015, 6, 26161–26176. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.L.; Wang, L.Y.; Yu, Y.L.; Chen, H.W.; Srivastava, S.; Petrovics, G.; Kung, H.J. A long noncoding RNA connects c-Myc to tumor metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 18697–18702. [Google Scholar] [CrossRef] [PubMed]
- Phan, L.; Chou, P.C.; Velazquez-Torres, G.; Samudio, I.; Parreno, K.; Huang, Y.; Tseng, C.; Vu, T.; Gully, C.; Su, C.H.; et al. The cell cycle regulator 14-3-3σ opposes and reverses cancer metabolic reprogramming. Nat. Commun. 2015, 6, 7530. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Feng, L.; Zhou, Y.; Carew, J.S.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Inhibition of mitochondrial respiration: A novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J. Biol. Chem. 2003, 278, 37832–37839. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Board, P.G.; Blackburn, A.C. Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells. Mol. Cancer 2011, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, H.S.; Bradley, H.L.; Tripathi, S.; Yu, W.M.; Tse, W.; Qu, C.K.; Bunting, K.D. Synergistic cell death in FLT3-ITD positive acute myeloid leukemia by combined treatment with metformin and 6-benzylthioinosine. Leuk. Res. 2016, 50, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Zuo, S.; Hellman, U.; Lundahl, P. On the oligomeric state of the red blood cell glucose transporter glut1. Biochim. Biophys. Acta 2003, 1618, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Kacsinta, A.D.; Dowdy, S.F. Current views on inducing synthetic lethal RNAi responses in the treatment of cancer. Expert Opin. Biol. Ther. 2016, 16, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.L.; Tian, M.; Li, X.; Li, J.J.; Huang, J.; Ouyang, L.; Zhang, Y.; Liu, B. Inhibition of bet bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 2015, 6, 5501–5516. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ivanov, A.A.; Su, R.; Gonzalez-Pecchi, V.; Qi, Q.; Liu, S.; Webber, P.; McMillan, E.; Rusnak, L.; Pham, C.; et al. The oncoppi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat. Commun. 2017, 8, 14356. [Google Scholar] [CrossRef] [PubMed]
- Edmunds, L.R.; Sharma, L.; Kang, A.; Lu, J.; Vockley, J.; Basu, S.; Uppala, R.; Goetzman, E.S.; Beck, M.E.; Scott, D.; et al. c-Myc programs fatty acid metabolism and dictates Acetyl-CoA abundance and fate. J. Biol. Chem. 2014, 289, 25382–25392. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Comerford, S.A.; Huang, Z.; Du, X.; Wang, Y.; Cai, L.; Witkiewicz, A.K.; Walters, H.; Tantawy, M.N.; Fu, A.; Manning, H.C.; et al. Acetate dependence of tumors. Cell 2014, 159, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Bradley, H.L.; Sabnis, H.; Pritchett, D.; Bunting, K.D. Nanoproteomic assays on hematopoietic stem cells. Methods Mol. Biol. 2014, 1185, 165–177. [Google Scholar] [PubMed]
- Sabnis, H.; Bradley, H.L.; Bunting, S.T.; Cooper, T.M.; Bunting, K.D. Capillary nano-immunoassay for AKT 1/2/3 and 4EBP1 phosphorylation in acute myeloid leukemia. J. Transl. Med. 2014, 12, 166. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Target in Glucose Metabolism | Inhibitor Name | References | Clinical Testing |
|---|---|---|---|
| GLUT-1 | Fasentin | [30,31,32] | Preclinical phase only, No current clinical trials |
| STF-31 | |||
| WZB117 | |||
| Hexokinase | 3-Bromopyruvate | [33,34] | 3-BP: Preclinical only, 2-DG: Multiple phase 1/2 clinical trials in lung, prostate, breast tumors |
| 2-Deoxyglucose | [37,38,39,40,41] | ||
| GAPDH | 3-Bromopyruvate | [35,36] | Preclinical phase only |
| Phosphoglucose Isomerase | 2-Deoxyglucose | [39,40] | Multiple phase 1/2 clinical trials in lung, prostate, breast tumors |
| AMPK | Metformin | [46,47] | Multiple phase 1 through 3 clinical trials in lung, pancreatic, ovarian tumors, leukemias |
| LDH | Galloflavin | [53] | Galloflavin: Preclinical only, Gossypol: Multiple phase 1/2 clinical trials in lung, prostate, brain, leukemias and lymphomas Oxamate & FX11: Preclinical only |
| Gossypol | [55] | ||
| Oxamate | [55] | ||
| FX11 | [55] | ||
| PDK1 | Dichloroacetate | [56] | Phase 1 clinical trials in breast, lung, brain, head & neck tumors |
| Unknown Target | Diclofenac | [59] | No specific cancer therapy trials |
| mTOR Inhibitor | Mechanism of Action | Protein Inhibition * | References | Current Clinical Timeline |
|---|---|---|---|---|
| Rapamycin | Destabilizes the mTOR-Raptor complex | MYC | [69,70] | Phase 1 through 4 clinical trials in multiple cancers (solid organ, hematopoietic cancers) |
| CCI-779 (Temsirolimus) | Cyclin-D1, Cyclin-D3, MYC | [64,71,72] | ||
| RAD001 (Everolimus) | MYC, Cyclin D1 | [66,73,74] | ||
| Icariside II | mTOR Kinase inhibitor | MYC | [60] | Preclinical testing only |
| BEZ235 | mTOR Kinase inhibitor | Cyclin A, Cyclin D1, Parp, Caspase 3, MYC | [75,76] | Phase 1 through 3 clinical trials in breast, prostate, renal tumors |
| MTI-31 | mTOR Kinase inhibitor | p-Akt, Cyclin D1, MYC | [77] | Preclinical testing only |
| AZD8055 | mTOR Kinase inhibitor | MYC, Mcl-1, c-Jun, Cyclin E | [78] | Phase 1/2 clinical trials in advanced solid tumors, lymphomas etc. |
| MLN0128 (INK128) | mTOR Kinase inhibitor | 4EBP1, p-S6K1, MYC | [79,80] | Phase 1/2 clinical trials in thyroid, lung, endometrial, breast, myeloma, lymphoma etc. |
| PI-103 | mTOR Kinase inhibitor | MYC, Cyclin D3, PI3K, p-Akt | [77,81] | Preclinical testing only |
| PP242 | mTOR Kinase inhibitor | MYC, Cyclin D1 | [82] | Preclinical testing only |
| OSI-027 | mTOR Kinase inhibitor | MYC | [80] | Phase 1 clinical trial in advanced solid tumors & lymphoma |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabnis, H.S.; Somasagara, R.R.; Bunting, K.D. Targeting MYC Dependence by Metabolic Inhibitors in Cancer. Genes 2017, 8, 114. https://doi.org/10.3390/genes8040114
Sabnis HS, Somasagara RR, Bunting KD. Targeting MYC Dependence by Metabolic Inhibitors in Cancer. Genes. 2017; 8(4):114. https://doi.org/10.3390/genes8040114
Chicago/Turabian StyleSabnis, Himalee S., Ranganatha R. Somasagara, and Kevin D. Bunting. 2017. "Targeting MYC Dependence by Metabolic Inhibitors in Cancer" Genes 8, no. 4: 114. https://doi.org/10.3390/genes8040114
APA StyleSabnis, H. S., Somasagara, R. R., & Bunting, K. D. (2017). Targeting MYC Dependence by Metabolic Inhibitors in Cancer. Genes, 8(4), 114. https://doi.org/10.3390/genes8040114