
Modeling and Targeting MYC Genes in Childhood Brain Tumors

{kind=link}
{kind=link}
Abstract
:1. Introduction
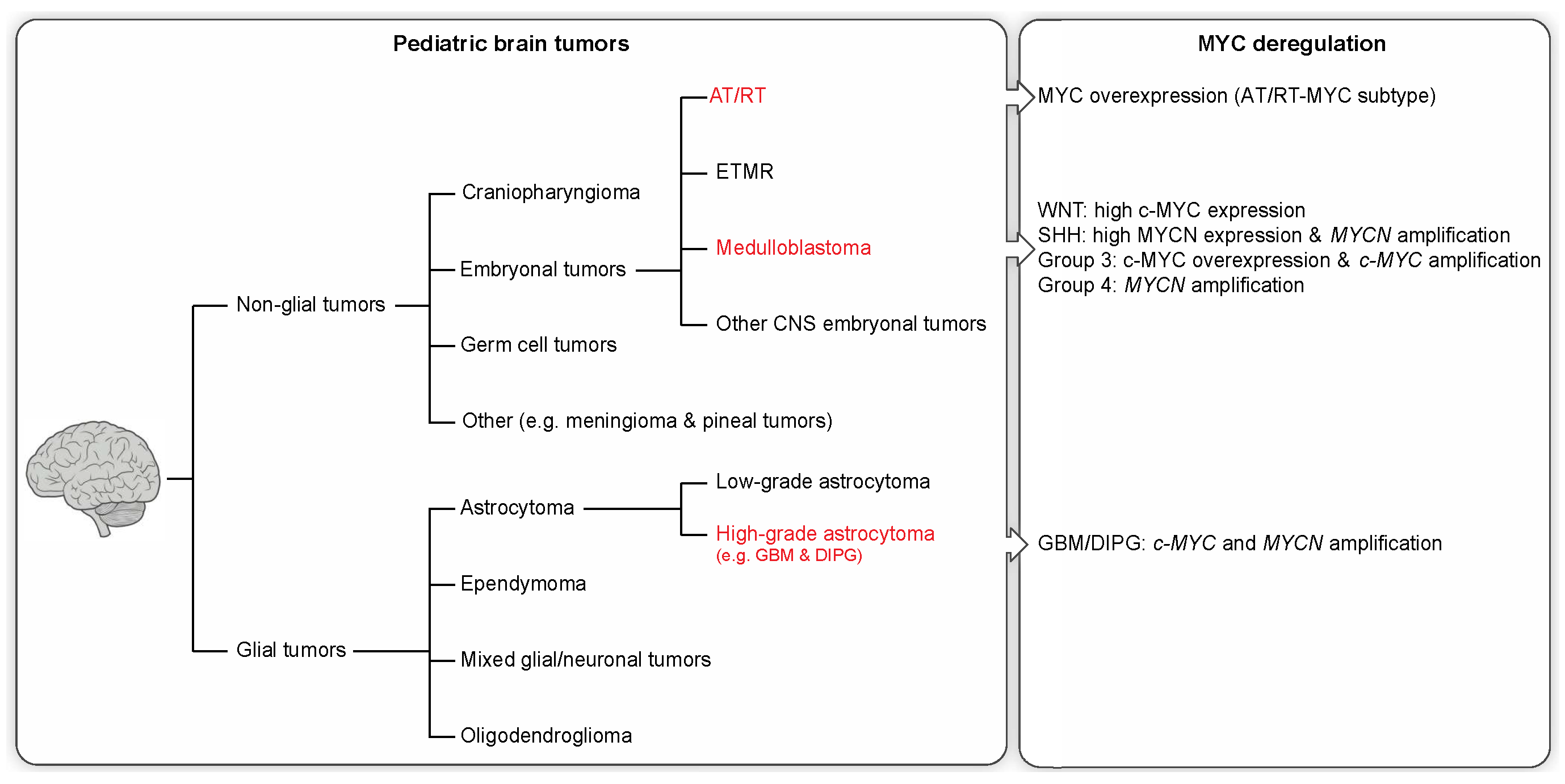
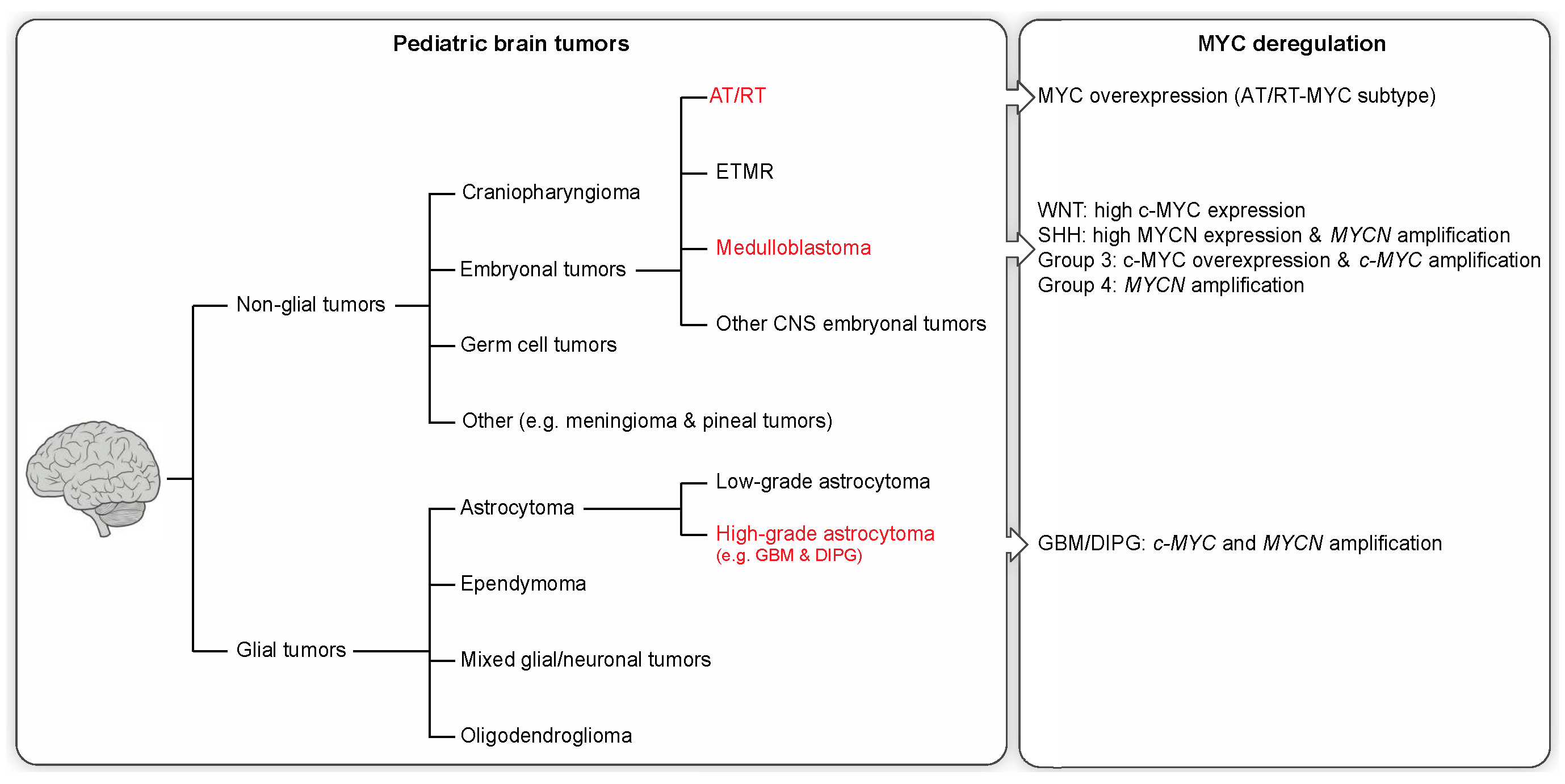
2. Pediatric Brain Tumors and Clinical Features
2.1. Non-Glial Tumors
2.2. Glial Tumors
3. Molecular Profiling and MYC Misregulation in Childhood Brain Tumors
4. Mechanisms Involved in MYC-Driven Brain Tumor Initiation
5. MYC-Driven Models of Brain Tumors
6. MYC Target Genes Involved in Tumor Maintenance and Recurrence
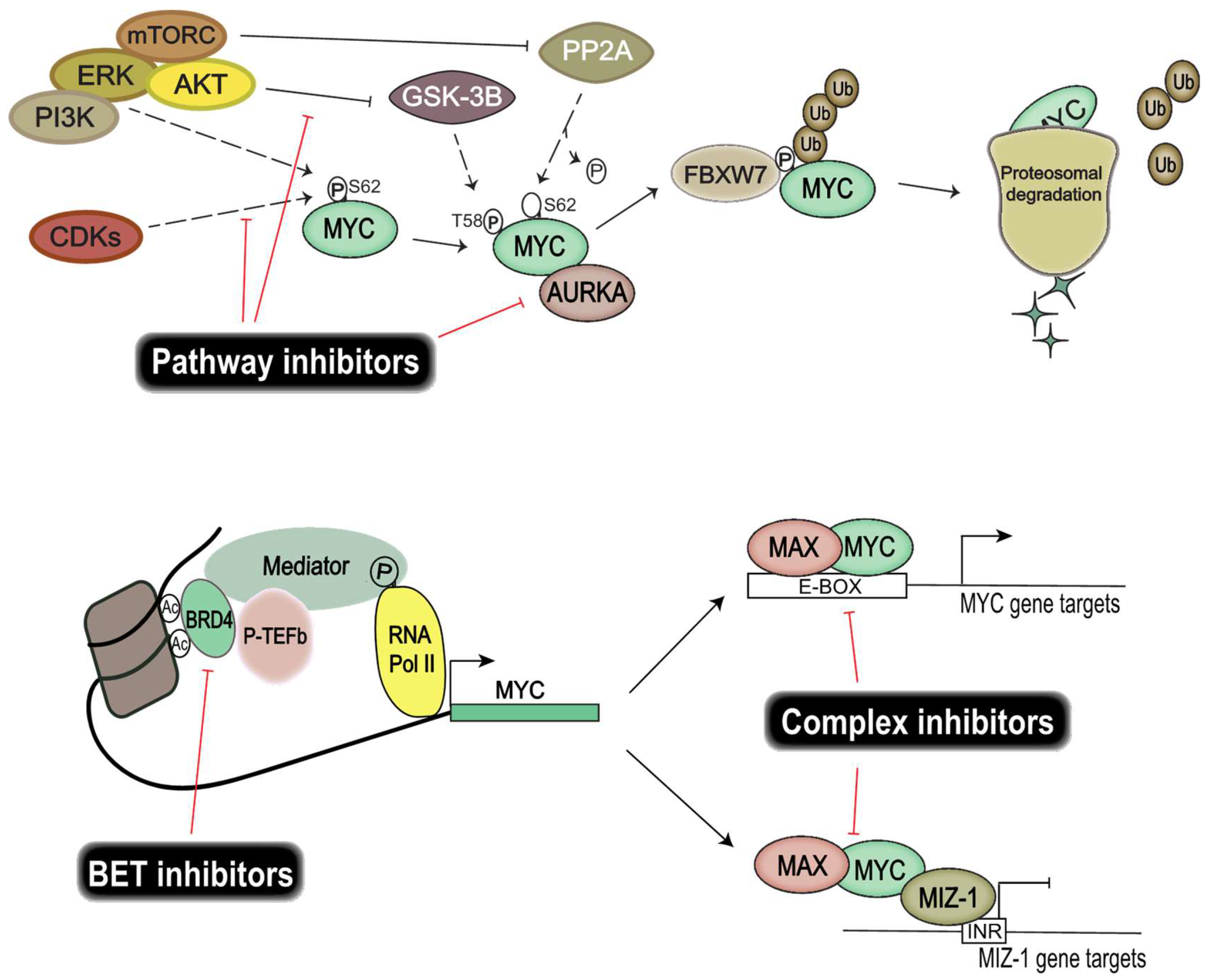
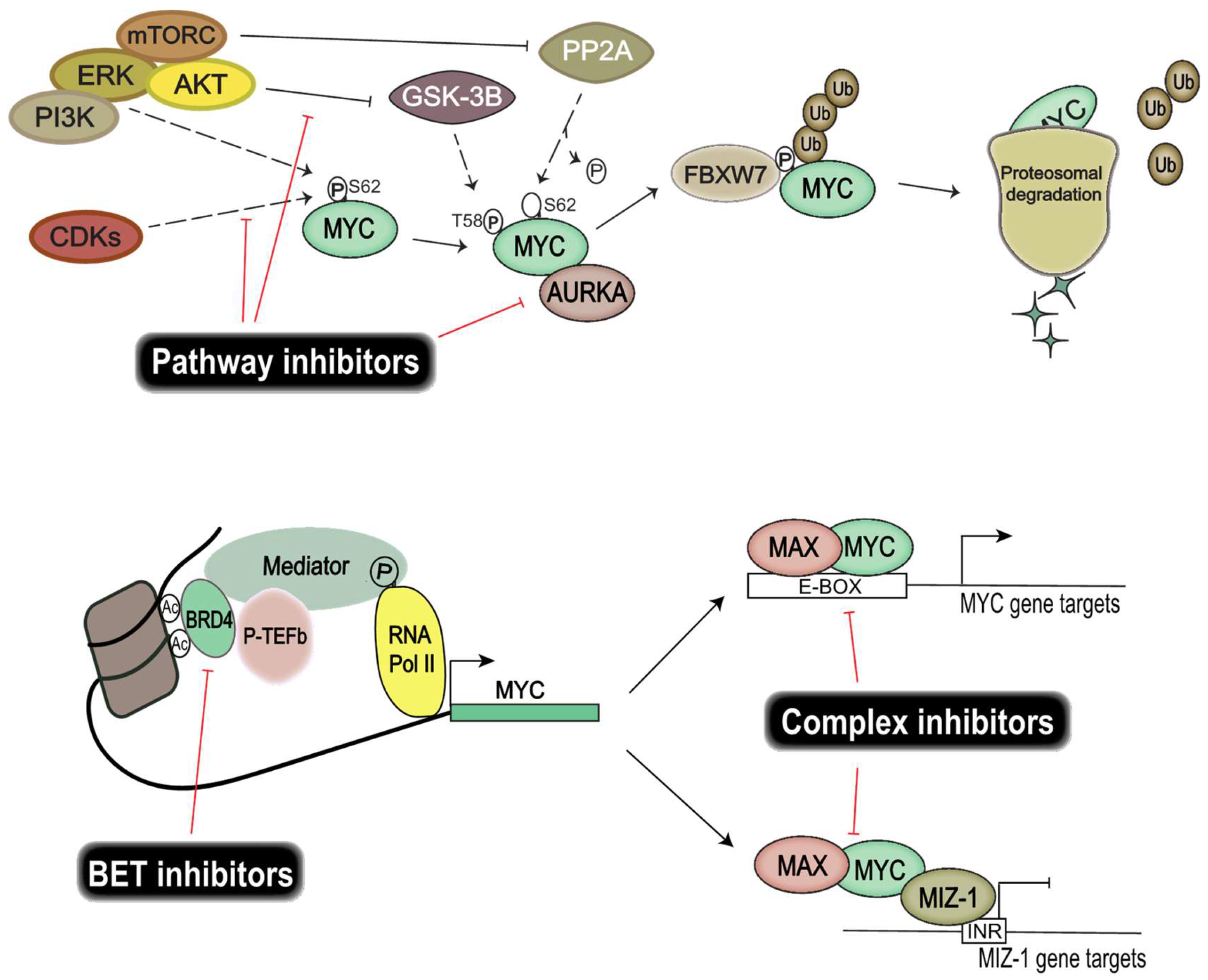
7. Pharmacological Inhibition of MYC Proteins and Their Transcriptional Targets
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roussel, M.F.; Robinson, G.W. Role of MYC in medulloblastoma. Cold Spring Harb. Perspect. Med. 2013, 5. [Google Scholar] [CrossRef]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.; Sturm, D.; Hermann, C.; Segura Wang, M.; et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Tansey, W.P. Mammalian MYC proteins and cancer. New J. Sci. 2014, 2014. [Google Scholar] [CrossRef]
- Wiese, K.E.; Walz, S.; von Eyss, B.; Wolf, E.; Athineos, D.; Sansom, O.; Eilers, M. The role of MIZ-1 in MYC-dependent tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014290. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, C.; Felsher, D.W. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin. Cancer Biol. 2006, 16, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Quickstats: Cancer death rates* for children and teens aged 1–19 years—United States, 1999, 2006, and 2014. Morb. Mortal. Wkly. Rep. 2016, 65, 1153.
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the united states in 2008–2012. Neuro Oncol. 2015, 17 (Suppl. 4), iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Ginn, K.F.; Gajjar, A. Atypical teratoid rhabdoid tumor: Current therapy and future directions. Front. Oncol. 2012, 2, 114. [Google Scholar] [PubMed]
- Korshunov, A.; Sturm, D.; Ryzhova, M.; Hovestadt, V.; Gessi, M.; Jones, D.T.; Remke, M.; Northcott, P.; Perry, A.; Picard, D.; et al. Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol. 2014, 128, 279–289. [Google Scholar] [CrossRef]
- Echevarria, M.E.; Fangusaro, J.; Goldman, S. Pediatric central nervous system germ cell tumors: A review. Oncologist 2008, 13, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Bender, S.; Jones, D.T.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Bjerke, L.; Mackay, A.; Nandhabalan, M.; Burford, A.; Jury, A.; Popov, S.; Bax, D.A.; Carvalho, D.; Taylor, K.R.; Vinci, M.; et al. Histone H3.3. Mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 2013, 3, 512–519.17. [Google Scholar] [CrossRef] [PubMed]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Vanan, M.I.; Eisenstat, D.D. Dipg in children—What can we learn from the past? Front. Oncol. 2015, 5, 237. [Google Scholar] [CrossRef]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Korshunov, A.; Pfister, S.M.; Taylor, M.D. The clinical implications of medulloblastoma subgroups. Nat. Rev. Neurol. 2012, 8, 340–351. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stutz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [PubMed]
- Lee, R.S.; Stewart, C.; Carter, S.L.; Ambrogio, L.; Cibulskis, K.; Sougnez, C.; Lawrence, M.S.; Auclair, D.; Mora, J.; Golub, T.R.; et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J. Clin. Investig. 2012, 122, 2983–2988. [Google Scholar] [PubMed]
- Hasselblatt, M.; Nagel, I.; Oyen, F.; Bartelheim, K.; Russell, R.B.; Schuller, U.; Junckerstorff, R.; Rosenblum, M.; Alassiri, A.H.; Rossi, S.; et al. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. 2014, 128, 453–456. [Google Scholar] [PubMed]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Jakacki, R.I.; Burger, P.C.; Kocak, M.; Boyett, J.M.; Goldwein, J.; Mehta, M.; Packer, R.J.; Tarbell, N.J.; Pollack, I.F. Outcome and prognostic factors for children with supratentorial primitive neuroectodermal tumors treated with carboplatin during radiotherapy: A report from the children’s oncology group. Pediatr. Blood Cancer 2015, 62, 776–783. [Google Scholar] [CrossRef]
- Sturm, D.; Orr, B.A.; Toprak, U.H.; Hovestadt, V.; Jones, D.T.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott, P.A.; Leis, I.; et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CPG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [PubMed]
- Bax, D.A.; Mackay, A.; Little, S.E.; Carvalho, D.; Viana-Pereira, M.; Tamber, N.; Grigoriadis, A.E.; Ashworth, A.; Reis, R.M.; Ellison, D.W.; et al. A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin. Cancer. Res. 2010, 16, 3368–3377. [Google Scholar] [PubMed]
- Jones, C.; Perryman, L.; Hargrave, D. Paediatric and adult malignant glioma: Close relatives or distant cousins? Nat. Rev. Clin. Oncol. 2012, 9, 400–413. [Google Scholar] [PubMed]
- Paugh, B.S.; Qu, C.; Jones, C.; Liu, Z.; Adamowicz-Brice, M.; Zhang, J.; Bax, D.A.; Coyle, B.; Barrow, J.; Hargrave, D.; et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 2010, 28, 3061–3068. [Google Scholar]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [PubMed]
- Swartling, F.J.; Savov, V.; Persson, A.I.; Chen, J.; Hackett, C.S.; Northcott, P.A.; Grimmer, M.R.; Lau, J.; Chesler, L.; Perry, A.; et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell 2012, 21, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [PubMed]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pages, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [PubMed]
- Khuong-Quang, D.A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in Grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar]
- Wiestler, B.; Capper, D.; Holland-Letz, T.; Korshunov, A.; von Deimling, A.; Pfister, S.M.; Platten, M.; Weller, M.; Wick, W. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 2013, 126, 443–451. [Google Scholar] [PubMed]
- Grimm, S.A.; Chamberlain, M.C. Anaplastic astrocytoma. CNS Oncol. 2016, 5, 145–157. [Google Scholar] [PubMed]
- Barrow, J.; Adamowicz-Brice, M.; Cartmill, M.; MacArthur, D.; Lowe, J.; Robson, K.; Brundler, M.A.; Walker, D.A.; Coyle, B.; Grundy, R. Homozygous loss of ADAM3A revealed by genome-wide analysis of pediatric high-grade glioma and diffuse intrinsic pontine gliomas. Neuro Oncol. 2011, 13, 212–222. [Google Scholar] [PubMed]
- Radke, J.; Bortolussi, G.; Pagenstecher, A. Akt and c-Myc induce stem-cell markers in mature primary p53-/- astrocytes and render these cells gliomagenic in the brain of immunocompetent mice. PLoS ONE 2013, 8, e56691. [Google Scholar]
- Bai, H.; Harmanci, A.S.; Erson-Omay, E.Z.; Li, J.; Coskun, S.; Simon, M.; Krischek, B.; Ozduman, K.; Omay, S.B.; Sorensen, E.A.; et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016, 48, 59–66. [Google Scholar]
- Kim, H.S.; Woolard, K.; Lai, C.; Bauer, P.O.; Maric, D.; Song, H.; Li, A.; Kotliarova, S.; Zhang, W.; Fine, H.A. Gliomagenesis arising from pten- and INK4A/ARF-deficient neural progenitor cells is mediated by the p53-FBXW7/CDC4 pathway, which controls c-MYC. Cancer Res. 2012, 72, 6065–6075. [Google Scholar] [PubMed]
- Felsher, D.W.; Zetterberg, A.; Zhu, J.; Tlsty, T.; Bishop, J.M. Overexpression of MYC causes p53-dependent g2 arrest of normal fibroblasts. Proc. Natl. Acad. Sci. USA 2000, 97, 10544–10548. [Google Scholar] [PubMed]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-MYC protein. Cell 1992, 69, 119–128. [Google Scholar]
- Nilsson, J.A.; Cleveland, J.L. MYC pathways provoking cell suicide and cancer. Oncogene 2003, 22, 9007–9021. [Google Scholar] [PubMed]
- Hoffman, B.; Liebermann, D.A. Apoptotic signaling by c-MYC. Oncogene 2008, 27, 6462–6472. [Google Scholar] [PubMed]
- Grandori, C.; Wu, K.J.; Fernandez, P.; Ngouenet, C.; Grim, J.; Clurman, B.E.; Moser, M.J.; Oshima, J.; Russell, D.W.; Swisshelm, K.; et al. Werner syndrome protein limits MYC-induced cellular senescence. Genes Dev. 2003, 17, 1569–1574. [Google Scholar] [PubMed]
- Land, H.; Parada, L.F.; Weinberg, R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983, 304, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. MYC signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar]
- Pei, Y.; Moore, C.E.; Wang, J.; Tewari, A.K.; Eroshkin, A.; Cho, Y.J.; Witt, H.; Korshunov, A.; Read, T.A.; Sun, J.L.; et al. An animal model of MYC-driven medulloblastoma. Cancer Cell 2012, 21, 155–167. [Google Scholar] [PubMed]
- Kawauchi, D.; Robinson, G.; Uziel, T.; Gibson, P.; Rehg, J.; Gao, C.; Finkelstein, D.; Qu, C.; Pounds, S.; Ellison, D.W.; et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 2012, 21, 168–180. [Google Scholar] [PubMed]
- Cho, Y.J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J. Clin. Oncol. 2011, 29, 1424–1430. [Google Scholar] [PubMed]
- Eischen, C.M.; Woo, D.; Roussel, M.F.; Cleveland, J.L. Apoptosis triggered by MYC-induced suppression of Bcl-XL or Bcl-2 is bypassed during lymphomagenesis. Mol. Cell. Biol. 2001, 21, 5063–5070. [Google Scholar] [CrossRef] [PubMed]
- Muthalagu, N.; Junttila, M.R.; Wiese, K.E.; Wolf, E.; Morton, J.; Bauer, B.; Evan, G.I.; Eilers, M.; Murphy, D.J. BIM is the primary mediator of MYC-induced apoptosis in multiple solid tissues. Cell Rep. 2014, 8, 1347–1353. [Google Scholar]
- Mitchell, K.O.; Ricci, M.S.; Miyashita, T.; Dicker, D.T.; Jin, Z.; Reed, J.C.; El-Deiry, W.S. Bax is a transcriptional target and mediator of c-MYC-induced apoptosis. Cancer Res. 2000, 60, 6318–6325. [Google Scholar] [PubMed]
- Jenkins, N.C.; Rao, G.; Eberhart, C.G.; Pedone, C.A.; Dubuc, A.M.; Fults, D.W. Somatic cell transfer of c-Myc and Bcl-2 induces large-cell anaplastic medulloblastomas in mice. J. Neurooncol. 2016, 126, 415–424. [Google Scholar] [PubMed]
- Eilers, M.; Schirm, S.; Bishop, J.M. The MYC protein activates transcription of the alpha-prothymosin gene. EMBO J. 1991, 10, 133–141. [Google Scholar] [PubMed]
- Bouchard, C.; Thieke, K.; Maier, A.; Saffrich, R.; Hanley-Hyde, J.; Ansorge, W.; Reed, S.; Sicinski, P.; Bartek, J.; Eilers, M. Direct induction of cyclin D2 by MYC contributes to cell cycle progression and sequestration of p27. EMBO J. 1999, 18, 5321–5333. [Google Scholar] [PubMed]
- Bretones, G.; Delgado, M.D.; Leon, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Felsher, D.W.; Bishop, J.M. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 3940–3944. [Google Scholar] [PubMed]
- Louis, S.F.; Vermolen, B.J.; Garini, Y.; Young, I.T.; Guffei, A.; Lichtensztejn, Z.; Kuttler, F.; Chuang, T.C.; Moshir, S.; Mougey, V.; et al. c-MYC induces chromosomal rearrangements through telomere and chromosome remodeling in the interphase nucleus. Proc. Natl. Acad. Sci. USA 2005, 102, 9613–9618. [Google Scholar] [PubMed]
- Kuzyk, A.; Mai, S. c-MYC-induced genomic instability. Cold Spring Harb. Perspect. Med. 2014, 4, a014373. [Google Scholar] [CrossRef] [PubMed]
- Mai, S. Overexpression of c-MYC precedes amplification of the gene encoding dihydrofolate reductase. Gene 1994, 148, 253–260. [Google Scholar]
- Karlsson, A.; Giuriato, S.; Tang, F.; Fung-Weier, J.; Levan, G.; Felsher, D.W. Genomically complex lymphomas undergo sustained tumor regression upon MYC inactivation unless they acquire novel chromosomal translocations. Blood 2003, 101, 2797–2803. [Google Scholar] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [PubMed]
- Hsieh, A.L.; Walton, Z.E.; Altman, B.J.; Stine, Z.E.; Dang, C.V. MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol. 2015, 43, 11–21. [Google Scholar]
- Barna, M.; Pusic, A.; Zollo, O.; Costa, M.; Kondrashov, N.; Rego, E.; Rao, P.H.; Ruggero, D. Suppression of MYC oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008, 456, 971–975. [Google Scholar] [PubMed]
- Janz, A.; Sevignani, C.; Kenyon, K.; Ngo, C.V.; Thomas-Tikhonenko, A. Activation of the MYC oncoprotein leads to increased turnover of thrombospondin-1 MRNA. Nucleic Acids Res. 2000, 28, 2268–2275. [Google Scholar] [PubMed]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. c-MYC is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef]
- Knies-Bamforth, U.E.; Fox, S.B.; Poulsom, R.; Evan, G.I.; Harris, A.L. c-MYC interacts with hypoxia to induce angiogenesis in vivo by a vascular endothelial growth factor-dependent mechanism. Cancer Res. 2004, 64, 6563–6570. [Google Scholar] [CrossRef] [PubMed]
- Shchors, K.; Shchors, E.; Rostker, F.; Lawlor, E.R.; Brown-Swigart, L.; Evan, G.I. The MYC-dependent angiogenic switch in tumors is mediated by interleukin 1β. Genes Dev. 2006, 20, 2527–2538. [Google Scholar] [PubMed]
- Giuriato, S.; Ryeom, S.; Fan, A.C.; Bachireddy, P.; Lynch, R.C.; Rioth, M.J.; van Riggelen, J.; Kopelman, A.M.; Passegue, E.; Tang, F.; et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc. Natl. Acad. Sci. USA 2006, 103, 16266–16271. [Google Scholar]
- Rakhra, K.; Bachireddy, P.; Zabuawala, T.; Zeiser, R.; Xu, L.; Kopelman, A.; Fan, A.C.; Yang, Q.; Braunstein, L.; Crosby, E.; et al. Cd4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 2010, 18, 485–498. [Google Scholar] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [PubMed]
- Fults, D.; Pedone, C.; Dai, C.; Holland, E.C. MYC expression promotes the proliferation of neural progenitor cells in culture and in vivo. Neoplasia 2002, 4, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.; Pedone, C.A.; Coffin, C.M.; Holland, E.C.; Fults, D.W. c-MYC enhances sonic hedgehog-induced medulloblastoma formation from nestin-expressing neural progenitors in mice. Neoplasia 2003, 5, 198–204. [Google Scholar] [PubMed]
- Browd, S.R.; Kenney, A.M.; Gottfried, O.N.; Yoon, J.W.; Walterhouse, D.; Pedone, C.A.; Fults, D.W. n-MYC can substitute for insulin-like growth factor signaling in a mouse model of sonic hedgehog-induced medulloblastoma. Cancer Res. 2006, 66, 2666–2672. [Google Scholar] [CrossRef]
- Su, X.; Gopalakrishnan, V.; Stearns, D.; Aldape, K.; Lang, F.F.; Fuller, G.; Snyder, E.; Eberhart, C.G.; Majumder, S. Abnormal expression of rest/nrsf and MYC in neural stem/progenitor cells causes cerebellar tumors by blocking neuronal differentiation. Mol. Cell. Biol. 2006, 26, 1666–1678. [Google Scholar] [PubMed]
- Zindy, F.; Uziel, T.; Ayrault, O.; Calabrese, C.; Valentine, M.; Rehg, J.E.; Gilbertson, R.J.; Sherr, C.J.; Roussel, M.F. Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 2007, 67, 2676–2684. [Google Scholar] [PubMed]
- Kessler, J.D.; Hasegawa, H.; Brun, S.N.; Emmenegger, B.A.; Yang, Z.J.; Dutton, J.W.; Wang, F.; Wechsler-Reya, R.J. N-myc alters the fate of preneoplastic cells in a mouse model of medulloblastoma. Genes Dev. 2009, 23, 157–170. [Google Scholar]
- Swartling, F.J.; Grimmer, M.R.; Hackett, C.S.; Northcott, P.A.; Fan, Q.W.; Goldenberg, D.D.; Lau, J.; Masic, S.; Nguyen, K.; Yakovenko, S.; et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010, 24, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.M.; Kuijper, S.; Lindsey, J.C.; Petrie, K.; Schwalbe, E.C.; Barker, K.; Boult, J.K.; Williamson, D.; Ahmad, Z.; Hallsworth, A.; et al. Combined MYC and p53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 2015, 27, 72–84. [Google Scholar] [PubMed]
- Poschl, J.; Stark, S.; Neumann, P.; Grobner, S.; Kawauchi, D.; Jones, D.T.; Northcott, P.A.; Lichter, P.; Pfister, S.M.; Kool, M.; et al. Genomic and transcriptomic analyses match medulloblastoma mouse models to their human counterparts. Acta Neuropathol. 2014, 128, 123–136. [Google Scholar] [PubMed]
- Jensen, N.A.; Pedersen, K.M.; Lihme, F.; Rask, L.; Nielsen, J.V.; Rasmussen, T.E.; Mitchelmore, C. Astroglial c-MYC overexpression predisposes mice to primary malignant gliomas. J. Biol. Chem. 2003, 278, 8300–8308. [Google Scholar] [CrossRef] [PubMed]
- Vo, B.T.; Wolf, E.; Kawauchi, D.; Gebhardt, A.; Rehg, J.E.; Finkelstein, D.; Walz, S.; Murphy, B.L.; Youn, Y.H.; Han, Y.G.; et al. The interaction of MYC with MIZ1 defines medulloblastoma subgroup identity. Cancer Cell 2016, 29, 5–16. [Google Scholar] [PubMed]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stutz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [PubMed]
- Van der Meer, L.T.; Jansen, J.H.; van der Reijden, B.A. GFI1 and Gfi1B: Key regulators of hematopoiesis. Leukemia 2010, 24, 1834–1843. [Google Scholar]
- Lin, C.Y.; Erkek, S.; Tong, Y.; Yin, L.; Federation, A.J.; Zapatka, M.; Haldipur, P.; Kawauchi, D.; Risch, T.; Warnatz, H.J.; et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 2016, 530, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.P.; Sawyers, C.L. Mechanisms of resistance to STI571 in Philadelphia chromosome-associated leukemias. Oncogene 2003, 22, 7389–7395. [Google Scholar] [CrossRef] [PubMed]
- Scognamiglio, R.; Cabezas-Wallscheid, N.; Thier, M.C.; Altamura, S.; Reyes, A.; Prendergast, A.M.; Baumgartner, D.; Carnevalli, L.S.; Atzberger, A.; Haas, S.; et al. Myc depletion induces a pluripotent dormant state mimicking diapause. Cell 2016, 164, 668–680. [Google Scholar] [CrossRef]
- Laurenti, E.; Varnum-Finney, B.; Wilson, A.; Ferrero, I.; Blanco-Bose, W.E.; Ehninger, A.; Knoepfler, P.S.; Cheng, P.F.; MacDonald, H.R.; Eisenman, R.N.; et al. Hematopoietic stem cell function and survival depend on c-MYC and N-Myc activity. Cell Stem Cell 2008, 3, 611–624. [Google Scholar] [CrossRef]
- Wey, A.; Knoepfler, P.S. c-myc and N-myc promote active stem cell metabolism and cycling as architects of the developing brain. Oncotarget 2010, 1, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Boxer, R.B.; Jang, J.W.; Sintasath, L.; Chodosh, L.A. Lack of sustained regression of c-MYC-induced mammary adenocarcinomas following brief or prolonged MYC inactivation. Cancer Cell 2004, 6, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef]
- Felsher, D.W.; Bishop, J.M. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell 1999, 4, 199–207. [Google Scholar]
- Pelengaris, S.; Littlewood, T.; Khan, M.; Elia, G.; Evan, G. Reversible activation of c-MYC in skin: Induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol. Cell 1999, 3, 565–577. [Google Scholar] [CrossRef]
- Jain, M.; Arvanitis, C.; Chu, K.; Dewey, W.; Leonhardt, E.; Trinh, M.; Sundberg, C.D.; Bishop, J.M.; Felsher, D.W. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002, 297, 102–104. [Google Scholar]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-MYC. Cell 2012, 151, 56–67. [Google Scholar]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar]
- Green, D.R. A MYC-induced apoptosis pathway surfaces. Science 1997, 278, 1246–1247. [Google Scholar] [PubMed]
- Schmitt, C.A.; Lowe, S.W. Bcl-2 mediates chemoresistance in matched pairs of primary Eμ-myc lymphomas in vivo. Blood Cells Mol. Dis. 2001, 27, 206–216. [Google Scholar]
- Sodir, N.M.; Swigart, L.B.; Karnezis, A.N.; Hanahan, D.; Evan, G.I.; Soucek, L. Endogenous MYC maintains the tumor microenvironment. Genes Dev. 2011, 25, 907–916. [Google Scholar] [CrossRef]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling MYC inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Masso-Valles, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of MYC family proteins eradicates kras-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513. [Google Scholar]
- Jung, M.; Russell, A.J.; Liu, B.; George, J.; Liu, P.Y.; Liu, T.; DeFazio, A.; Bowtell, D.D.; Oberthuer, A.; London, W.B.; et al. A MYC activity signature predicts poor clinical outcomes in MYC-associated cancers. Cancer Res. 2017, 77, 971–981. [Google Scholar]
- Mollaoglu, G.; Guthrie, M.R.; Bohm, S.; Bragelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to Aurora kinase inhibition. Cancer Cell 2017, 31, 270–285. [Google Scholar] [CrossRef]
- Rye, M.B.; Bertilsson, H.; Drablos, F.; Angelsen, A.; Bathen, T.F.; Tessem, M.B. Gene signatures ESC, MYC and ERG-fusion are early markers of a potentially dangerous subtype of prostate cancer. BMC Med. Genom. 2014, 7, 50. [Google Scholar] [CrossRef]
- Gogas, H.; Kotoula, V.; Alexopoulou, Z.; Christodoulou, C.; Kostopoulos, I.; Bobos, M.; Raptou, G.; Charalambous, E.; Tsolaki, E.; Xanthakis, I.; et al. MYC copy gain, chromosomal instability and PI3K activation as potential markers of unfavourable outcome in trastuzumab-treated patients with metastatic breast cancer. J. Transl. Med. 2016, 14. [Google Scholar] [CrossRef]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef]
- Wade, M.A.; Sunter, N.J.; Fordham, S.E.; Long, A.; Masic, D.; Russell, L.J.; Harrison, C.J.; Rand, V.; Elstob, C.; Bown, N.; et al. c-MYC is a radiosensitive locus in human breast cells. Oncogene 2015, 34, 4985–4994. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-MYC. Cell 2011, 146, 904–917. [Google Scholar]
- Milde, T.; Lodrini, M.; Savelyeva, L.; Korshunov, A.; Kool, M.; Brueckner, L.M.; Antunes, A.S.; Oehme, I.; Pekrun, A.; Pfister, S.M.; et al. HD-MB03 is a novel Group 3 medulloblastoma model demonstrating sensitivity to histone deacetylase inhibitor treatment. J. Neurooncol. 2012, 110, 335–348. [Google Scholar] [CrossRef]
- Wang, H.; Teriete, P.; Hu, A.; Raveendra-Panickar, D.; Pendelton, K.; Lazo, J.S.; Eiseman, J.; Holien, T.; Misund, K.; Oliynyk, G.; et al. Direct inhibition of c-MYC-max heterodimers by celastrol and celastrol-inspired triterpenoids. Oncotarget 2015, 6, 32380–32395. [Google Scholar] [PubMed]
- Goga, A.; Yang, D.; Tward, A.D.; Morgan, D.O.; Bishop, J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007, 13, 820–827. [Google Scholar]
- Amati, B.; Alevizopoulos, K.; Vlach, J. MYC and the cell cycle. Front. Biosci. 1998, 3, d250–d268. [Google Scholar]
- Jansen-Durr, P.; Meichle, A.; Steiner, P.; Pagano, M.; Finke, K.; Botz, J.; Wessbecher, J.; Draetta, G.; Eilers, M. Differential modulation of cyclin gene expression by MYC. Proc. Natl. Acad. Sci. USA 1993, 90, 3685–3689. [Google Scholar]
- Campaner, S.; Doni, M.; Hydbring, P.; Verrecchia, A.; Bianchi, L.; Sardella, D.; Schleker, T.; Perna, D.; Tronnersjo, S.; Murga, M.; et al. CDK2 suppresses cellular senescence induced by the c-MYC oncogene. Nat. Cell Biol. 2010, 12, 54–59. [Google Scholar]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef]
- Walker, A.J.; Wedam, S.; Amiri-Kordestani, L.; Bloomquist, E.; Tang, S.; Sridhara, R.; Chen, W.; Palmby, T.R.; Fourie Zirkelbach, J.; Fu, W.; et al. FDA approval of palbociclib in combination with fulvestrant for the treatment of hormone receptor-positive, HER2-negative metastatic breast cancer. Clin. Cancer Res. 2016, 22, 4968–4972. [Google Scholar]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar]
- Dean, J.L.; McClendon, A.K.; Hickey, T.E.; Butler, L.M.; Tilley, W.D.; Witkiewicz, A.K.; Knudsen, E.S. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 2012, 11, 2756–2761. [Google Scholar] [CrossRef]
- Barton, K.L.; Misuraca, K.; Cordero, F.; Dobrikova, E.; Min, H.D.; Gromeier, M.; Kirsch, D.G.; Becher, O.J. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS ONE 2013, 8, e77639. [Google Scholar] [CrossRef]
- Michaud, K.; Solomon, D.A.; Oermann, E.; Kim, J.S.; Zhong, W.Z.; Prados, M.D.; Ozawa, T.; James, C.D.; Waldman, T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010, 70, 3228–3238. [Google Scholar] [CrossRef]
- Whiteway, S.L.; Harris, P.S.; Venkataraman, S.; Alimova, I.; Birks, D.K.; Donson, A.M.; Foreman, N.K.; Vibhakar, R. Inhibition of cyclin-dependent kinase 6 suppresses cell proliferation and enhances radiation sensitivity in medulloblastoma cells. J. Neurooncol. 2013, 111, 113–121. [Google Scholar]
- Weiss, G.J.; Hidalgo, M.; Borad, M.J.; Laheru, D.; Tibes, R.; Ramanathan, R.K.; Blaydorn, L.; Jameson, G.; Jimeno, A.; Isaacs, J.D.; et al. Phase I study of the safety, tolerability and pharmacokinetics of PHA-848125AC, a dual tropomyosin receptor kinase A and cyclin-dependent kinase inhibitor, in patients with advanced solid malignancies. Investig. New Drugs 2012, 30, 2334–2343. [Google Scholar]
- Molenaar, J.J.; Ebus, M.E.; Geerts, D.; Koster, J.; Lamers, F.; Valentijn, L.J.; Westerhout, E.M.; Versteeg, R.; Caron, H.N. Inactivation of CDK2 is synthetically lethal to MYCN over-expressing cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12968–12973. [Google Scholar]
- Den Hollander, J.; Rimpi, S.; Doherty, J.R.; Rudelius, M.; Buck, A.; Hoellein, A.; Kremer, M.; Graf, N.; Scheerer, M.; Hall, M.A.; et al. Aurora kinases A and B are up-regulated by MYC and are essential for maintenance of the malignant state. Blood 2010, 116, 1498–1505. [Google Scholar] [CrossRef]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schuttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc is a critical function of Aurora a in human neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar]
- El-Sheikh, A.; Fan, R.; Birks, D.; Donson, A.; Foreman, N.K.; Vibhakar, R. Inhibition of Aurora kinase A enhances chemosensitivity of medulloblastoma cell lines. Pediatr. Blood Cancer 2010, 55, 35–41. [Google Scholar]
- Ahmad, Z.; Jasnos, L.; Gil, V.; Howell, L.; Hallsworth, A.; Petrie, K.; Sawado, T.; Chesler, L. Molecular and in vivo characterization of cancer-propagating cells derived from MYCN-dependent medulloblastoma. PLoS ONE 2015, 10, e0119834. [Google Scholar]
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.C.; Kang, T.W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L.; et al. A MYC-Aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753. [Google Scholar]
- Vaughan, L.; Clarke, P.A.; Barker, K.; Chanthery, Y.; Gustafson, C.W.; Tucker, E.; Renshaw, J.; Raynaud, F.; Li, X.; Burke, R.; et al. Inhibition of mTOR-kinase destabilizes MYCN and is a potential therapy for MYCN-dependent tumors. Oncotarget 2016, 7, 57525–57544. [Google Scholar]
- Segerstrom, L.; Baryawno, N.; Sveinbjornsson, B.; Wickstrom, M.; Elfman, L.; Kogner, P.; Johnsen, J.I. Effects of small molecule inhibitors of PI3K/AKT/MTOR signaling on neuroblastoma growth in vitro and in vivo. Int. J. Cancer 2011, 129, 2958–2965. [Google Scholar] [CrossRef]
- Baryawno, N.; Sveinbjornsson, B.; Eksborg, S.; Chen, C.S.; Kogner, P.; Johnsen, J.I. Small-molecule inhibitors of phosphatidylinositol 3-kinase/AKT signaling inhibit WNT/beta-catenin pathway cross-talk and suppress medulloblastoma growth. Cancer Res. 2010, 70, 266–276. [Google Scholar]
- Diveshkumar, K.V.; Sakrikar, S.; Rosu, F.; Harikrishna, S.; Gabelica, V.; Pradeepkumar, P.I. Specific stabilization of c-MYC and c-KIT G-quadruplex DNA structures by indolylmethyleneindanone scaffolds. Biochemistry 2016, 55, 3571–3585. [Google Scholar]
- Sun, K.; Atoyan, R.; Borek, M.A.; DellaRocca, S.; Samson, M.E.; Ma, A.W.; Xu, G.X.; Patterson, T.; Tuck, D.P.; Viner, J.L.; et al. Dual HDAC and PI3K inhibitor CUDC-907 downregulates MYC and suppresses growth of MYC-dependent cancers. Mol. Cancer Ther. 2016. [Google Scholar] [CrossRef]
- Pei, Y.; Liu, K.W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K antagonists cooperate to inhibit growth of MYC-driven medulloblastoma. Cancer Cell 2016, 29, 311–323. [Google Scholar]
- Peukert, K.; Staller, P.; Schneider, A.; Carmichael, G.; Hanel, F.; Eilers, M. An alternative pathway for gene regulation by MYC. EMBO J. 1997, 16, 5672–5686. [Google Scholar]
- Van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updats 2015, 19, 1–12. [Google Scholar]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma genotype dictates blood brain barrier phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hutter, S.; Bolin, S.; Weishaupt, H.; Swartling, F.J. Modeling and Targeting MYC Genes in Childhood Brain Tumors. Genes 2017, 8, 107. https://doi.org/10.3390/genes8040107
Hutter S, Bolin S, Weishaupt H, Swartling FJ. Modeling and Targeting MYC Genes in Childhood Brain Tumors. Genes. 2017; 8(4):107. https://doi.org/10.3390/genes8040107
Chicago/Turabian StyleHutter, Sonja, Sara Bolin, Holger Weishaupt, and Fredrik J. Swartling. 2017. "Modeling and Targeting MYC Genes in Childhood Brain Tumors" Genes 8, no. 4: 107. https://doi.org/10.3390/genes8040107
APA StyleHutter, S., Bolin, S., Weishaupt, H., & Swartling, F. J. (2017). Modeling and Targeting MYC Genes in Childhood Brain Tumors. Genes, 8(4), 107. https://doi.org/10.3390/genes8040107