
Anatomy of Mammalian Replication Domains

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
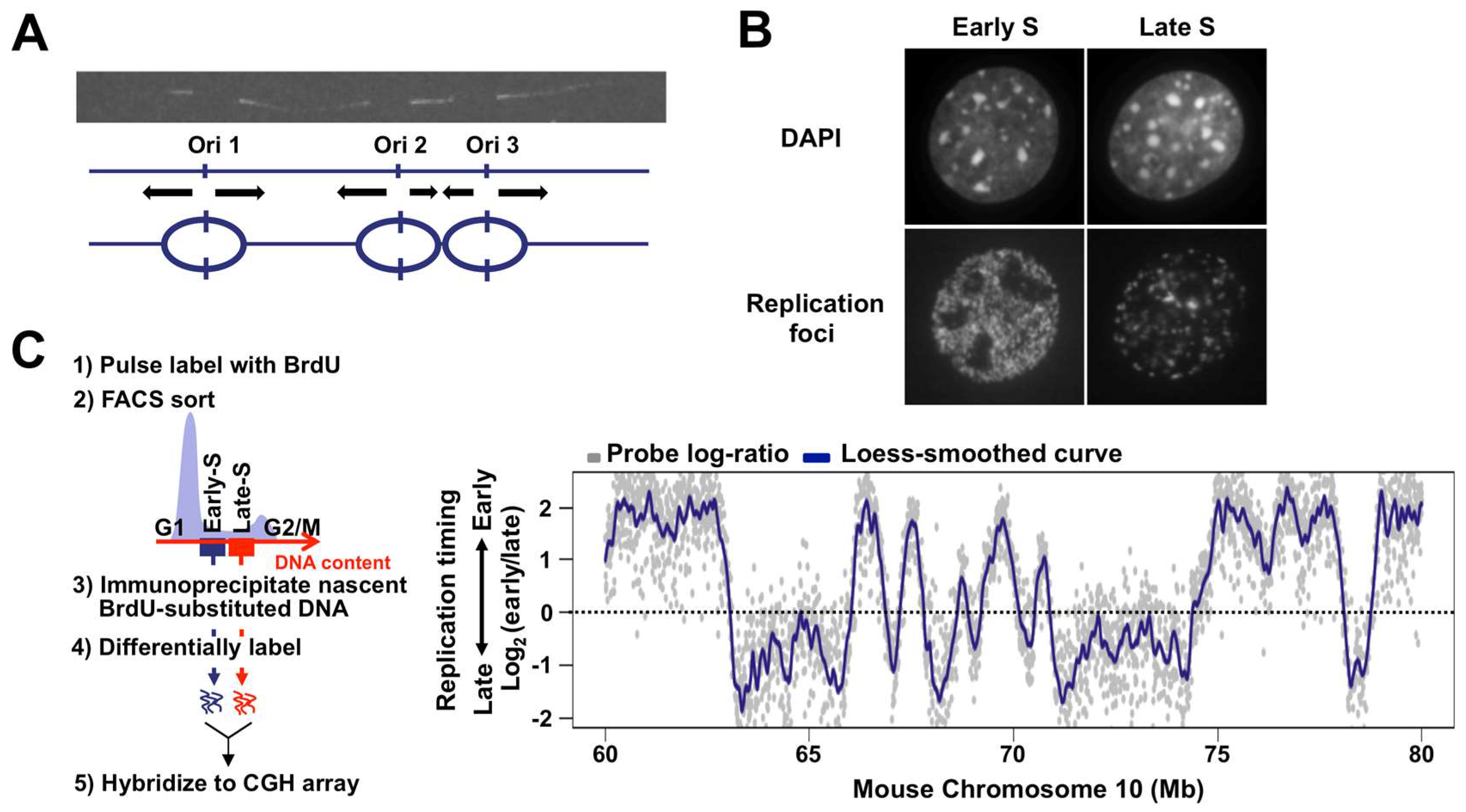
2. The Mammalian Replication Domain Comes into Focus
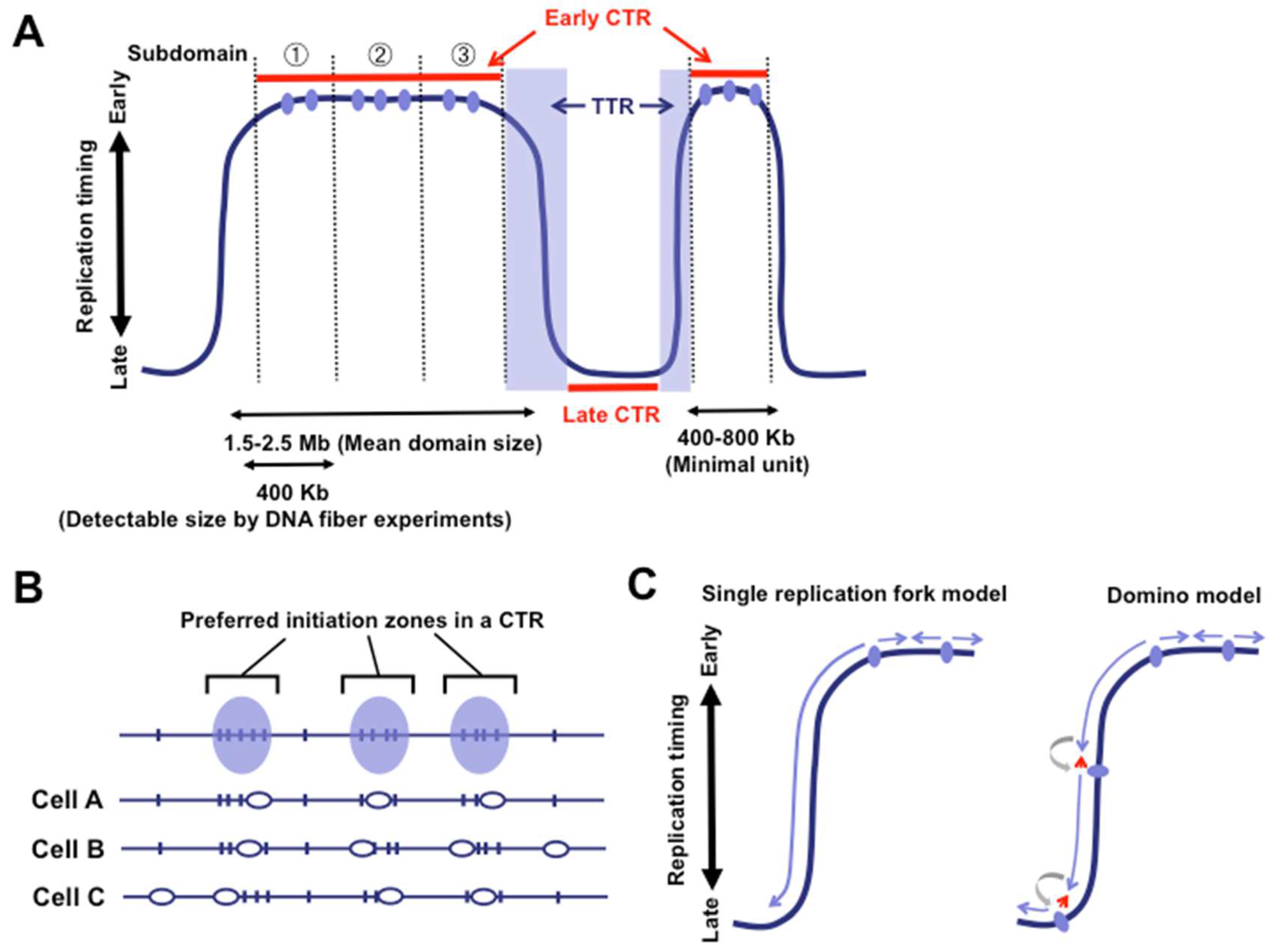
3. Flexible Nature of Replication Origins in CTRs
4. TTRs: One-Way Roads?
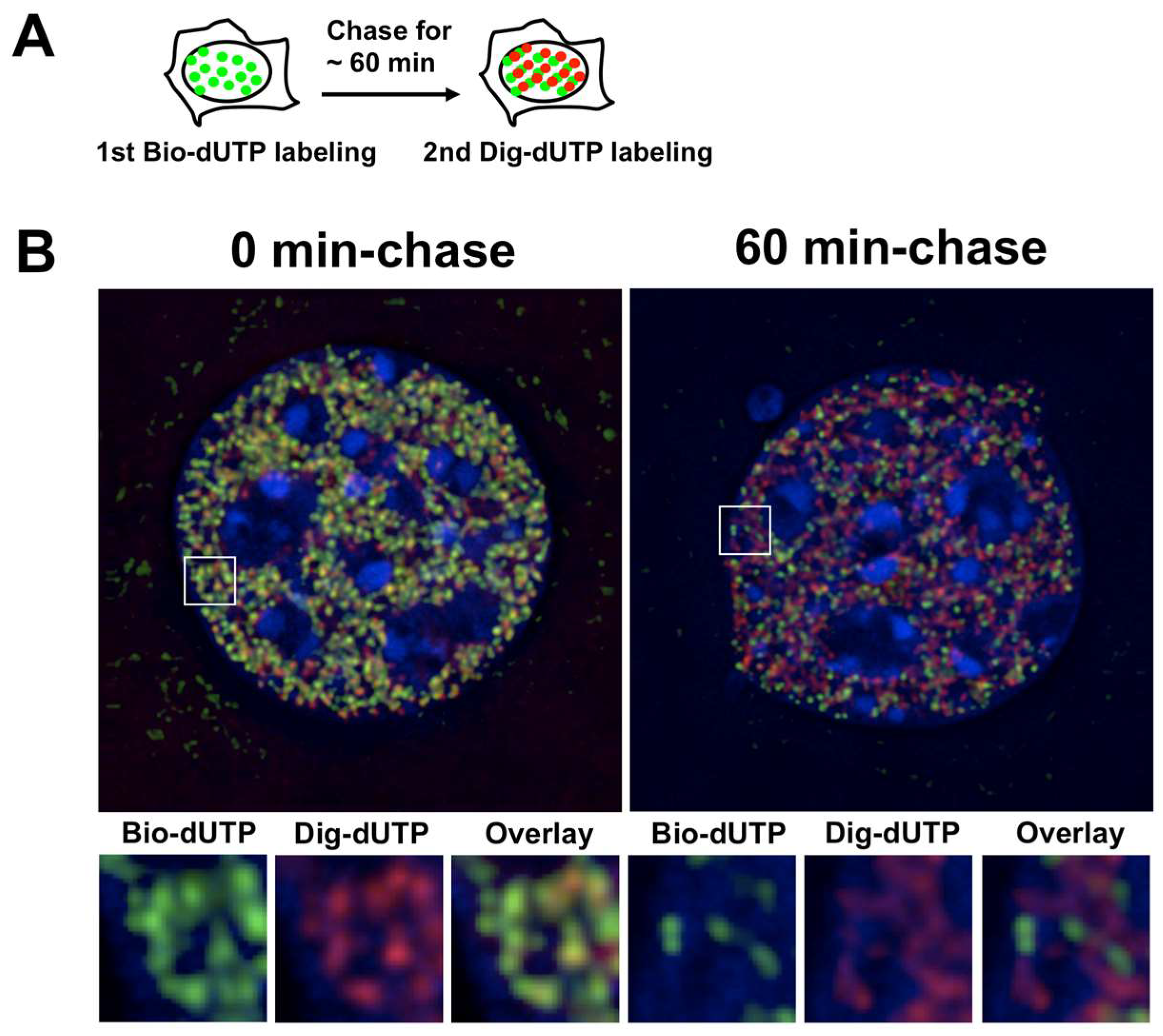
5. Dynamic Properties of Replication Domains
6. Replication Domains, Replication Foci, and Replication Origins: Making All Things Consistent
7. Close Relationship between Replication Domains and Three-Dimensional (3D) Genome Organization
8. A New Step toward Understanding the Biological Significance of Replication Domain Regulation
9. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Berezney, R.; Dubey, D.D.; Huberman, J.A. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 2000, 108, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Huberman, J.A.; Riggs, A.D. On the mechanism of DNA replication in mammalian chromosomes. J. Mol. Biol. 1968, 32, 327–341. [Google Scholar] [CrossRef]
- Jackson, D.A.; Pombo, A. Replicon clusters are stable units of chromosome structure: Evidence that nuclear organization contributes to the efficient activation and propagation of s phase in human cells. J. Cell Biol. 1998, 140, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.R.; Gilbert, D.M. A distinct g1 step required to specify the chinese hamster dhfr replication origin. Science 1996, 271, 1270–1272. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, D.S.; Gilbert, D.M. The spatial position and replication timing of chromosomal domains are both established in early g1 phase. Mol. Cell 1999, 4, 983–993. [Google Scholar] [CrossRef]
- Rivera-Mulia, J.C.; Gilbert, D.M. Replicating large genomes: Divide and conquer. Mol. Cell 2016, 62, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, D.S.; Berezney, R. The spatio-temporal organization of DNA replication sites is identical in primary, immortalized and transformed mammalian cells. J. Cell Sci. 2002, 115, 4037–4051. [Google Scholar] [CrossRef] [PubMed]
- Nakayasu, H.; Berezney, R. Mapping replicational sites in the eucaryotic cell nucleus. J. Cell Biol. 1989, 108, 1–11. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, R.T.; Henderson, S.C.; Spector, D.L. Dynamic organization of DNA replication in mammalian cell nuclei: Spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J. Cell Biol. 1992, 116, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Handsaker, R.E.; Kamitaki, N.; Karlic, R.; Ghosh, S.; Polak, P.; Eggan, K.; McCarroll, S.A. Genetic variation in human DNA replication timing. Cell 2014, 159, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Farkash-Amar, S.; Lipson, D.; Polten, A.; Goren, A.; Helmstetter, C.; Yakhini, Z.; Simon, I. Global organization of replication time zones of the mouse genome. Genome Res. 2008, 18, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, I.; Ryba, T.; Itoh, M.; Yokochi, T.; Schwaiger, M.; Chang, C.W.; Lyou, Y.; Townes, T.M.; Schubeler, D.; Gilbert, D.M. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008, 6, e245. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.S.; Thomas, S.; Sandstrom, R.; Canfield, T.K.; Thurman, R.E.; Weaver, M.; Dorschner, M.O.; Gartler, S.M.; Stamatoyannopoulos, J.A. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. USA 2010, 107, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, S.I.; Manders, E.M.; Kimura, H.; Taguchi, H.; Okumura, K. Mapping sites where replication initiates in mammalian cells using DNA fibers. Exp. Cell Res. 2001, 271, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, S.; Sugimura, K.; Saito, T.; Sato, C.; Fukushima, Y.; Taguchi, H.; Okumura, K. Regulation of replication at the r/g chromosomal band boundary and pericentromeric heterochromatin of mammalian cells. Exp. Cell Res. 2005, 304, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Battaglia, D.; Pope, B.D.; Hiratani, I.; Gilbert, D.M. Genome-scale analysis of replication timing: From bench to bioinformatics. Nat. Protoc. 2011, 6, 870–895. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Buckley, Q.; Sasaki, T.; Zimmerman, J.; Didier, R.A.; Nazor, K.; Loring, J.F.; Lian, Z.; Weissman, S.; Robins, A.J.; et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 2015, 25, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, S.; Lei, I.; Ryba, T.; Sasaki, T.; Dileep, V.; Battaglia, D.; Gao, X.; Fang, P.; Fan, Y.; Esteban, M.A.; et al. Murine esbaf chromatin remodeling complex subunits baf250a and brg1 are necessary to maintain and reprogram pluripotency-specific replication timing of select replication domains. Epigenetics Chromatin 2013, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Cayrou, C.; Coulombe, P.; Vigneron, A.; Stanojcic, S.; Ganier, O.; Peiffer, I.; Rivals, E.; Puy, A.; Laurent-Chabalier, S.; Desprat, R.; et al. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 2011, 21, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Norio, P.; Kosiyatrakul, S.; Yang, Q.; Guan, Z.; Brown, N.M.; Thomas, S.; Riblet, R.; Schildkraut, C.L. Progressive activation of DNA replication initiation in large domains of the immunoglobulin heavy chain locus during b cell development. Mol. Cell 2005, 20, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Lebofsky, R.; Heilig, R.; Sonnleitner, M.; Weissenbach, J.; Bensimon, A. DNA replication origin interference increases the spacing between initiation events in human cells. Mol. Biol. Cell 2006, 17, 5337–5345. [Google Scholar] [CrossRef] [PubMed]
- Techer, H.; Koundrioukoff, S.; Azar, D.; Wilhelm, T.; Carignon, S.; Brison, O.; Debatisse, M.; Le Tallec, B. Replication dynamics: Biases and robustness of DNA fiber analysis. J. Mol. Biol. 2013, 425, 4845–4855. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Martin, M.M.; Regairaz, M.; Huang, L.; You, Y.; Lin, C.M.; Ryan, M.; Kim, R.; Shimura, T.; Pommier, Y.; et al. The DNA repair endonuclease mus81 facilitates fast DNA replication in the absence of exogenous damage. Nat. Commun. 2015, 6, 6746. [Google Scholar] [CrossRef] [PubMed]
- Besnard, E.; Babled, A.; Lapasset, L.; Milhavet, O.; Parrinello, H.; Dantec, C.; Marin, J.M.; Lemaitre, J.M. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with g-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 2012, 19, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; McCarroll, S.A. Random replication of the inactive x chromosome. Genome Res. 2014, 24, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Petryk, N.; Kahli, M.; d’Aubenton-Carafa, Y.; Jaszczyszyn, Y.; Shen, Y.; Silvain, M.; Thermes, C.; Chen, C.L.; Hyrien, O. Replication landscape of the human genome. Nat. Commun. 2016, 7, 10208. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Alm, E.J.; Arkin, A.P. Interruptions in gene expression drive highly expressed operons to the leading strand of DNA replication. Nucleic Acids Res. 2005, 33, 3224–3234. [Google Scholar] [CrossRef] [PubMed]
- Bechhoefer, J.; Rhind, N. Replication timing and its emergence from stochastic processes. Trends Genet. 2012, 28, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Hayano, M.; Masai, H. Replication timing regulation of eukaryotic replicons: Rif1 as a global regulator of replication timing. Trends Genet. 2013, 29, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Knott, S.R.; Peace, J.M.; Ostrow, A.Z.; Gan, Y.; Rex, A.E.; Viggiani, C.J.; Tavare, S.; Aparicio, O.M. Forkhead transcription factors establish origin timing and long-range clustering in s. Cerevisiae. Cell 2012, 148, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Goren, A.; Tabib, A.; Hecht, M.; Cedar, H. DNA replication timing of the human beta-globin domain is controlled by histone modification at the origin. Genes Dev. 2008, 22, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Hughes, C.M.; Kosiyatrakul, S.; Norio, P.; Sen, R.; Fiering, S.; Allis, C.D.; Bouhassira, E.E.; Schildkraut, C.L. Decreased replication origin activity in temporal transition regions. J. Cell Biol. 2009, 187, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, G.; Rappailles, A.; Baker, A.; Chen, C.L.; Arneodo, A.; Goldar, A.; d’Aubenton-Carafa, Y.; Thermes, C.; Audit, B.; Hyrien, O. Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput. Biol. 2011, 7, e1002322. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, I.; Ryba, T.; Itoh, M.; Rathjen, J.; Kulik, M.; Papp, B.; Fussner, E.; Bazett-Jones, D.P.; Plath, K.; Dalton, S.; et al. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res. 2010, 20, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Hiratani, I.; Sasaki, T.; Battaglia, D.; Kulik, M.; Zhang, J.; Dalton, S.; Gilbert, D.M. Replication timing: A fingerprint for cell identity and pluripotency. PLoS Comput. Biol. 2011, 7, e1002225. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, N.; Boyle, S.; Fiegler, H.; Woodfine, K.; Carter, N.P.; Bickmore, W.A. Chromatin architecture of the human genome: Gene-rich domains are enriched in open chromatin fibers. Cell 2004, 118, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, I.; Takebayashi, S.; Lu, J.; Gilbert, D.M. Replication timing and transcriptional control: Beyond cause and effect-part ii. Curr. Opin. Genet. Dev. 2009, 19, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Gilbert, D.M. Replication timing and transcriptional control: Beyond cause and effect-part iii. Curr. Opin. Cell Biol. 2016, 40, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, S.; Dileep, V.; Ryba, T.; Dennis, J.H.; Gilbert, D.M. Chromatin-interaction compartment switch at developmentally regulated chromosomal domains reveals an unusual principle of chromatin folding. Proc. Natl. Acad. Sci. USA 2012, 109, 12574–12579. [Google Scholar] [CrossRef] [PubMed]
- Yokochi, T.; Poduch, K.; Ryba, T.; Lu, J.; Hiratani, I.; Tachibana, M.; Shinkai, Y.; Gilbert, D.M. G9a selectively represses a class of late-replicating genes at the nuclear periphery. Proc. Natl. Acad. Sci. USA 2009, 106, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Samarabandu, J.; Devdhar, R.S.; Acharya, R.; Cheng, P.C.; Meng, C.; Berezney, R. Spatial and temporal dynamics of DNA replication sites in mammalian cells. J. Cell Biol. 1998, 143, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Manders, E.M.; Stap, J.; Brakenhoff, G.J.; van Driel, R.; Aten, J.A. Dynamics of three-dimensional replication patterns during the s-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 1992, 103 Pt 3, 857–862. [Google Scholar] [PubMed]
- Manders, E.M.; Stap, J.; Strackee, J.; van Driel, R.; Aten, J.A. Dynamic behavior of DNA replication domains. Exp. Cell Res. 1996, 226, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Taddei, A.; Roche, D.; Sibarita, J.B.; Turner, B.M.; Almouzni, G. Duplication and maintenance of heterochromatin domains. J. Cell Biol. 1999, 147, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Baddeley, D.; Chagin, V.O.; Schermelleh, L.; Martin, S.; Pombo, A.; Carlton, P.M.; Gahl, A.; Domaing, P.; Birk, U.; Leonhardt, H.; et al. Measurement of replication structures at the nanometer scale using super-resolution light microscopy. Nucleic Acids Res. 2010, 38, e8. [Google Scholar] [CrossRef] [PubMed]
- Cseresnyes, Z.; Schwarz, U.; Green, C.M. Analysis of replication factories in human cells by super-resolution light microscopy. BMC Cell Biol. 2009, 10, 88. [Google Scholar] [CrossRef] [PubMed]
- Chagin, V.O.; Casas-Delucchi, C.S.; Reinhart, M.; Schermelleh, L.; Markaki, Y.; Maiser, A.; Bolius, J.J.; Bensimon, A.; Fillies, M.; Domaing, P.; et al. 4D visualization of replication foci in mammalian cells corresponding to individual replicons. Nat. Commun. 2016, 7, 11231. [Google Scholar] [CrossRef] [PubMed]
- Lob, D.; Lengert, N.; Chagin, V.O.; Reinhart, M.; Casas-Delucchi, C.S.; Cardoso, M.C.; Drossel, B. 3D replicon distributions arise from stochastic initiation and domino-like DNA replication progression. Nat. Commun. 2016, 7, 11207. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Olivares-Chauvet, P.; Shaw, A.; Jackson, D.A. S phase progression in human cells is dictated by the genetic continuity of DNA foci. PLoS Genet. 2010, 6, e1000900. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, S.; Department of Biochemistry and Proteomics, Graduate School of Medicine, Mie University, Tsu, Mie, Japan. Personal communication, 2017.
- Dekker, J.; Marti-Renom, M.A.; Mirny, L.A. Exploring the three-dimensional organization of genomes: Interpreting chromatin interaction data. Nat. Rev. Genet. 2013, 14, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Dileep, V.; Ay, F.; Sima, J.; Vera, D.L.; Noble, W.S.; Gilbert, D.M. Topologically associating domains and their long-range contacts are established during early g1 coincident with the establishment of the replication-timing program. Genome Res. 2015, 25, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Dileep, V.; Rivera-Mulia, J.C.; Sima, J.; Gilbert, D.M. Large-scale chromatin structure-function relationships during the cell cycle and development: Insights from replication timing. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Ishii, A.; Kanoh, Y.; Oda, M.; Nishito, Y.; Masai, H. Rif1 regulates the replication timing domains on the human genome. EMBO J. 2012, 31, 3667–3677. [Google Scholar] [CrossRef] [PubMed]
- Cornacchia, D.; Dileep, V.; Quivy, J.P.; Foti, R.; Tili, F.; Santarella-Mellwig, R.; Anthony, C.; Almouzni, G.; Gilbert, D.M.; Buonomo, S.B. Mouse rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 2012, 31, 3678–3690. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear architecture organized by rif1 underpins the replication-timing program. Mol. Cell 2016, 61, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, F.; Hashimshony, T.; Keshet, I.; Cedar, H. Establishment of transcriptional competence in early and late s phase. Nature 2002, 420, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Selig, S.; Okumura, K.; Ward, D.C.; Cedar, H. Delineation of DNA replication time zones by fluorescence in situ hybridization. EMBO J. 1992, 11, 1217–1225. [Google Scholar] [PubMed]
- Kagotani, K.; Takebayashi, S.; Kohda, A.; Taguchi, H.; Paulsen, M.; Walter, J.; Reik, W.; Okumura, K. Replication timing properties within the mouse distal chromosome 7 imprinting cluster. Biosci. Biotechnol. Biochem. 2002, 66, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Van der Aa, N.; Cheng, J.; Mateiu, L.; Zamani Esteki, M.; Kumar, P.; Dimitriadou, E.; Vanneste, E.; Moreau, Y.; Vermeesch, J.R.; Voet, T. Genome-wide copy number profiling of single cells in s-phase reveals DNA-replication domains. Nucleic Acids Res. 2013, 41, e66. [Google Scholar] [CrossRef] [PubMed]
- Manukjan, G.; Tauscher, M.; Steinemann, D. Replication timing influences DNA copy number determination by array-cgh. Biotechniques 2013, 55, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.S.; Kester, L.; Spanjaard, B.; Bienko, M.; van Oudenaarden, A. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015, 33, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Andrey, G.; Montavon, T.; Mascrez, B.; Gonzalez, F.; Noordermeer, D.; Leleu, M.; Trono, D.; Spitz, F.; Duboule, D. A switch between topological domains underlies hoxd genes collinearity in mouse limbs. Science 2013, 340, 1234167. [Google Scholar] [CrossRef] [PubMed]
- Lupianez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [PubMed]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takebayashi, S.-i.; Ogata, M.; Okumura, K. Anatomy of Mammalian Replication Domains. Genes 2017, 8, 110. https://doi.org/10.3390/genes8040110
Takebayashi S-i, Ogata M, Okumura K. Anatomy of Mammalian Replication Domains. Genes. 2017; 8(4):110. https://doi.org/10.3390/genes8040110
Chicago/Turabian StyleTakebayashi, Shin-ichiro, Masato Ogata, and Katsuzumi Okumura. 2017. "Anatomy of Mammalian Replication Domains" Genes 8, no. 4: 110. https://doi.org/10.3390/genes8040110
APA StyleTakebayashi, S.-i., Ogata, M., & Okumura, K. (2017). Anatomy of Mammalian Replication Domains. Genes, 8(4), 110. https://doi.org/10.3390/genes8040110