Comparative Transcriptome Analysis of Male and Female Conelets and Development of Microsatellite Markers in Pinus bungeana, an Endemic Conifer in China

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Isolation and Quality Assay
2.3. Transcriptome Sequencing
2.4. Data Processing and Assembly
2.5. Differential Expression Analysis (DEG)
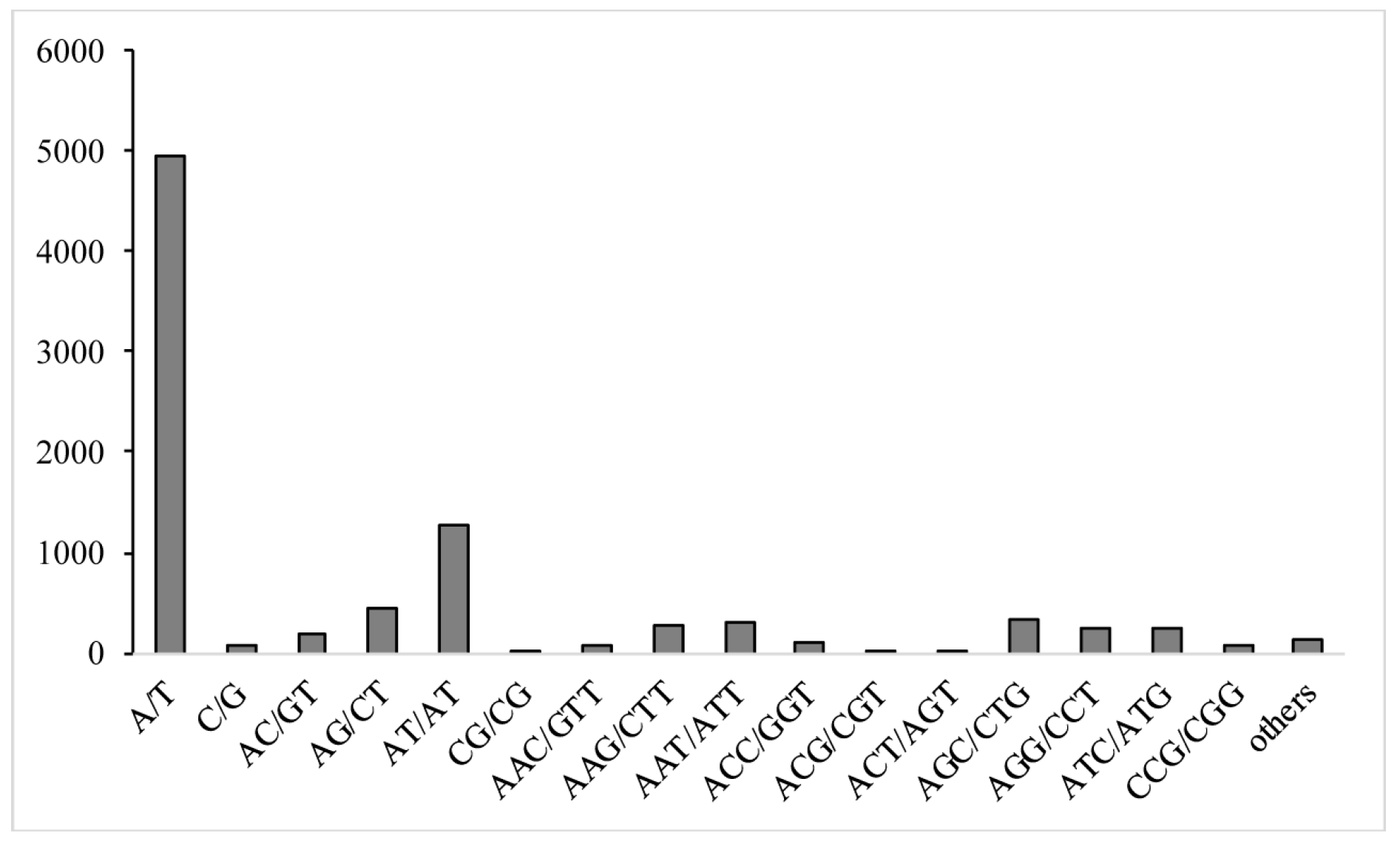
2.6. Identification of Expressed Sequence Tag-Simple Sequence Repeats (EST-SSRs)
2.7. Validation of EST-SSR Markers and Population Genetic Analysis
3. Results
3.1. Transcriptome Sequencing and De Novo Assembly of Pinus bungeana
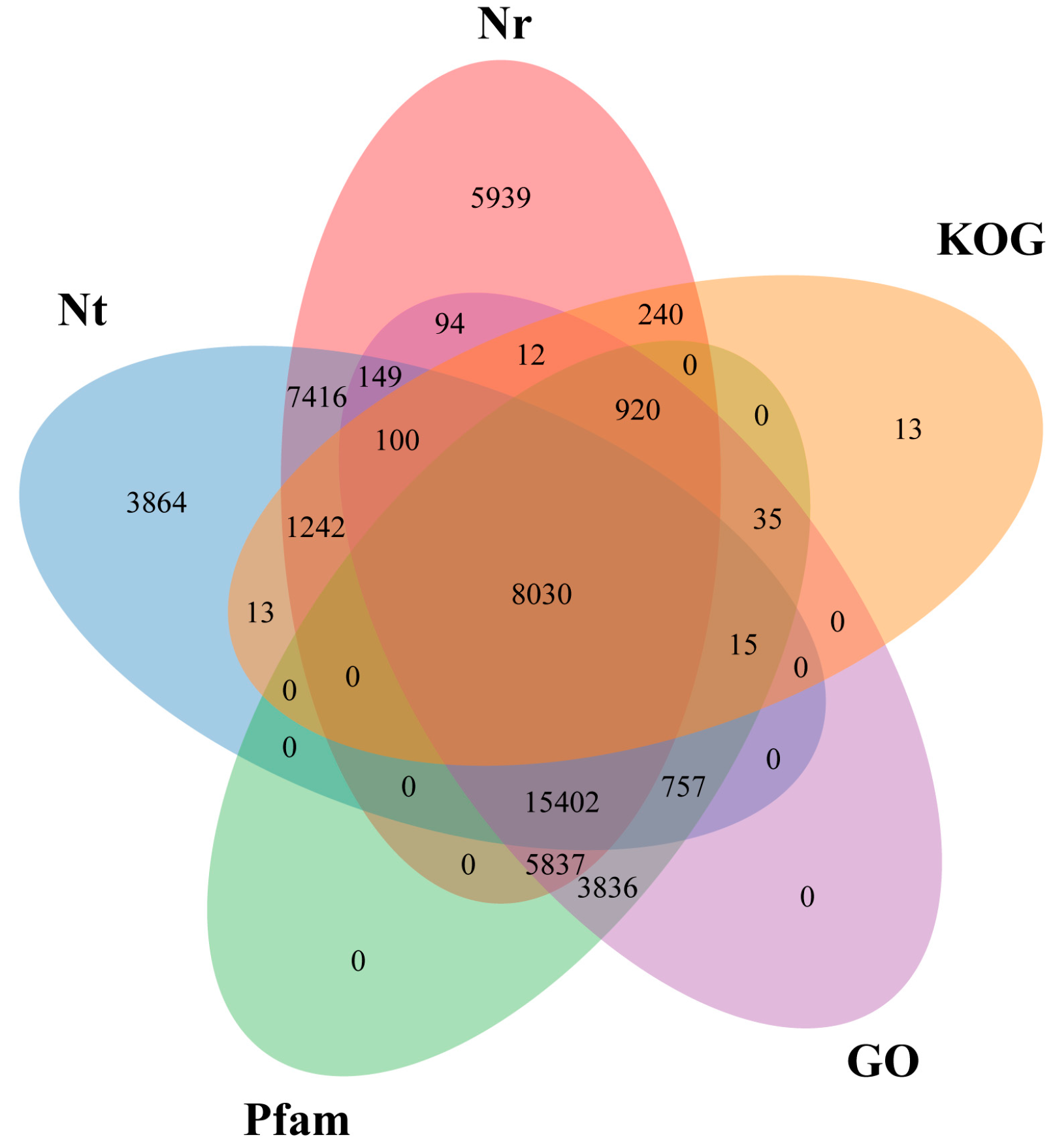
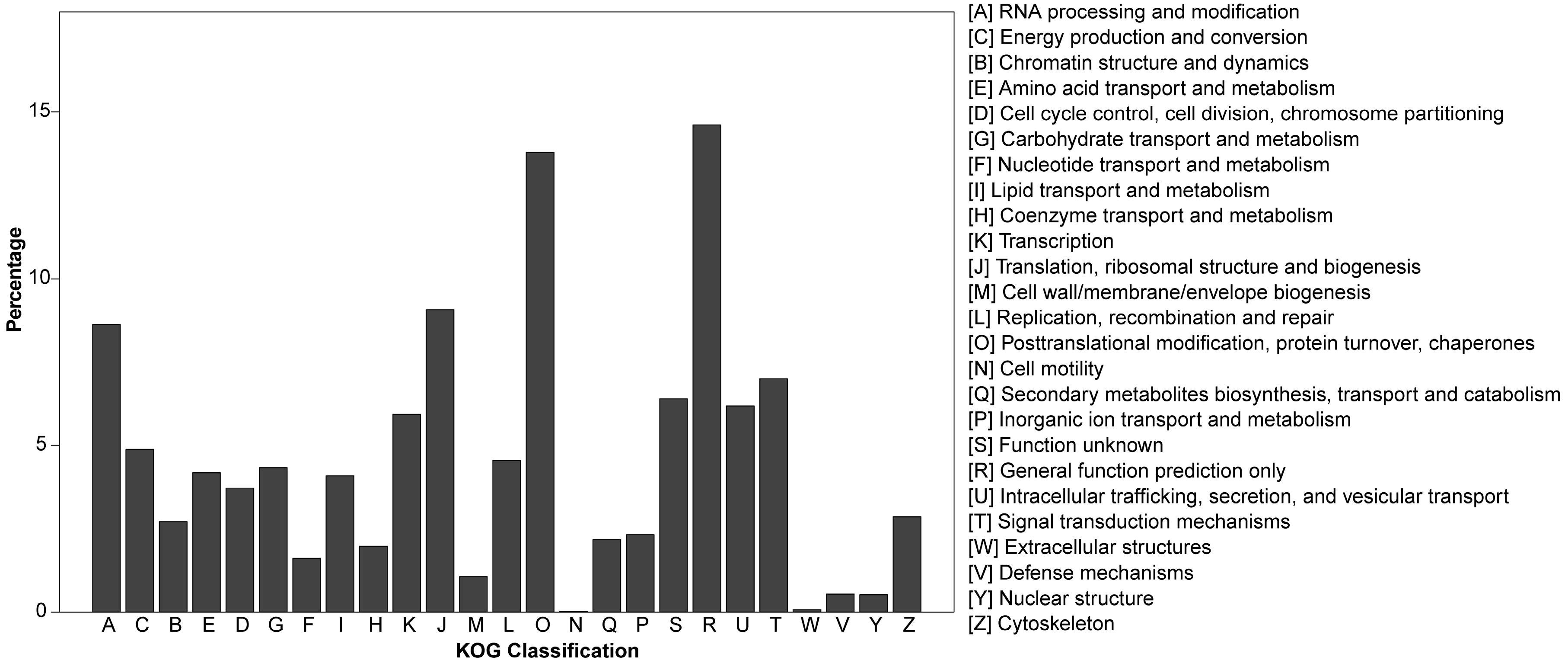
3.2. Gene Annotation of Pinus bungeana
3.3. Analysis of Differently Expressed Genes in Male and Female Conelets of Pinus bungeana
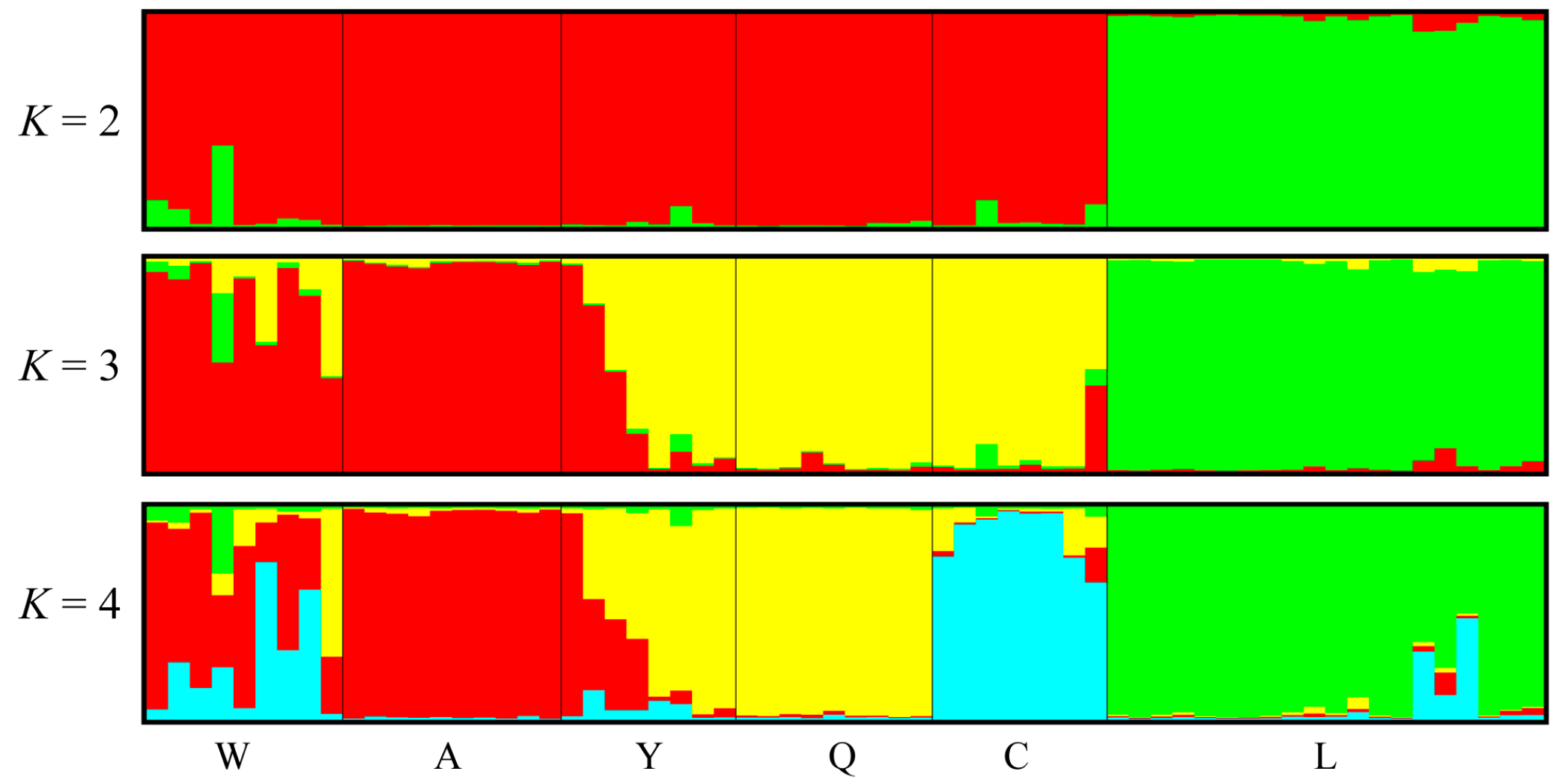
3.4. Polymorphism of EST-SSR Markers and Population Genetic Structure
4. Discussion
4.1. Transcriptome Characterization
4.2. The Pathway Analysis of Differentially Expressed Genes
4.2.1. The Plant Hormones Pathway Analysis
4.2.2. Tryptophan Metabolism
4.2.3. Cysteine and Methionine Metabolism
4.2.4. Zeatin Biosynthesis
4.2.5. Other Important Plant Hormones
4.2.6. Photosynthesis Metabolism Pathway
4.3. Polymorphism of EST-SSR Markers and Population Structure of Pinus bungeana
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walbot, V.; Evans, M.M.S. Unique features of the plant life cycle and their consequences. Nat. Rev. Genet. 2003, 4, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Wellmer, F.; Graciet, E.; Riechmann, J.L. Specification of floral organs in Arabidopsis. J. Exp. Bot. 2014, 65, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Harkess, A.; Mercati, F.; Shan, H.Y.; Sunseri, F.; Falavigna, A.; Leebens-Mack, J. Sex-biased gene expression in dioecious garden asparagus (Asparagus officinalis). New Phytol. 2015, 207, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Stobdan, T.; Srivastava, R.B.; Jaiswal, V.; Chauhan, R.S.; Kant, A. Sex-biased temporal gene expression in male and female floral buds of seabuckthorn (Hippophae rhamnoides). PLoS ONE 2015, 10, e0124890. [Google Scholar] [CrossRef] [PubMed]
- Rocheta, M.; Sobral, R.; Magalhães, J.; Amorim, M.I.; Ribeiro, T.; Pinheiro, M.; Egas, C.; Morais-Cecílio, L.; Costa, M.M.R. Comparative transcriptomic analysis of male and female flowers of monoecious Quercus suber. Front. Plant Sci. 2014, 5, 599. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Yuan, H.; Sun, X.; Porth, I.; Li, Y.; El-Kassaby, Y.A.; Li, W. A transcriptomics investigation into pine reproductive organ development. New Phytol. 2016, 209, 1278–1289. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.G.; Zheng, Y.; Joung, J.G.; Liu, S.; Zhang, Z.; Crasta, O.R.; Sobral, B.W.; Xu, Y.; Huang, S.; Fei, Z. Transcriptome sequencing and comparative analysis of cucumber flowers with different sex types. BMC Genom. 2010, 11, 384. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yin, T.; Ye, N.; Chen, Y.; Yin, T.; Liu, M.; Hassani, D. Transcriptome analysis of the differentially expressed genes in the male and female shrub willows (Salix suchowensis). PLoS ONE 2013, 8, e60181. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Qin, Z.; Zhou, X.; Feng, Z.; Du, Y. Transcriptome profile analysis of floral sex determination in cucumber. J. Plant Physiol. 2010, 167, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Uebbing, S.; Gyllenstrand, N.; Lagercrantz, U.; Lascoux, M.; Källman, T. Sequencing of the needle transcriptome from Norway spruce (Picea abies Karst L.) reveals lower substitution rates, but similar selective constraints in gymnosperms and angiosperms. BMC Genom. 2012, 13, 589. [Google Scholar] [CrossRef] [PubMed]
- Syring, J.; Farrell, K.; Businský, R.; Cronn, R.; Liston, A. Widespread genealogical nonmonophyly in species of Pinus subgenus. Strobus. Syst. Biol. 2007, 56, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Yang, K.Z.; Wei, X.X.; Wang, X.Q. Revisiting the phosphatidylethanolamine-binding protein (PEBP) gene family reveals cryptic FLOWERING LOCUS T gene homologs in gymnosperms and sheds new light on functional evolution. New Phytol. 2016, 212, 730–744. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.R.; Tsumura, Y.; Yoshimaru, H.; Nagasaka, K.; Szmidt, A.E. Phylogenetic relationship of Eurasian pines (Pinus, Pinaceae) based on chloroplast rbcL, matK, rpl20-rps18 spacer, and trnV intron sequences. Am. J. Bot. 1999, 86, 1742–1753. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.S.; Wang, X.R. Mitochondrial DNA capture and divergence in Pinus provide new insights into the evolution of the genus. Mol. Phylogenet. Evol. 2014, 80, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.Z.; Liu, Y.Y.; Nazaire, M.; Wei, X.X.; Wang, X.Q. Molecular phylogenetics and evolutionary history of sect. Quinquefoliae (Pinus): Implications for Northern Hemisphere biogeography. Mol. Phylogenet. Evol. 2015, 87, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.X.; Wang, M.L.; Liu, Z.L.; Zhu, J.; Yan, M.Y.; Li, Z.H. Nucleotide polymorphism and phylogeographic history of an endangered conifer species Pinus bungeana. Biochem. Syst. Ecol. 2016, 64, 89–96. [Google Scholar] [CrossRef]
- Zhang, L.; Yan, H.F.; Wu, W.; Yu, H.; Ge, X.J. Comparative transcriptome analysis and marker development of two closely related Primrose species (Primula poissonii and Primula wilsonill). BMC Genom. 2013, 14, 329. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cai, C.F.; Cheng, F.Y.; Cui, H.L.; Zhou, H. Characterisation and development of EST-SSR markers in tree peony using transcriptome sequences. Mol. Breed. 2014, 34, 1853–1866. [Google Scholar] [CrossRef]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. Solexa QA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [PubMed]
- Kitts, P.A.; Church, D.M.; Thibaud-Nissen, F.; Choi, J.; Hem, V.; Sapojnikov, V.; Smith, R.G.; Tatusova, T.; Xiang, C.; Zherikov, A.; et al. Assembly: A resource for assembled genomes at NCBI. Nucleic Acids Res. 2016, 44, D73–D80. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Lane, L.; Boutet, E.; Lieberherr, D.; Tognolli, M.; Bougueleret, L.; Bairoch, A. The UniProtKB/Swiss-Prot knowledgebase and its Plant Proteome Annotation Program. J. Proteom. 2009, 72, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, J. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Word, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.B.; Xie, Y.H.; Sun, X.M. Development and characterization of polymorphic genic-SSR markers in Larix kaempferi. Molecules 2015, 20, 6060–6067. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2007, 1, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.C.; Yang, R.C.; Boyle, T. POPGENE. Microsoft Windows-Based Freeware for Population Genetic Analysis Release 1.31; University of Alberta: Edmonton, AB, Canada, 1999. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis using Likelihood, Distance, and Parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Earl, D.; vonHoldt, B. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomezcabrero, D.; Cervera, A.; Mcpherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.G.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, I.M.; Stam, M.E.; van Tunen, A.J.; Mol, J.N.; Stuitje, A.R. Antisense inhibition of flavonoid biosynthesis in petunia anthers results in male sterility. Plant Cell 1992, 4, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.M. Hormonal regulation of plant growth and development. PLoS Biol. 2004, 2, e311. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, E.; Little, H.A.; Hammar, S.A.; Grumet, R. Effect of modified endogenous ethylene production on sex expression, bisexual flower development and fruit production in melon (Cucumis melo L.). Sex. Plant Reprod. 2005, 18, 131–142. [Google Scholar] [CrossRef]
- Little, H.A.; Papadopoulou, E.; Hammar, S.A.; Grumet, R. The influence of ethylene perception on sex expression in melon (Cucumis melo L.) as assessed by expression of the mutant ethylene receptor, At-etr1-1, under the control of constitutive and floral targeted promoters. Sex. Plant Reprod. 2007, 20, 123–136. [Google Scholar] [CrossRef]
- Diggle, P.K.; Di Stilio, V.S.; Gschwend, A.R.; Golenberg, E.M.; Moore, R.C.; Russell, J.R.W.; Sinclair, J.P. Multiple developmental processes underlie sex differentiation in angiosperms. Trends Genet. 2011, 27, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Pharis, R.P. Manipulation of flowering in conifers through the use of plant hormones. In Modern Methods in Forest Genetics; Springer: Berlin, Germany, 1976; pp. 265–282. [Google Scholar]
- Pharis, R.P.; Kuo, C.G. Physiology of gibberellins in conifers. Can. J. Forest Res. 1997, 7, 299–325. [Google Scholar] [CrossRef]
- Pharis, R.P.; Ross, S.D.; Wample, R.L.; Owens, J.N. Promotion of flowering in conifers of the Pinaceae by certain of the gibberellins. In ISHS Acta Horticulturae 56: Symposium on Juvenility in Woody Perennials; ISHS: Leuven, Belgium, 1976. [Google Scholar]
- Galoch, E. The hormonal control of sex differentiation in dioecious plants of hemp (Cannabis sativa). The influence of plant growth regulators on sex expression in male and female plants. Acta Soc. Bot. Pol. 1978, 47, 153. [Google Scholar] [CrossRef]
- Mano, Y.; Nemoto, K. The pathway of auxin biosynthesis in plants. J. Exp. Bot. 2012, 63, 2853–2872. [Google Scholar] [CrossRef] [PubMed]
- Hagen, G.; Guilfoyle, T. Auxin-responsive gene expression: Genes, promoters and regulatory factors. Plant Mol. Biol. 2002, 49, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Péret, B.; Swarup, R. AUX/LAX genes encode a family of auxin influx transporters that perform distinct functions during Arabidopsis development. Plant Cell 2012, 24, 2874–2885. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wu, S.; Van, H.J.; Wang, Y.; Ding, B.; Fei, Z.; Clarke, T.H.; Reed, J.W.; van der Knaap, E. Down-regulation of auxin response factors 6 and 8 by microRNA 167 leads to floral development defects and female sterility in tomato. J. Exp. Bot. 2014, 65, 2507–2520. [Google Scholar] [CrossRef] [PubMed]
- Boualem, A.; Fergany, M.; Fernandez, R.; Troadec, C.; Martin, A.; Morin, H.; Sari, M.A.; Collin, F.; Flowers, J.M.; Pitrat, M.; et al. A conserved mutation in an ethylene biosynthesis enzyme leads to andromonoecy in melons. Science 2008, 321, 836–838. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Khan, N.A.; Ferrante, A.; Trivellini, A.; Francini, A.; Khan, M.I.R. Ethylene role in plant growth, development and senescence: Interaction with other phytohormones. Front. Plant Sci. 2017, 8, 475. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Hua, J.; Chen, Q.G.; Chang, C.; Medrano, L.J.; Bleecker, A.B.; Meyerowitz, E.M. Etr2 is an etr1-like gene involved in ethylene signaling in Arabidopsis. Proc. Natl. Acad. Sci. USA 1998, 95, 5812–5817. [Google Scholar] [CrossRef] [PubMed]
- Bartrina, I.; Jensen, H.; Novak, O.; Strnad, M.; Werner, T.; Schmülling, T. Gain-of-function mutants of the cytokinin receptors AHK2 and AHK3 regulate plant organ size, flowering time and plant longevity. Plant Physiol. 2017, 173, 1783–1797. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita-Tsujimura, K.; Kakimoto, T. Cytokinin receptors in sporophytes are essential for male and female functions in Arabidopsis thaliana. Plant Signal. Behav. 2011, 6, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.W.; Oh, S.I.; Kim, Y.Y.; Yoo, K.S.; Cui, M.H.; Shin, J.S. Arabidopsis histidine-containing phosphotransfer factor 4 (AHP4) negatively regulates secondary wall thickening of the anther endothecium during flowering. Mol. Cells 2008, 25, 294–300. [Google Scholar] [PubMed]
- Irish, E.E.; Nelson, T. Sex determination in monoecious and dioecious plants. Plant Cell 1989, 1, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.E.; King, K.E.; Ait-Ali, T.; Harberd, N.P. How gibberellin regulates plant growth and development: A molecular genetic analysis of gibberellin signaling. Annu. Rev. Plant. Physiol. Plant. Mol. Biol. 2001, 52, 67–88. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.; Atsmon, D.; Galun, E. Sexual differentiation in cucumber: The effects of abscisic acid and other growth regulators on various sex genotypes. Plant Cell Physiol. 1977, 18, 261–269. [Google Scholar]
- Rudich, J.; Halevy, A.H. Involvement of abscisic acid in the regulation of sex expression in the cucumber. Plant Cell Physiol. 1974, 15, 635–642. [Google Scholar] [CrossRef]
- Park, S.Y.; Fung, P.; Nishimura, N.; Jensen, D.R.; Fujii, H.; Zhao, Y.; Lumba, S.; Santiago, J.; Rodrigues, A.; Chow, T.F.; et al. Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science 2009, 324, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Guzman, M.; Pizzio, G.A.; Antoni, R.; Vera-Sirera, F.; Merilo, E.; Bassel, G.W.; Fernández, M.A.; Holdsworth, M.J.; Perez-Amador, M.A.; Kollist, H.; et al. Arabidopsis PYR/PYL/RCAR receptors play a major role in quantitative regulation of stomatal aperture and transcriptional response to abscisic acid. Plant Cell 2012, 24, 2483–2496. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Quinn, J.A. Tests of a mechanistic model of one hormone regulating both sexes in Cucumis sativus (Cucurbitaceae). Am. J. Bot. 1995, 82, 1537–1546. [Google Scholar] [CrossRef]
- Ei-Gizawy, A.M.; EI-Oksh, I.; Sharaf, A.; EI-Habar, M. Effect of gibberellic acid and alar on flowering and seed yield of spinach. Egypt. J. Hortic. 1992, 19, 191–200. [Google Scholar]
- Gallego-Giraldo, C.; Hu, J.; Urbez, C.; Gomez, M.D.; Sun, T.; Perez-Amador, M.A. Role of the gibberellin receptors GID1 during fruit-set in Arabidopsis. Plant J. 2014, 79, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Ueguchi-Tanaka, M.; Hirano, K.; Hasegawa, Y.; Kitano, H.; Matsuoka, M. Release of the repressive activity of rice DELLA protein SLR1 by gibberellin does not require SLR1 degradation in the gid2 mutant. Plant Cell 2008, 20, 2437–2446. [Google Scholar] [CrossRef] [PubMed]
- Benschop, J.J.; Millenaar, F.F.; Smeet, M.E.; van Zanten, M.; Voesenek, L.A.; Peeters, A.J. Abscisic acid antagonizes ethylene-induced hyponastic growth in Arabidopsis. Plant Physiol. 2007, 143, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Domagalska, M.A.; Schomburg, F.M.; Amasino, R.M.; Vierstra, R.D.; Nagy, F.; Davis, S.J. Attenuation of brassinosteroid signaling enhances FLC expression and delays flowering. Development 2007, 134, 2841–2850. [Google Scholar] [CrossRef] [PubMed]
- Vick, B.A.; Zimmerman, D.C. The biosynthesis of jasmonic acid: A physiological role for plant lipoxygenase. Biochem. Biophys. Res. Commun. 1983, 111, 470–477. [Google Scholar] [CrossRef]
- Stintzi, A.; Browse, J. The Arabidopsis male-sterile mutant, opr3, lacks the 12-oxophytodienoic acid reductase required for jasmonate synthesis. Proc. Natl. Acad. Sci. USA 2000, 97, 10625–10630. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, S.; Kawai-Oda, A.; Ueda, J.; Nishida, I.; Okada, K. The DEFECTIVE IN ANTHER DEHISCENCE1 gene encodes a novel phospholipase A1 catalyzing the initial step of jasmonic acid biosynthesis, which synchronizes pollen maturation, anther dehiscence, and flower opening in Arabidopsis. Plant Cell 2001, 13, 2191–2209. [Google Scholar] [CrossRef] [PubMed]
- Caldelari, D.; Wang, G.; Farmer, E.E.; Dong, X. Arabidopsis lox3 lox4 double mutants are male sterile and defective in global proliferative arrest. Plant Mol. Biol. 2011, 75, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Li, J.J.; Song, R.; Messing, J.; Chen, X. CARPEL FACTORY, a dicer homolog, and HEN1, a novel protein, act in microRNA in metabolism in Arabidopsis thaliana. Curr. Biol. 2002, 12, 1484–1495. [Google Scholar] [CrossRef]
- Chung, H.S.; Howe, G.A. A critical role for the TIFY motif in repression of jasmonate signaling by a stabilized splice variant of the JASMONATE ZIM-domain protein JAZ10 in Arabidopsis. Plant Cell 2009, 21, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Qi, T.; Huang, H.; Ren, Q.; Wu, D.; Chang, C.; Peng, W.; Liu, Y.; Peng, J.; Xie, D. The Jasmonate-ZIM domain proteins interact with the R2R3-MYB transcription factors MYB21 and MYB24 to affect Jasmonate-regulated stamen development in Arabidopsis. Plant Cell 2011, 23, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Kazan, K.; Manners, J.M. MYC2: The master in action. Mol. Plant 2013, 6, 686–703. [Google Scholar] [CrossRef] [PubMed]
- Dellaporta, S.L.; Calderon-Urrea, A. Sex determination in flowering plants. Plant Cell. 1993, 5, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.F.; Gill, G.P.; Fraser, L.G.; McNeilage, M.A. Sex determination in Actinidia. 1. Sex-linked markers and progeny sex ratio in diploid A. chinensis. Sex. Plant Reprod. 1997, 10, 149–154. [Google Scholar] [CrossRef]
- Yang, Q.; Ren, L.; Du, G. Effects of ethephon, GA3 and nutrient elements on sex expression of Chinese chestnut. Sci. Horti. 1985, 26, 209–215. [Google Scholar]
- Weng, J.K.; Mo, H.; Chapple, C. Over-expression of F5H in COMT-deficient Arabidopsis leads to enrichment of an unusual lignin and disruption of pollen wall formation. Plant J. 2010, 64, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Tsuchiya, T.; Kishitani, S.; Tanaka, Y.; Toriyama, K. Partial male sterility in transgenic tobacco carrying antisense and sense PAL cDNA under the control of a tapetum-specific promoter. Plant Cell Physiol. 1996, 37, 215–222. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, S.; Lian, C. De novo transcriptome sequencing analysis of cDNA library and large-scale unigene assembly in Japanese red pine (Pinus densiflora). Int. J. Mol. Sci. 2015, 16, 29047–29059. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.X.; Lin, F.R.; Huang, P.; Zheng, Y.Q. Moderate genetic diversity and genetic differentiation in the relict tree Liquidambar formosana Hance revealed by genic simple sequence repeat markers. Front. Plant Sci. 2016, 7, 1411. [Google Scholar] [CrossRef] [PubMed]
- Mayr, E. Systematics and the Origin of Species; Columbia University Press: New York, NY, USA, 1942. [Google Scholar]
- Hewitt, G.M. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, B.; Mao, J.F.; Ingvarsson, P.; Zeng, Q.Y.; Wang, X.R. Demography and speciation history of the homoploid hybrid pine Pinus densata on the Tibetan plateau. Mol. Ecol. 2012, 21, 4811–4827. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhao, W.; Wang, X.R.; Mao, J.F.; Gao, J. Colonization of the Tibetan Plateau by the homoploid hybrid pine Pinus densata. Mol. Ecol. 2011, 20, 3769–3811. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.I.; Gout, B.S. Molecular population genetics and the search for adaptive evolution in plants. Mol. Biol. Evol. 2005, 22, 506–519. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Location | Longitude (E) | Latitude (N) | Altitude (m) | N |
|---|---|---|---|---|---|
| W | Wuzi Mountain, Shaanxi | 107.83 | 32.93 | 700 | 9 |
| A | Ankang, Shaanxi | 108.94 | 32.97 | 577 | 10 |
| Y | Huozhou, Shanxi | 111.87 | 36.60 | 900 | 8 |
| Q | Qinyang, Henan | 112.79 | 35.22 | 972 | 9 |
| C | Taiyuan, Shanxi | 112.32 | 37.72 | 1100 | 8 |
| L | Mianyang, Sichuan | 105.09 | 32.22 | 778 | 20 |
| Sample | Raw Reads | Clean Reads | Clean Bases (bp) | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|
| Female 1 | 56000000 | 54951878 | 6.87G | 96.14 | 92.25 | 44.26 |
| Male 1 | 53506322 | 52712306 | 6.59G | 96.64 | 93.36 | 44.05 |
| Female 2 | 56000000 | 55262192 | 6.91G | 96.74 | 93.48 | 44.28 |
| Male 2 | 54024824 | 53089856 | 6.64G | 96.63 | 93.28 | 44.32 |
| Female 3 | 41215900 | 39528272 | 5.93G | 96.69 | 91.84 | 45.35 |
| Male3 | 46220380 | 44510796 | 6.68G | 96.59 | 91.64 | 44.63 |
| Primers | N | NA | NE | HE | HO | P |
|---|---|---|---|---|---|---|
| 5358 | 64 | 1.5 | 1.216 | 0.134 | 0.188 | 0.518 |
| 7309 | 64 | 1.833 | 1.528 | 0.294 | 0.208 | 0.021 * |
| 24177 | 64 | 1.667 | 1.236 | 0.173 | 0.164 | 0.235 |
| 67970 | 64 | 1.333 | 1.148 | 0.097 | 0.126 | 0.634 |
| 10335 | 64 | 1.5 | 1.108 | 0.087 | 0.033 | 0.000 *** |
| 73317 | 64 | 2 | 1.568 | 0.350 | 0.489 | 0.009 ** |
| 72763 | 64 | 1.5 | 1.32 | 0.189 | 0.208 | 0.033 * |
| 66538 | 64 | 2 | 1.169 | 0.112 | 0.045 | 0.000 *** |
| 60339 | 64 | 1.5 | 1.078 | 0.067 | 0.072 | 0.835 |
| 34533 | 64 | 1.667 | 1.494 | 0.262 | 0.413 | 0.049 * |
| 33255 | 64 | 1.333 | 1.314 | 0.161 | 0.158 | 0.000 *** |
| 10373 | 64 | 1.5 | 1.205 | 0.130 | 0.135 | 0.025 * |
| 11371 | 64 | 1.667 | 1.191 | 0.117 | 0.056 | 0.000 *** |
| 19808 | 64 | 1.833 | 1.583 | 0.339 | 0.492 | 0.008 ** |
| 7028 | 64 | 1.667 | 1.096 | 0.083 | 0.089 | 0.000 *** |
| 6545 | 64 | 3 | 2.404 | 0.455 | 0.345 | 0.072 |
| 7029 | 64 | 1.833 | 1.559 | 0.321 | 0.240 | 0.988 |
| Mean | 1.754 | 1.383 | 0.205 | 0.206 |
| Population | N | NA | NE | HE | HO |
|---|---|---|---|---|---|
| W | 9 | 1.789 | 1.450 | 0.231 | 0.231 |
| A | 10 | 1.632 | 1.310 | 0.189 | 0.256 |
| Y | 8 | 1.842 | 1.510 | 0.240 | 0.255 |
| Q | 9 | 1.842 | 1.436 | 0.221 | 0.187 |
| C | 8 | 1.579 | 1.292 | 0.168 | 0.197 |
| L | 20 | 1.842 | 1.298 | 0.181 | 0.112 |
| Mean | 1.754 | 1.383 | 0.205 | 0.206 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, D.; Jia, Y.; Yang, J.; Li, Z.-H. Comparative Transcriptome Analysis of Male and Female Conelets and Development of Microsatellite Markers in Pinus bungeana, an Endemic Conifer in China. Genes 2017, 8, 393. https://doi.org/10.3390/genes8120393
Duan D, Jia Y, Yang J, Li Z-H. Comparative Transcriptome Analysis of Male and Female Conelets and Development of Microsatellite Markers in Pinus bungeana, an Endemic Conifer in China. Genes. 2017; 8(12):393. https://doi.org/10.3390/genes8120393
Chicago/Turabian StyleDuan, Dong, Yun Jia, Jie Yang, and Zhong-Hu Li. 2017. "Comparative Transcriptome Analysis of Male and Female Conelets and Development of Microsatellite Markers in Pinus bungeana, an Endemic Conifer in China" Genes 8, no. 12: 393. https://doi.org/10.3390/genes8120393
APA StyleDuan, D., Jia, Y., Yang, J., & Li, Z.-H. (2017). Comparative Transcriptome Analysis of Male and Female Conelets and Development of Microsatellite Markers in Pinus bungeana, an Endemic Conifer in China. Genes, 8(12), 393. https://doi.org/10.3390/genes8120393