
Mitochondria and α-Synuclein: Friends or Foes in the Pathogenesis of Parkinson’s Disease?

 , , , ,
, , , , 
{kind=link}
Abstract
1. Introduction
2. Parkinson Disease α-Synuclein’s Pathology and Its Relation with Neuronal Degeneration
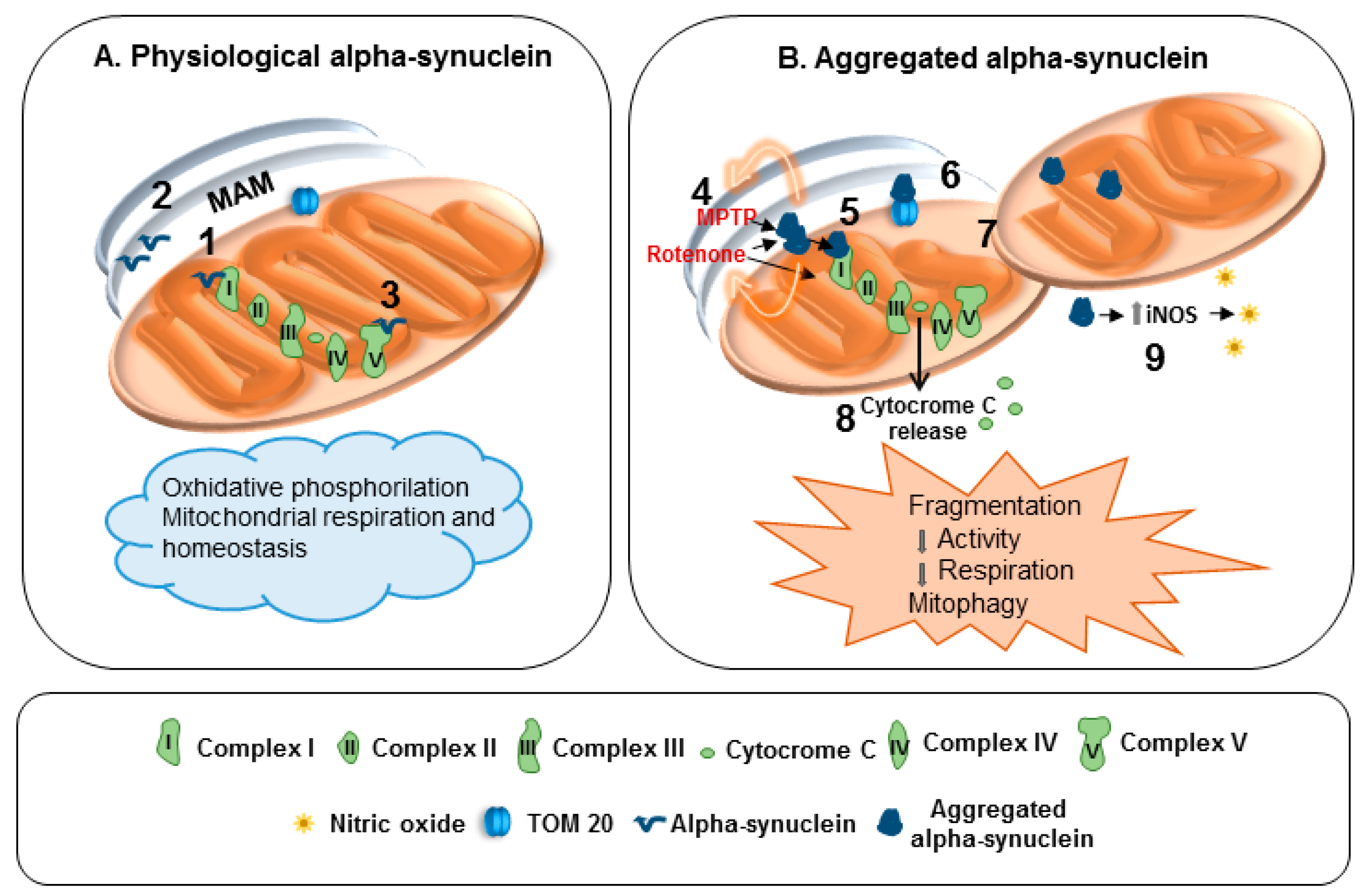
3. Mitochondria Alterations in Parkinson’s Disease
4. Mitochondria and α-Synuclein: Reciprocal Modulation
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Calo, L.; Wegrzynowicz, M.; Santivañez-Perez, J.; Grazia Spillantini, M. Synaptic failure and α-Synuclein. Mov. Disord. 2016, 31, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. The α-Synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann. N. Y. Acad. Sci. 2000, 920, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Dev, K.K.; Hofele, K.; Barbieri, S.; Buchman, V.L.; van der Putten, H. Part II: α-Synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology 2003, 45, 14–44. [Google Scholar] [CrossRef]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Missale, C.; Spano, P. α-Synuclein synaptic pathology and its implications in the development of novel therapeutic approaches to cure Parkinson’s disease. Brain Res. 2012, 1432, 95–113. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.Y.; Röyttä, M.; Rinne, J.O.; Collan, Y.; Rinne, U.K. Correlation between neuromorphometry in the substantia nigra and clinical features in Parkinson’s disease using disector counts. J. Neurol. Sci. 1997, 151, 83–87. [Google Scholar] [CrossRef]
- Bellucci, A.; Mercuri, N.B.; Venneri, A.; Faustini, G.; Longhena, F.; Pizzi, M.; Missale, C.; Spano, P. Review: Parkinson’s disease: From synaptic loss to connectome dysfunction. Neuropathol. Appl. Neurobiol. 2016, 42, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Antonini, A.; Pizzi, M.; Spano, P. The End Is the Beginning: Parkinson’s Disease in the Light of Brain Imaging. Front. Aging Neurosci. 2017, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Schaeffer, W.J. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Schon, E.A.; Przedborski, S. A new role for α-Synuclein in Parkinson’s disease: Alteration of ER-mitochondrial communication. Mov. Disord. 2015, 30, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [PubMed]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Halliday, G.M.; Simuni, T. Calcium, mitochondrial dysfunction and slowing the progression of Parkinson’s disease. Exp. Neurol. 2017, 298, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; Giguère, N.; Bourque, M.-J.; Lévesque, M.; Slack, R.S.; Trudeau, L.-É. Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R.; Xiromerisiou, G.; Singleton, A. How genetics research in Parkinson’s disease is enhancing understanding of the common idiopathic forms of the disease. Curr. Opin. Neurol. 2005, 18, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R.; Hardy, J.; Lewis, P.A. Genetic neuropathology of Parkinson’s disease. Int. J. Clin. Exp. Pathol. 2008, 1, 217–231. [Google Scholar] [PubMed]
- Kramer, M.L.; Behrens, C.; Schulz-Schaeffer, W.J. Selective detection, quantification, and subcellular location of α-Synuclein aggregates with a protein aggregate filtration assay. Biotechniques 2008, 44, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar] [PubMed]
- Tofaris, G.K.; Razzaq, A.; Ghetti, B.; Lilley, K.S.; Spillantini, M.G. Ubiquitination of α-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 2003, 278, 44405–44411. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Beach, T.G.; Hedreen, J.; Richfield, E.K. Critical role of truncated α-synuclein and aggregates in Parkinson’s disease and incidental Lewy body disease. Brain Pathol. 2012, 22, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Crowther, R.A.; Jakes, R.; Spillantini, M.G.; Goedert, M. Synthetic filaments assembled from C-terminally truncated α-synuclein. FEBS Lett. 1998, 436, 309–312. [Google Scholar] [CrossRef]
- Garcia-Reitbock, P.; Anichtchik, O.; Bellucci, A.; Iovino, M.; Ballini, C.; Fineberg, E.; Ghetti, B.; Della Corte, L.; Spano, P.; Tofaris, G.K.; et al. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain 2010, 133 Pt 7, 2032–2044. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Navarria, L.; Falarti, E.; Zaltieri, M.; Bono, F.; Collo, G.; Grazia, M.; Missale, C.; Spano, P. Redistribution of DAT/α-synuclein complexes visualized by “in situ” proximity ligation assay in transgenic mice modelling early Parkinson’s disease. PLoS ONE 2011, 6, e27959. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Ikeda, T.; Takasaki, J.; Yamada, M. Familial Parkinson disease mutations influence α-synuclein assembly. Neurobiol. Dis. 2011, 43, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Olgiati, S.; Thomas, A.; Quadri, M.; Breedveld, G.J.; Graafland, J.; Eussen, H.; Douben, H.; de Klein, A.; Onofrj, M.; Bonifati, V. Early-onset parkinsonism caused by α-synuclein gene triplication: Clinical and genetic findings in a novel family. Parkinsonism Relat. Disord. 2015, 21, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-Synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Gao, F.; Yang, J.; Wang, D.; Li, C.; Fu, Y.; Wang, H.; He, W.; Zhang, J. Mitophagy in Parkinson’s disease: Pathogenic and therapeutic implications. Front. Neurol. 2017, 8, 527. [Google Scholar] [CrossRef] [PubMed]
- Pozo Devoto, V.M.; Falzone, T.L. Mitochondrial dynamics in Parkinson’s disease: A role for α-synuclein? Dis. Model Mech. 2017, 10, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Kirik, D.; Rosenblad, C.; Burger, C.; Lundberg, C.; Johansen, T.E.; Muzyczka, N.; Mandel, R.J.; Björklund, A. Parkinson-like neurodegeneration induced by targeted overexpression of α-synuclein in the nigrostriatal system. J. Neurosci. 2002, 22, 2780–2791. [Google Scholar] [PubMed]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.; Yang, S.; Sauchanka, O.; Spillantini, M.G.; Anichtchik, O. Behavioural deficits in transgenic mice expressing human truncated (1–120 amino acid) α-synuclein. Exp. Neurol. 2015, 264, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Matsumoto, K.; Takayama, K.; Yoshimoto, M.; Takahashi, H. NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson’s disease. Neurosci. Lett. 1997, 239, 45–48. [Google Scholar] [CrossRef]
- Braak, H.; Sastre, M.; Bohl, J.R.; de Vos, R.A.; Del Tredici, K. Parkinson’s disease: Lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol. 2007, 113, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Cersosimo, M.G.; Raina, G.B.; Pecci, C.; Pellene, A.; Calandra, C.R.; Gutiérrez, C.; Micheli, F.E.; Benarroch, E.E. Gastrointestinal manifestations in Parkinson’s disease: Prevalence and occurrence before motor symptoms. J. Neurol. 2013, 260, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Sumikura, H.; Takao, M.; Hatsuta, H.; Ito, S.; Nakano, Y.; Uchino, A.; Nogami, A.; Saito, Y.; Mochizuki, H.; Murayama, S. Distribution of α-synuclein in the spinal cord and dorsal root ganglia in an autopsy cohort of elderly persons. Acta Neuropathol. Commun. 2015, 3, 57. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H. Parkinson’s disease and aging: Same or different process? Mov. Disord. 2008, 23, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Engelender, S.; Isacson, O. The Threshold Theory for Parkinson’s Disease. Trends Neurosci. 2017, 40, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Parkinson’s disease is not simply a prion disorder. J. Neurosci. 2017, 37, 9799–9807. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.A.; Gardner, K.; Chinnery, P.F. Mitochondrial DNA mutations in ageing and disease: Implications for HIV? Antivir. Ther. 2015, 20, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I.; Cuervo, A.M. Proteostasis and the aging proteome in health and disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S33–S38. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef] [PubMed]
- Bindoff, L.A.; Birch-Machin, M.; Cartlidge, N.E.; Parker, W.D., Jr.; Turnbull, D.M. Mitochondrial function in Parkinson’s disease. Lancet 1989, 2, 49. [Google Scholar]
- Blin, O.; Desnuelle, C.; Rascol, O.; Borg, M.; Peyro Saint Paul, H.; Azulay, J.P.; Billé, F.; Figarella, D.; Coulom, F.; Pellissier, J.F. Mitochondrial respiratory failure in skeletal muscle from patients with Parkinson’s disease and multiple system atrophy. J. Neurol. Sci. 1994, 125, 95–101. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Boyson, S.J.; Parks, J.K. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.H.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R.; Shults, C.W. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann. Neurol. 1995, 37, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Mytilineou, C.; Werner, P.; Molinari, S.; Di Rocco, A.; Cohen, G.; Yahr, M.D. Impaired oxidative decarboxylation of pyruvate in fibroblasts from patients with Parkinson’s disease. J. Neural. Transm. Park. Dis. Dement. Sect. 1994, 8, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Ohta, S.; Tanaka, M.; Takamiya, S.; Suzuki, K.; Sato, T.; Oya, H.; Ozawa, T.; Kagawa, Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res. Commun. 1989, 163, 1450–1455. [Google Scholar] [CrossRef]
- Autere, J.; Moilanen, J.S.; Finnilä, S.; Soininen, H.; Mannermaa, A.; Hartikainen, P.; Hallikainen, M.; Majamaa, K. Mitochondrial DNA polymorphisms as risk factors for Parkinson’s disease and Parkinson’s disease dementia. Hum. Genet. 2004, 115, 29–35. [Google Scholar] [PubMed]
- Luoma, P.; Melberg, A.; Rinne, J.O.; Kaukonen, J.A.; Nupponen, N.N.; Chalmers, R.M.; Oldfors, A.; Rautakorpi, I.; Peltonen, L.; Majamaa, K.; et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet 2004, 364, 875–882. [Google Scholar] [CrossRef]
- Sterky, F.H.; Hoffman, A.F.; Milenkovic, D.; Bao, B.; Paganelli, A.; Edgar, D.; Wibom, R.; Lupica, C.R.; Olson, L.; Larsson, N.G. Altered dopamine metabolism and increased vulnerability to MPTP in mice with partial deficiency of mitochondrial complex I in dopamine neurons. Hum. Mol. Genet. 2012, 21, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A. Monogenic Parkinson’s disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat. Disord. 2013, 19, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. α-Synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Pozo Devoto, V.M.; Dimopoulos, N.; Alloatti, M.; Pardi, M.B.; Saez, T.M.; Otero, M.G.; Cromberg, L.E.; Marín-Burgin, A.; Scassa, M.E.; Stokin, G.B.; et al. α-Synuclein control of mitochondrial homeostasis in human-derived neurons is disrupted by mutations associated with Parkinson’s disease. Sci. Rep. 2017, 7, 5042. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 216–231. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.S.; Parihar, A.; Fujita, M.; Hashimoto, M.; Ghafourifar, P. Mitochondrial association of α-synuclein causes oxidative stress. Cell. Mol. Life Sci. 2008, 65, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Tapias, V.; Hu, X.; Luk, K.C.; Sanders, L.H.; Lee, V.M.; Greenamyre, J.T. Synthetic α-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell. Mol. Life Sci. 2017, 74, 2851–2874. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, R.E.; King, A.E.; Dickson, T.C. α-Synuclein protects neurons from apoptosis downstream of free-radical production through modulation of the MAPK signalling pathway. Neurotox. Res. 2013, 23, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, A.; Moisan, F. Update in the epidemiology of Parkinson’s disease. Curr. Opin. Neurol. 2008, 21, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef] [PubMed]
- Nistico, R.; Mehdawy, B.; Piccirilli, S.; Mercuri, N. Paraquat- and rotenone-induced models of Parkinson’s disease. Int. J. Immunopathol. Pharmacol. 2011, 24, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Lewis, V.; Blesa, J.; Przedborski, S. Animal models of Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. 1), S183–S185. [Google Scholar] [CrossRef]
- Maturana, M.G.; Pinheiro, A.S.; de Souza, T.L.; Follmer, C. Unveiling the role of the pesticides paraquat and rotenone on α-synuclein fibrillation in vitro. Neurotoxicology 2015, 46, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.; Gil-Bea, F.J.; Dalfó, E.; Cuadrado, M.; Cabodevilla, F.; Sánchez, B.; Catena, S.; Sesma, T.; Ribé, E.; Ferrer, I.; et al. Increased sensitivity to MPTP in human α-synuclein A30P transgenic mice. Neurobiol. Aging 2006, 27, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Klivenyi, P.; Siwek, D.; Gardian, G.; Yang, L.; Starkov, A.; Cleren, C.; Ferrante, R.J.; Kowall, N.W.; Abeliovich, A.; Beal, M.F. Mice lacking α-synuclein are resistant to mitochondrial toxins. Neurobiol. Dis. 2006, 21, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Kholodilov, N.; Vila, M.; Trillat, A.C.; Goodchild, R.; Larsen, K.E.; Staal, R.; Tieu, K.; Schmitz, Y.; Yuan, C.A. Resistance of α-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. USA 2002, 99, 14524–14529. [Google Scholar] [CrossRef] [PubMed]
- Fountaine, T.M.; Wade-Martins, R. RNA interference-mediated knockdown of α-synuclein protects human dopaminergic neuroblastoma cells from MPP(+) toxicity and reduces dopamine transport. J. Neurosci. Res. 2007, 85, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.E.; Murphy, E.J.; Mitchell, D.C.; Golovko, M.Y.; Scaglia, F.; Barceló-Coblijn, G.C.; Nussbaum, R.L. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking α-synuclein. Mol. Cell. Biol. 2005, 25, 10190–10201. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric alpha-synuclein exerts a physiological role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faustini, G.; Bono, F.; Valerio, A.; Pizzi, M.; Spano, P.; Bellucci, A. Mitochondria and α-Synuclein: Friends or Foes in the Pathogenesis of Parkinson’s Disease? Genes 2017, 8, 377. https://doi.org/10.3390/genes8120377
Faustini G, Bono F, Valerio A, Pizzi M, Spano P, Bellucci A. Mitochondria and α-Synuclein: Friends or Foes in the Pathogenesis of Parkinson’s Disease? Genes. 2017; 8(12):377. https://doi.org/10.3390/genes8120377
Chicago/Turabian StyleFaustini, Gaia, Federica Bono, Alessandra Valerio, Marina Pizzi, PierFranco Spano, and Arianna Bellucci. 2017. "Mitochondria and α-Synuclein: Friends or Foes in the Pathogenesis of Parkinson’s Disease?" Genes 8, no. 12: 377. https://doi.org/10.3390/genes8120377
APA StyleFaustini, G., Bono, F., Valerio, A., Pizzi, M., Spano, P., & Bellucci, A. (2017). Mitochondria and α-Synuclein: Friends or Foes in the Pathogenesis of Parkinson’s Disease? Genes, 8(12), 377. https://doi.org/10.3390/genes8120377